

ABSTRACT
Tryptophan (TRP) metabolism through the kynurenine pathway generates multiple biologically active metabolites with diverse immunomodulatory effects, but their roles in glomerulonephritis (GN), particularly in innate immunity, remain poorly understood. Using a nephrotoxic serum‐induced GN (NTS‐GN) model, we first analyzed mice deficient in key TRP‐metabolizing enzymes of the kynurenine pathway: Indoleamine 2,3‐dioxygenase 1 and 2 (IDO1 and IDO2), and kynurenine 3‐monooxygenase (KMO), and found that Ido1‐deficient mice exhibited exacerbated kidney injury and glomerular neutrophil infiltration, whereas Ido2 deficiency had no significant impact. In contrast, Kmo‐deficient mice showed reduced crescent formation. Unexpectedly, the concentration of kynurenic acid (KYNA), a downstream metabolite of IDO1, was elevated in the kidney cortex of Ido1‐deficient mice. Exogenous KYNA administration improved survival, ameliorated renal injury, and reduced neutrophil infiltration in Ido1‐deficient mice, indicating its protective effect against antibody‐mediated injury. Moreover, KYNA suppressed immune complex‐mediated neutrophil spreading, attenuated FcγR–dependent Syk phosphorylation, and reduced VEGF secretion in vitro. Our results position KYNA as a key modulator of neutrophil‐driven inflammation in antibody‐mediated GN. This study uncovers distinct roles for kynurenine pathway enzymes and highlights the TRP–KYNA pathway as a promising immunometabolic target for controlling innate immune responses in GN.
Keywords: glomerulonephritis; Indoleamine 2,3‐dioxygenase; kynurenic acid; neutrophils; RRID:AB_2337118; RRID:IMSR_JAX:005867; RRID:MGI:2159965; RRID:MGI:3028467; RRID:MGI:5759308; tryptophan
The graphical abstract illustrates that Ido1 deficiency exacerbates antibody‐mediated glomerulonephritis through enhanced neutrophil activation, whereas kynurenic acid suppresses neutrophil spreading, FcγR–Syk signaling, and VEGF release, thereby protecting against glomerular injury.

1. Introduction
Tryptophan (TRP) is an essential amino acid, with over 95% of it metabolized via the kynurenine pathway, which is the sole de novo pathway for the synthesis of nicotinamide adenine dinucleotide (NAD) in the body (Figure 1) [1]. Indoleamine 2,3‐dioxygenase (IDO), which has two isoforms—IDO1 and IDO2—serves as the rate‐limiting enzyme in the kynurenine pathway [2, 3]. Recent research has increasingly highlighted the complex relationship between the kynurenine pathway and immune function, particularly with the discovery of IDO's role in promoting maternal immune tolerance during pregnancy [4]. Subsequent studies have revealed the immunomodulatory properties of IDO and the kynurenine pathway, such as the regulation of T cell activation by IDO‐expressing antigen‐presenting cells through tryptophan catabolism [5, 6, 7], the alteration of macrophage immune function in aging and inflammation due to reduced cell‐intrinsic NAD production [8], and immune evasion by malignant tumors via IDO1 expression [9]. In contrast, IDO2 is thought to play a crucial role in B cells, facilitating the development of autoimmunity [10].
FIGURE 1.

Kynurenine pathway.
Focusing on the relationship between TRP metabolism and kidney disease, it has been observed that the deficiency in kynurenine 3‐monooxygenase (KMO), one of the enzymes in the kynurenine pathway, protects mice against acute kidney injury (AKI) in an ischemia–reperfusion injury model [11]. Additionally, quinolinate phosphoribosyl transferase (QPRT), the terminal enzyme in the de novo NAD biosynthesis in the kynurenine pathway, is crucial for preserving renal NAD levels, which contribute to resistance against AKI [12]. However, the understanding of TRP metabolism in the context of immune‐mediated glomerulonephritis remains limited, with only a few scattered reports addressing this area. Hou et al. reported that 1‐methyl‐tryptophan, a non‐selective IDO inhibitor, exacerbated kidney function, corroborated with histopathological changes in a mouse model of nephrotoxic serum glomerulonephritis (NTS‐GN) [13], but the individual roles of IDO1 versus IDO2 and the contributions of downstream metabolites in the kynurenic pathway remain unclear. Additionally, little is known about how TRP metabolism affects neutrophils—key effectors in the early phase of antibody‐mediated GN [14, 15, 16, 17]. This represents a critical gap in our understanding. To address this gap, the present study aims to clarify the distinct contributions of key kynurenine pathway enzymes and their downstream metabolites to the development of antibody‐mediated glomerulonephritis by leveraging a series of knockout mouse strains in a nephrotoxic serum‐induced glomerulonephritis model.
2. Materials and Methods
2.1. Animals
All mice were mature males aged 8 weeks in a specific pathogen‐free environment. Ido1‐deficient mice on a C57BL/6J background were obtained from Jackson Laboratory (RRID:IMSR_JAX:005867, Bar Harbor, ME, USA), and Ido2‐ and Kmo‐deficient mice on a C57BL/6N background were obtained from the Knockout Mouse Project (RRID:IMSR_EUMMCR:20750 for Ido2‐deficient and RRID:MGI:5759308 for Kmo‐deficient mice, KOMP, CA, USA) repository. Kynurenine aminotransferase 2 (Kat2)‐deficient mice, generated by deleting a 37 234 bp genomic region between Exon 2 (chr8:60507258) and the intron between Exons 12 and 13 (chr8:60544492) using CRISPR/Cas9 on a C57BL/6N background, were generated and maintained in our facility. We purchased WT C57BL/6J (RRID:MGI:3028467) and C57BL/6N (RRID:MGI:2159965) mice from Charles River Laboratories (Yokohama, Japan). Mice were randomly assigned to experimental groups. Histological assessments were performed in a blinded manner. All animal experiments were approved by the Institutional Animal Care and Use Committee of Fujita Health University (Approval No. AP19052) and conducted in accordance with the Guidelines for Proper Conduct of Animal Experiments of the Science Council of Japan and the ARRIVE guidelines.
2.2. Experimental Mouse Model of Antibody‐Mediated Glomerulonephritis
Rabbit anti‐mouse GBM serum was generated following established protocols [18]. Glomeruli from the renal cortex of C57BL/6 mice were isolated using a series of sieves with progressively smaller pore sizes (150‐, 106‐, and 45‐μm mesh) and subsequently disrupted by sonication. GBM fractions were collected via centrifugation. For immunization, GBM protein was emulsified with TiterMax Gold Adjuvant (Cat# T2684, Sigma‐Aldrich Co. LLC, St. Louis, MO, USA), and the subcutaneous administration to New Zealand white rabbits, as well as the production of anti‐mouse GBM serum, were performed by a commercial vendor (Scrum Inc.), using glomerular sonicates prepared at our institution. To induce NTS‐GN, mice were pre‐immunized with rabbit IgG (RRID: AB_2337118, Jackson ImmunoResearch) and Complete Freund's Adjuvant (Cat# 7001, Chondrex Inc.) on day −3, followed by intravenous administration of rabbit anti‐mouse glomerular basement membrane serum. Kynurenic acid (KYNA, Cat# K3375, Sigma‐Aldrich Co. LLC, St. Louis, MO, USA) was administered intraperitoneally at a dose of 10 μg/g every other day, starting on the day of NTS administration and continuing until sacrifice.
2.3. Functional and Histological Assessment of Diseased Kidney
Urine samples were collected using metabolic cages prior to sacrifice, and serum was obtained from the inferior vena cava at the time of sacrifice. The analysis of urinary albumin, urinary creatinine, and serum creatinine was outsourced to Oriental Kobo Co. Ltd. Glomerular crescent formation was evaluated on 3‐μm paraffin sections stained with periodic acid‐Schiff (PAS) reagent. Crescents were defined as glomeruli showing two or more layers of cells within Bowman's space occupying more than 10% of the circumference. The percentage of crescent formation was calculated by examining all glomeruli in each kidney section. The deposit index was determined by assessing the proportion of PAS‐positive area in the glomerular capillary loops within each section. For each glomerulus, a score of 0 was assigned if no deposits were observed; 1 for deposits covering up to 25% of the capillary loops; 2 for 25%–50%; 3 for 50%–75%; and 4 for 75%–100%, using a previously reported method with slight modifications [19]. The average score of all glomeruli within the section was calculated to determine the deposit index for that tissue. Neutrophil infiltration into glomeruli was evaluated on 3 μm paraffin sections using esterase staining, and the number of positively stained cells per glomerulus was counted and compared among groups.
2.4. Immune Complex‐Mediated Neutrophil Activation
Immune complexes (ICs) were formed by adding anti‐BSA antibodies (Sigma‐Aldrich Co. LLC, St. Louis, MO, USA) to cell culture plates pre‐coated with bovine serum albumin (BSA, Cat# B2901, RRID:AB_258533, Sigma‐Aldrich Co. LLC, St. Louis, MO, USA) [20]. Bone marrow‐derived neutrophils from WT and each TRP‐metabolizing enzyme‐deficient mouse were then seeded onto these plates. Neutrophil morphological changes were monitored from 5 to 60 min. The effect of KYNA was assessed in Ido1‐deficient neutrophils cultured with or without KYNA (45 μM), with WT neutrophils included as controls. Neutrophils were stained with DAPI (Cat# D1306, Sigma‐Aldrich Co. LLC, St. Louis, MO, USA) for nuclei staining and Rhodamine Phalloidin (Cat# R415, Thermo Fisher Scientific, Waltham, MA, USA) for actin staining. Using the Opera Phenix high‐content screening system (PerkinElmer, Waltham, MA, USA), the nucleus and cytoplasm of neutrophils were automatically recognized (Figure S1). Neutrophils were classified based on the difference in signal intensity of Rhodamine Phalloidin between the nucleus and the perinuclear region. We evaluated numbers and areas of the cells exhibiting symmetric actin staining as “spread cells” and those that failed to meet the criteria as “round cells.” When the neutrophils were activated by ICs, the polarization and rearrangement of the actin cytoskeleton altered over time.
For cytokine measurement, culture supernatants from neutrophils on IC‐coated plates were collected at 0, 6, and 12 h. Cytokine levels were analyzed using the Mouse Cytokine Array Q1 (Quantibody Mouse Cytokine Array 1 Kit; Cat# QAM‐CYT‐1) via Full Testing Service by RayBiotech Inc. (Peachtree Corners, GA, USA).
Western blot analysis for Fcγ receptor–dependent signaling was performed as previously reported [21]: neutrophils were pretreated with anti‐CD16/32 antibody (clone 93, eBioscience, Invitrogen, Cat# 15246827) on ice for 30 min, followed by warming at 37°C for 20 min in the presence or absence of KYNA. FcγR cross‐linking was induced by adding AffiniPure Goat Anti‐Rat IgG, F(ab′)2 fragment specific antibody (Jackson ImmunoResearch, Cat# 112‐005‐072). Cells were lysed at 0, 30, and 60 s after stimulation, and the cleared lysates were subjected to SDS–PAGE and immunoblotting. Membranes were probed with anti–phospho‐Syk (Tyr525/526) antibody (Cell Signaling Technology, Cat# 2711), total Syk antibody (Syk [D3Z1E] XP Rabbit mAb, Cell Signaling Technology, Cat# 13198, RRID:AB_2687924), and HRP‐conjugated anti‐rabbit IgG (Cell Signaling Technology, Cat# 7074). Band intensities were quantified from replicate experiments. All experiments were performed with the same number of neutrophils (5 × 106 cells).
2.5. Measurement of the Concentration of TRP Metabolites
The metabolites in TRP–KYN metabolism were measured as described previously [22]. For TRP, KYN, and KYNA, kidney cortex was weighed and homogenized (1:3, w/v) in 10% perchloric acid (Cat# 311413, Wako Pure Chemical Co., Osaka, Japan) by ultrasonic homogenizer (MICROTEC Co., Chiba, Japan). After mixing, the precipitated proteins were removed by centrifugation (15 300 g × 15 min, 4°C). The supernatants (50 μL) were injected into a high‐performance liquid chromatography system (HPLC; SHIMADZU Co., Kyoto, Japan).
2.6. Statistical Analysis
No data were excluded from analysis. Sample sizes were determined empirically based on prior studies of similar models. Wild‐type (WT) mice were included in larger numbers than knockout strains to serve as a shared control group across multiple experimental comparisons, in order to reduce total animal use and enhance statistical power.
Group comparisons for kidney function, histological parameters, and metabolite concentrations were performed using the Mann–Whitney U test. When multiple comparisons were conducted within a single set of measurements (e.g., across more than two genotypes), p‐values were adjusted using the Benjamini–Hochberg method. For survival analysis, Kaplan–Meier curves were generated and compared using the log‐rank test. Time‐dependent morphological changes, cytokine production in neutrophils, and Western blot band intensities were analyzed using one‐way analysis of variance (ANOVA) at each time point, followed by Tukey's honestly significant difference (HSD) test for post hoc pairwise comparisons. For each mouse in the neutrophil experiments, values were averaged across five randomly selected microscopic fields per time point and used as one biological replicate. All statistical analyses were performed using Python version 3.11.9 (Python Software Foundation, https://www.python.org/) [23]. Two‐sided p‐values < 0.05 were considered statistically significant. Exact sample sizes and p‐values are provided in figure legends and Supporting Information Tables.
3. Results
3.1. Deficiency of Ido1 and Kmo Alters Kidney Injury Severity in NTS‐GN
To evaluate disease severity in NTS‐GN (Figure 2A), we assessed kidney function and histopathological changes in mice deficient in tryptophan‐metabolizing enzymes IDO1, IDO2, and KMO (Figure 2B). Serum creatinine and urinary albumin‐to‐creatinine ratio (UACR) were measured on days 0, 7, and 14, accompanied by histological assessment (Figure 2C–F). Ido1‐deficient mice exhibited significantly worsened kidney dysfunction compared to wild‐type (WT) mice at both time points, with elevated serum creatinine and UACR (e.g., p = 0.0048 and p = 0.0057 on day 14). These mice also showed a marked increase in glomerular crescent formation and PAS‐positive glomerular deposits. In contrast, Ido2‐deficient mice displayed no significant differences from WT in either kidney function or histological parameters. Kmo‐deficient mice exhibited a significant reduction in glomerular crescent formation on day 14 (p = 0.0022), while other parameters remained like WT. The intensity of glomerular antibody deposition did not differ among WT, Ido1‐, Ido2‐, and Kmo‐deficient mice (Figure S2A,B).
FIGURE 2.

Tryptophan metabolism and renal injury in enzyme‐deficient mice with NTS‐GN. (A) Experimental timeline and protocol for NTS‐GN induction. Mice were pre‐immunized with rabbit IgG and Complete Freund's Adjuvant on day −3, followed by intravenous injection of nephrotoxic serum on day 0, and were sacrificed on days 7 and 14 for sample collection. (B) Representative PAS‐stained kidney sections on day 14 after NTS‐GN induction in Ido1‐deficient, Ido2‐deficient, Kmo‐deficient, and WT mice. Red arrowheads indicate glomerular crescent formation, and yellow arrows indicate PAS‐positive deposits. Scale bars, 100 μm. (C–F) Kidney function and histological parameters on days 0, 7, and 14 after NTS‐GN induction: (C) serum creatinine, (D) urinary albumin‐to‐creatinine ratio (UACR), (E) crescent formation rate, (F) PAS‐positive glomerular deposit scores. On day 14, Ido1‐deficient mice showed significantly elevated serum creatinine (p = 0.0048), UACR (p = 0.0057), crescent formation (p = 0.0022), and glomerular deposit scores (p = 0.0045) compared with WT mice (n = 8–17 per group). Group comparisons were performed using the Mann–Whitney U test with Benjamini–Hochberg correction. See also Table S1 for complete statistical results. KAT2, kynurenine aminotransferase 2; KMO, kynurenine 3‐monooxygenase; KYNU, kynureninase; TDO, tryptophan 2,3‐dioxygenase; QPRT, quinolinic acid phosphoribosyl transferase; 3‐HAO, 3‐hydroxyanthranilate 3,4‐dioxygenase.
3.2. Deficiency of TRP‐Metabolizing Enzymes Affects Glomerular Neutrophil Infiltration and Morphological Changes in NTS‐GN
Disruption of the glomerular basement membrane and crescent formation are characteristic features of antibody‐mediated glomerular injury. However, the earliest phase of this process is driven by neutrophil activation via Fc receptors and integrins, followed by adhesion to glomerular endothelial cells. Because neutrophils express the aryl hydrocarbon receptor (AhR), a ligand‐activated transcription factor responsive to TRP‐derived metabolites, we hypothesized that TRP metabolism may influence early neutrophil‐mediated glomerular inflammation. Neutrophil infiltration into glomeruli, evaluated by esterase staining (Figure 3A), was significantly higher in Ido1‐deficient mice than in WT on day 7 (Figure 3B). In contrast, Ido2‐deficient mice showed no significant differences from WT on either day 7 or day 14. Although not statistically significant, Kmo‐deficient mice exhibited a trend toward reduced neutrophil infiltration at both time points compared to WT. Next, we evaluated the chronological morphological activation of neutrophils stimulated by ICs using the Opera Phenix high‐content imaging system. Neutrophils isolated from Ido1‐deficient, Kmo‐deficient, and WT mice were plated on IC‐coated surfaces and observed over time (Figure 3C). Neutrophils from Ido1‐deficient mice exhibited earlier activation and sustained morphological spreading compared to those from WT and Kmo‐deficient mice. At 60 min, the spread cell area was also significantly greater in Ido1‐deficient neutrophils compared to both WT and Kmo‐deficient cells (Figure 3D, p = 0.0070 and p = 0.0025, respectively). At 30 and 60 min, the percentage of spread‐form neutrophils was significantly higher in Ido1‐deficient mice than in WT (Figure 3E, p = 0.018 and p = 0.021, respectively).
FIGURE 3.
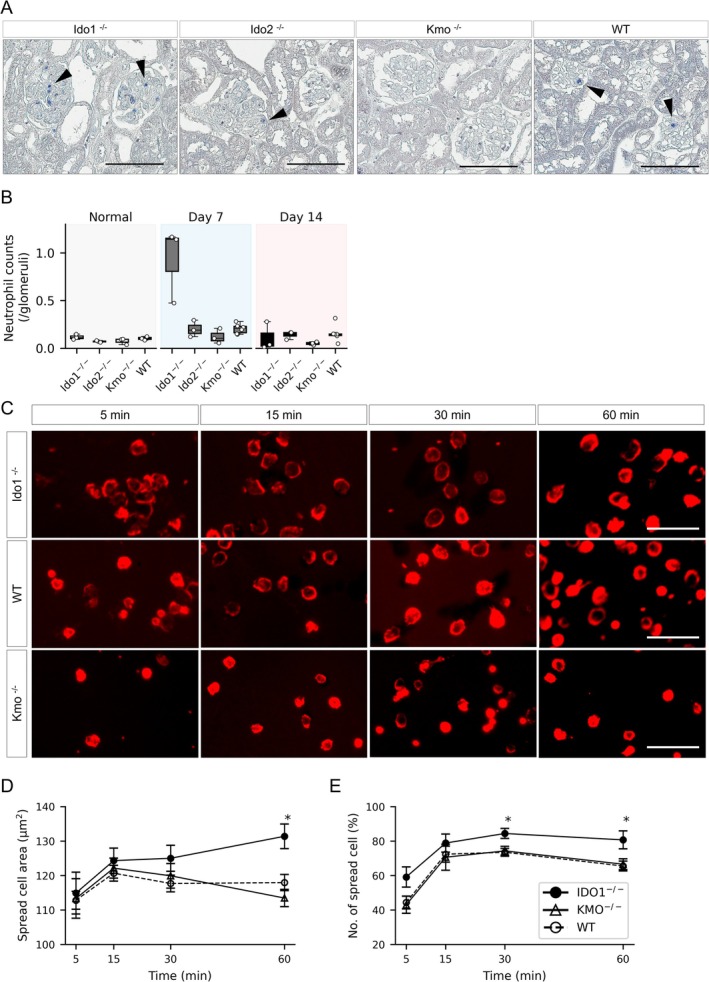
Glomerular neutrophil infiltration and time‐dependent morphological changes in enzyme‐deficient mice with NTS‐GN. (A) Representative esterase‐stained kidney sections on day 7 after NTS‐GN induction in Ido1‐deficient, Ido2‐deficient, Kmo‐deficient, and WT mice. Blue‐stained cells indicate neutrophil infiltration within glomeruli. Scale bars: 100 μm. (B) Quantification of glomerular neutrophil infiltration on days 0, 7, and 14. Neutrophil counts were significantly increased in Ido1‐deficient mice on day 7 compared with WT (p = 0.024, Mann–Whitney U test). No significant differences were observed in Ido2‐ or Kmo‐deficient mice at either time point. n = 3 per knockout group (Ido1, Ido2, Kmo); n = 6 for WT at days 7 and 14. n = 3 in each group at day 0. (C) Time‐course images of neutrophil morphological changes following immune complex stimulation. Bone marrow‐derived neutrophils from Ido1‐deficient (top row), WT (middle row), and Kmo‐deficient (bottom row) mice were seeded onto plates coated with BSA/anti‐BSA immune complexes and stained with rhodamine phalloidin to visualize F‐actin. Images were captured at 5, 15, 30, and 60 min after stimulation (left to right). (D,E) Quantification of neutrophil morphological changes over time in Ido1‐deficient (n = 6), Kmo‐deficient (n = 5), and WT (n = 10) mice. The percentage of spread‐form cells was significantly higher in Ido1‐deficient mice than in WT at 30 and 60 min (p = 0.018 and 0.021, respectively). At 60 min, the spread area was significantly larger in Ido1‐deficient neutrophils than in both WT and Kmo‐deficient neutrophils (p = 0.0070 and 0.0025, respectively). Left: Spread area (μm2); right: Percentage of spread‐form neutrophils (%). Asterisks (*) indicate statistically significant differences (p < 0.05, respectively).
3.3. Altered Levels of Kynurenine Pathway Metabolites in the Kidney Cortex of TRP‐Enzyme‐Deficient Mice
We analyzed the concentrations of TRP, KYN, kynurenic acid (KYNA), and 3‐hydroxyanthranilic acid (3HAA) in the kidney cortex of NTS‐GN mice. TRP levels increased after disease induction in all groups; however, there were no significant differences among Ido1‐, Ido2, Kmo‐deficient, and WT mice under either normal or nephritic conditions (Figure 4A). In Ido1‐deficient mice prior to NTS‐GN induction, KYN levels were significantly lower (p = 0.0086), and the KYN/TRP ratio was also reduced (p = 0.0040) compared to WT. After nephritis induction, KYN gradually increased and was comparable to WT on day 14 (Figure 4C,D). KYNA levels were similar between Ido1‐deficient and WT mice under normal conditions (p = 0.88). However, on day 7, KYNA levels were significantly elevated in Ido1‐deficient mice compared to WT (p < 0.001), despite the absence of functional IDO1 (Figure 4B). On day 14, KYNA levels in Ido1‐deficient mice remained higher than those in WT, although the difference did not reach statistical significance (p = 0.058). Notably, the KYNA/KYN ratio progressively increased in Ido1‐deficient mice, whereas the 3HAA/KYN ratio did not show a corresponding rise (Figure 4E,F). These data indicate that TRP metabolism was preferentially shifted toward KYNA rather than the 3HAA branch of the pathway. In Ido2‐deficient mice, KYN levels and the KYN/TRP ratio were comparable to those in WT under both normal and nephritic conditions, further supporting that IDO1 is the dominant enzyme driving TRP metabolism in this setting. Both KYN and KYNA levels were markedly elevated in Kmo‐deficient mice under both conditions.
FIGURE 4.

Concentrations of tryptophan and its metabolites in the kidney cortex. (A–F) Box plots showing (A) tryptophan (TRP), (B) kynurenic acid (KYNA), (C) kynurenine (KYN), (D) the kynurenine‐to‐tryptophan ratio (KYN/TRP), (E) KYNA to KYN ratio (KYNA/KYN), and (F) 3‐hydroxyanthranilic acid (3HAA) to KYN ratio in Ido1‐deficient, Kmo‐deficient, and WT mice under normal conditions and on days 0, 7, and 14 after NTS‐GN induction. TRP levels increased after disease induction in all groups, with no significant differences between genotypes. Ido1‐deficient mice exhibited reduced KYN and KYN/TRP at baseline, and a significant elevation of KYNA on day 7 despite lacking functional IDO1. KYNA levels in Ido1‐deficient mice remained higher than WT on day 14, although the difference was not statistically significant. The KYNA/KYN ratio (E) progressively increased in Ido1‐deficient mice, whereas the 3HAA/KYN ratio (F) did not show a corresponding rise. Kmo‐deficient mice consistently showed markedly elevated KYN, KYNA, and KYN/TRP levels. Statistical comparisons were performed using the Mann–Whitney U test with Benjamini–Hochberg correction (n = 6–20 per group). See also Table S2 for full results.
3.4. Exogenous KYNA Administration Improves Survival and Kidney Injury in Ido1‐Deficient NTS‐GN Mice
Given the unexpected elevation of KYNA in Ido1‐deficient mice during the early inflammatory phase, we next examined whether exogenous KYNA could exert a protective effect in antibody‐mediated kidney injury. KYNA or phosphate‐buffered saline (PBS) was administered intraperitoneally into Ido1‐deficient NTS‐GN mice every other day throughout the observation period (Figure 5A). All mice in the PBS‐treated group died by day 14, whereas KYNA administration significantly improved survival (log‐rank test, p = 0.015; Figure 5B). Consistent with this protective effect, glomerular infiltration of neutrophils was significantly reduced in KYNA‐treated mice compared with PBS‐treated controls on both day 4 and 7 (Figure 5C, p = 0.029 and p = 0.032, respectively). KYNA‐treated mice showed significantly improved kidney function on day 7, with lower serum creatinine (p = 0.046) and UACR (p = 0.0028) compared to PBS‐treated controls (Figure 5D). KYNA also reduced glomerular crescent formation (p < 0.001). In the kidney cortex, KYNA concentration was significantly increased in the KYNA‐treated group (Figure 5D). Additionally, the KYN/TRP ratio was higher in the KYNA group, indicating enhanced TRP‐to‐KYN conversion and suggesting that KYNA treatment alters local tryptophan metabolism. Antibody deposition in glomeruli was comparable between PBS‐ and KYNA‐treated Ido1‐deficient mice (Figure S2C).
FIGURE 5.
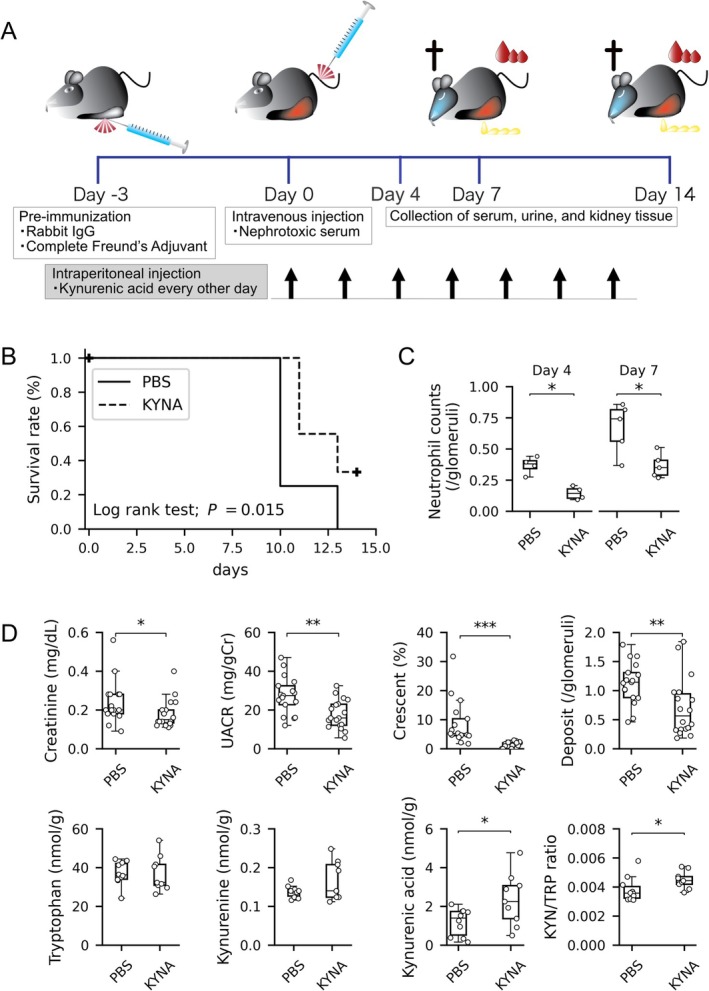
Effect of KYNA administration in Ido1‐deficient mice with NTS‐GN. (A) Experimental timeline for KYNA administration in the NTS‐GN model. KYNA or PBS was administered intraperitoneally every other day into Ido1‐deficient mice. (B) Kaplan–Meier survival curves for KYNA‐treated (dashed line, n = 9) and phosphate‐buffered saline (PBS)‐treated (solid line, n = 8) Ido1‐deficient mice. KYNA treatment significantly improved survival (log‐rank test, p = 0.015). (C) Quantification of glomerular neutrophil infiltration on days 4 and 7. Neutrophil counts were significantly decreased in Ido1‐deficient mice with KYNA injection on days 4 and 7 compared with PBS‐treated control (p = 0.029 and p = 0.032, respectively, Mann–Whitney U test). n = 4 for each group at day 4 and n = 5 at day 7. (D) Box plots showing therapeutic effects of KYNA on kidney injury (top row: Serum creatinine, UACR, crescent formation rate, PAS‐positive deposit scores; n = 16–18 per group) and on tryptophan metabolism in the kidney cortex (bottom row: TRP, KYN, KYNA, and KYN/TRP ratio; n = 9–10 per group) at day 7. KYNA‐treated mice showed significantly improved renal parameters and increased KYNA and KYN/TRP ratio (p = 0.046 and p = 0.031, respectively; Mann–Whitney U test; p < 0.05 for all significant comparisons). Asterisks (*, **, ***) indicate statistically significant differences (p < 0.05, p < 0.01, p < 0.001, respectively).
3.5. Accelerated Neutrophil Activation in Ido1‐Deficient Mice Is Reversed by KYNA
Given that exogenous KYNA ameliorated glomerular injury and reduced neutrophil infiltration in vivo, we next examined its direct effect on neutrophil activation in vitro. The addition of KYNA to Ido1‐deficient neutrophils stimulated with ICs suppressed their activation and spreading, resulting in a significantly smaller spread cell area at 60 min compared with untreated cells (p = 0.041; Figure 6A,B). Consistent with these morphological findings, Western blot analysis of FcγR signaling showed that phosphorylation of Syk was rapidly and strongly induced in Ido1‐deficient neutrophils after FcγR cross‐linking at 60 s, and this response was attenuated by KYNA treatment (Figure 6C, p = 0.001). We next assessed cytokine release from IC‐stimulated neutrophils under the same conditions as the morphological assays, with supernatants collected at 0, 6, and 12 h. Among the 20 cytokines analyzed using the RayBiotech Mouse Cytokine Array Q1, most were below the detection limit. VEGF and TNFα were the only cytokines consistently detectable. VEGF secretion was significantly increased in Ido1‐deficient neutrophils at 6 h compared with WT, and this increase was significantly suppressed by KYNA treatment (Figure 6D, p = 0.008 and p = 0.038, respectively). In contrast, TNFα secretion did not differ significantly among groups at any time point (Figure 6E).
FIGURE 6.

Time‐dependent morphological activation of neutrophils stimulated with immune complexes. (A, B) Effect of KYNA on neutrophil spreading in Ido1‐deficient mice. At 60 min, KYNA‐treated Ido1‐deficient neutrophils (n = 5) showed a significantly reduced spread area compared to untreated Ido1‐deficient neutrophils (n = 6) (p = 0.041) and remained significantly larger than WT (n = 10) (p = 0.017). For each mouse, five random sections were analyzed per time point; the average of these was used as a single data point. The same set of mice was used across all time points, with independent measurements conducted at each time. Data are presented as mean ± SEM. Statistical analysis was performed using one‐way ANOVA followed by Tukey's post hoc test. (C) Western blot analysis of FcγR–Syk signaling. In Ido1‐deficient neutrophils, phosphorylation of Syk was rapidly and strongly induced at 30–60 s after IC cross‐linking, whereas this response was attenuated by KYNA treatment. Representative blots and total Syk controls are shown. Band intensities from four independent experiments were quantified; at 60 s, phospho‐Syk levels were significantly lower in KYNA‐treated Ido1‐deficient neutrophils compared with untreated Ido1‐deficient neutrophils (p = 0.001). (D) Cytokine secretion from IC‐stimulated neutrophils. Supernatants were collected at 0, 6, and 12 h and analyzed using a cytokine array. (D) VEGF secretion was significantly increased in Ido1‐deficient neutrophils at 6 h compared with WT, and was significantly reduced by KYNA treatment. (E) TNFα secretion was detectable but did not differ significantly among groups at any time point. Asterisks (*, **) indicate statistically significant differences (p < 0.05, p < 0.01, p < 0.001, respectively).
3.6. Kat2 Deficiency Aggravates Proteinuria in NTS‐GN
The preceding therapeutic findings suggest a protective role of KYNA in antibody‐mediated glomerular inflammation. Since KAT2 is a key enzyme that converts KYN to KYNA, we next examined the effects of Kat2 deficiency in the NTS‐GN model. In Kat2‐deficient mice, KYNA levels in the kidney cortex were below the detection limit on day 14 after disease induction. These mice exhibited significantly higher UACR compared to WT (p = 0.0073), whereas serum creatinine levels remained comparable (Figure S3).
4. Discussion
Our study demonstrated that Ido1 deficiency exacerbates renal dysfunction and pathological glomerular damage in the NTS‐GN model. Notably, KYNA administration effectively alleviated the renal impairment and corrected neutrophil dysregulation caused by Ido1 deficiency, including morphological spreading, FcγR–dependent Syk signaling, and VEGF secretion, highlighting its therapeutic potential via alteration of neutrophil function. Meanwhile, Ido2 deficiency had no significant impact on renal injury, and Kmo deficiency resulted in reduced crescent formation. Together, these findings underscore the critical role of TRP metabolism, with distinct contributions from specific metabolic enzymes, and position KYNA as a promising therapeutic target for modulating immune responses and protecting against the progression of antibody‐mediated glomerulonephritis.
The phenotypes observed in NTS‐GN models with Ido1‐, Ido2‐, and Kmo‐deficient mice point to the distinct roles of these enzymes, and reported expression patterns provide biological context for these differences. Ido1 deficiency significantly exacerbates NTS‐GN, accompanied by enhanced neutrophil activation. The detrimental effect of Ido1 deficiency is mediated not only through impaired NAD synthesis [8, 12] but also through the loss of TRP metabolite–mediated regulation of neutrophil function. Consistent with this role, Ido1 is scarcely detectable under steady‐state conditions but is robustly induced by inflammatory cytokines such as IFN‐γ, both in renal endothelial cells and in immune cells including antigen‐presenting cells and macrophages [24], and also in neutrophils [25]. By contrast, Ido2 deficiency had little impact on disease progression. This limited effect may reflect both its relatively weak enzymatic activity and its expression pattern. Indeed, the KYN/TRP ratio, a marker of IDO activity, in Ido2‐deficient mice was significantly higher than that in Ido1‐deficient mice during the first week after NTS‐GN induction. Given that NTS‐GN is primarily a neutrophil‐driven model, it is also relevant that IDO2 is expressed at low levels in B cells and selected dendritic‐cell subsets and has been reported in renal proximal tubular cells, but there is little evidence for expression in neutrophils [26, 27, 28, 29, 30], which is consistent with the minimal phenotype. Kmo deficiency was associated with a reduced crescent formation rate. While previous studies have shown that Kmo deficiency mitigates renal interstitial injury in ischemia–reperfusion models [11], this study provides evidence that KMO aggravates glomerular injury. High concentrations of KYN and KYNA were observed in the renal tissues of Kmo‐deficient mice, which could have contributed to the renoprotective effects observed.
Despite being the final metabolite in the TRP pathway and having no contribution to NAD synthesis, KYNA exhibited a marked renoprotective effect in our NTS‐GN model. Prior studies have shown that kynurenine‐pathway metabolites such as KYN and 3‐HAA have immunoregulatory properties, including suppression of NK and pro‐inflammatory T‐cell responses and promotion of Th2 and regulatory T cells [31, 32, 33, 34]. Some of the immunological effects are mediated by activation of the AhR, which is expressed in both innate and adaptive immune cells, and KYNA itself has been reported to activate AhR [35, 36, 37, 38]. In addition, KYNA is also a ligand for G protein‐coupled receptor 35 [39], and its interaction contributes to the reduction of LPS‐induced inflammatory responses in monocytes and macrophages [40] as well as the regulation of cytokine release in human iNKT cells [41]. These mechanisms provide a plausible framework for KYNA's anti‐inflammatory activity in immune‐mediated diseases, including antibody‐mediated glomerulonephritis.
Building on these findings, our study newly demonstrates that KYNA protects against glomerular injury through direct modulation of neutrophil responses. In Ido1‐deficient mice, increased tissue infiltration of neutrophils was observed in the early stage of NTS‐GN, suggesting their role in exacerbating renal injury. Additionally, neutrophils isolated from the bone marrow of Ido1‐deficient mice showed sustained morphological changes to the “Spread form” in vitro, accompanied by enhanced FcγR–Syk phosphorylation and increased VEGF secretion, which may reflect their heightened activation state. KYNA administration alleviated both the in vivo and in vitro abnormalities, supporting its role in suppressing neutrophil‐driven injury. Mechanistically, KYNA may exert this effect via AhR activation, as supported by reports that another AhR ligand, TCDD (2,3,7,8‐tetrachlorodibenzo‐p‐dioxin), suppresses neutrophil infiltration in silica‐induced lung injury [38]. Moreover, studies showing that KYNA enhances TSG‐6 expression through AhR activation [42, 43], together with our results, suggest that KYNA may suppress neutrophil chemotaxis through this pathway. These findings highlight KYNA as a unique TRP metabolite that confers renoprotection not only through broad anti‐inflammatory actions but also through the mechanism of neutrophil modulation.
Interestingly, despite the absence of functional IDO1, KYNA levels were increased in Ido1‐deficient mice. Together with the progressive rise of the KYNA/KYN ratio and the unchanged 3HAA/KYN ratio, this finding suggests that KYNA production can be enhanced in an IDO1‐independent manner. A likely explanation is preferential diversion of tryptophan metabolism toward the KYNA branch under inflammatory conditions. This mechanism may account for the paradoxical elevation of KYNA observed in Ido1‐deficient mice and further supports the complexity of TRP metabolism in antibody‐mediated glomerulonephritis.
Additional analyses of alternative mechanisms did not indicate a major contribution in this model. Glomerular antibody deposition was comparable among genotypes and between KYNA‐ and PBS‐treated mice, indicating that differences in disease severity were not attributable to altered immune complex formation (Figure S2). We also examined macrophage activation since previous studies have linked IDO to macrophage function [8]. In our experiments, bone marrow–derived macrophages from Ido1‐deficient and wild‐type mice showed comparable CD86 induction after M1 stimulation (Figure S4). This discrepancy from prior reports may reflect the use of bone marrow–derived rather than peritoneal macrophages, or the fact that these cells may not fully reproduce the behavior of tissue‐resident macrophages in vivo. Thus, our results do not exclude a role for macrophages, but rather suggest that neutrophil activation represents the more prominent mechanism under the present conditions.
This study provides further insights into the role of TRP metabolism in the progression of renal pathology. Notably, KYNA exhibited unique anti‐inflammatory effects independent of NAD, alleviating NTS‐GN. Furthermore, while Ido1 deficiency exacerbated renal injury, the impacts of Ido2 and Kmo deficiencies revealed distinct roles of these enzymes, underscoring the individual contributions of enzymes in the TRP metabolic pathway. These findings suggest the potential of KYNA‐centered immunomodulatory strategies as a promising therapeutic approach, not only for NTS‐GN but also for other immune‐mediated diseases in particular through neutrophil modulation demonstrated in this study. Further research is anticipated to clarify the molecular mechanisms of KYNA and its potential clinical applications.
Author Contributions
R.U. conducted all in vivo and in vitro experiments, performed data analysis, and drafted the manuscript. Y.I. contributed to in vitro analyses. Y.Ya. and K.S. generated and provided genetically modified mouse strains and measured the concentrations of tryptophan metabolites. N.T. supervised the entire study, including project planning, experimental design, and data interpretation. M.H. and H.H. performed histopathological evaluations. S.M. was responsible for statistical analysis. K.T. and S.K. contributed to sample processing and data acquisition. Y.Yu. provided critical review of the manuscript. All authors read and approved the final revised version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
FIGURES S1‐S4: fsb271076‐sup‐0001‐FigureS1‐S4.pdf.
Table S1: fsb271076‐sup‐0002‐TableS1.pdf.
Table S2: fsb271076‐sup‐0003‐TableS2.pdf.
Acknowledgments
We thank Ms. Okada, a research assistant at Fujita Health University, for her valuable administrative and technical support. We also acknowledge generous contributions to this research from the following medical institutions: Mizuno Clinic (Hosui‐kai Medical Corporation), Shinsei‐kai Daiichi Hospital (Nagoya Memorial Foundation), and Nagoya Sakae Clinic. This work was supported by a Grant‐in‐Aid for Scientific Research (C, #22K08344) from the Ministry of Education, Culture, Sports, Science, and Technology (N.T.); a grant from the Aichi Kidney Foundation (2020–2022, R.U.); a research grant from GSK Japan (2019, Y.I.); and a faculty research grant from Fujita Health University (N.T.).
Umeda R., Ito Y., Minatoguchi S., et al., “Investigation of the Impact of Tryptophan‐Metabolizing Enzymes and Kynurenic Acid on Antibody‐Mediated Glomerulonephritis,” The FASEB Journal 39, no. 18 (2025): e71076, 10.1096/fj.202501800R.
Funding: This work was supported by Japan Society for the Promotion of Science London (JSPS), C,#22K08344. Aichi Kidney Foundation. GlaxoSmithKline Japan (GSK). Fujita Health University.
Data Availability Statement
The datasets and analysis code supporting this study are available from the corresponding author upon request. We plan to deposit all relevant materials in a public repository (e.g., Zenodo) and make them publicly accessible by the time of manuscript acceptance.
References
- 1. Badawy A. A., “Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects,” International Journal of Tryptophan Research: IJTR 10 (2017): 1178646917691938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shimizu T., Nomiyama S., Hirata F., and Hayaishi O., “Indoleamine 2,3‐Dioxygenase. Purification and Some Properties,” Journal of Biological Chemistry 253, no. 13 (1978): 4700–4706. [PubMed] [Google Scholar]
- 3. Ball H. J., Sanchez‐Perez A., Weiser S., et al., “Characterization of an Indoleamine 2,3‐Dioxygenase‐Like Protein Found in Humans and Mice,” Gene 396, no. 1 (2007): 203–213. [DOI] [PubMed] [Google Scholar]
- 4. Munn D. H., Zhou M., Attwood J. T., et al., “Prevention of Allogeneic Fetal Rejection by Tryptophan Catabolism,” Science 281, no. 5380 (1998): 1191–1193. [DOI] [PubMed] [Google Scholar]
- 5. Munn D. H., Shafizadeh E., Attwood J. T., Bondarev I., Pashine A., and Mellor A. L., “Inhibition of T Cell Proliferation by Macrophage Tryptophan Catabolism,” Journal of Experimental Medicine 189, no. 9 (1999): 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hwu P., Du M. X., Lapointe R., Do M., Taylor M. W., and Young H. A., “Indoleamine 2,3‐Dioxygenase Production by Human Dendritic Cells Results in the Inhibition of T Cell Proliferation,” Journal of Immunology 164, no. 7 (2000): 3596–3599. [DOI] [PubMed] [Google Scholar]
- 7. Baban B., Chandler P. R., Sharma M. D., et al., “IDO Activates Regulatory T Cells and Blocks Their Conversion Into Th17‐Like T Cells,” Journal of Immunology 183, no. 4 (2009): 2475–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Minhas P. S., Liu L., Moon P. K., et al., “Macrophage de Novo NAD(+) Synthesis Specifies Immune Function in Aging and Inflammation,” Nature Immunology 20, no. 1 (2019): 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Muller A. J. and Scherle P. A., “Targeting the Mechanisms of Tumoral Immune Tolerance With Small‐Molecule Inhibitors,” Nature Reviews. Cancer 6, no. 8 (2006): 613–625. [DOI] [PubMed] [Google Scholar]
- 10. Merlo L. M., DuHadaway J. B., Grabler S., Prendergast G. C., Muller A. J., and Mandik‐Nayak L., “IDO2 Modulates T Cell‐Dependent Autoimmune Responses Through a B Cell‐Intrinsic Mechanism,” Journal of Immunology 196, no. 11 (2016): 4487–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng X., Zhang A., Binnie M., et al., “Kynurenine 3‐Monooxygenase Is a Critical Regulator of Renal Ischemia‐Reperfusion Injury,” Experimental & Molecular Medicine 51, no. 2 (2019): 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Poyan Mehr A., Tran M. T., Ralto K. M., et al., “De Novo NAD(+) Biosynthetic Impairment in Acute Kidney Injury in Humans,” Nature Medicine 24, no. 9 (2018): 1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hou W., Li S., Wu Y., Du X., and Yuan F., “Inhibition of Indoleamine 2, 3‐Dioxygenase‐Mediated Tryptophan Catabolism Accelerates Crescentic Glomerulonephritis,” Clinical and Experimental Immunology 156, no. 2 (2009): 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang M. S., Hsu Y. L., Yeh I. J., Liu K. T., and Yen M. C., “The Expression Profile of mRNA and tRNA Genes in Splenocytes and Neutrophils After In Vivo Delivery of Antitumor Short Hairpin RNA of Indoleamine 2,3‐ Dioxygenase,” International Journal of Molecular Sciences 21, no. 18 (2020): 6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu K. T., Liu Y. H., Liu H. L., Chong I. W., Yen M. C., and Kuo P. L., “Neutrophils Are Essential in Short Hairpin RNA of Indoleamine 2,3‐ Dioxygenase Mediated‐Antitumor Efficiency,” Molecular Therapy ‐ Nucleic Acids 5, no. 12 (2016): e397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Loughman J. A., Yarbrough M. L., Tiemann K. M., and Hunstad D. A., “Local Generation of Kynurenines Mediates Inhibition of Neutrophil Chemotaxis by Uropathogenic Escherichia coli ,” Infection and Immunity 84, no. 4 (2016): 1176–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. El‐Zaatari M., Chang Y. M., Zhang M., et al., “Tryptophan Catabolism Restricts IFN‐γ‐Expressing Neutrophils and Clostridium difficile Immunopathology,” Journal of Immunology 193, no. 2 (2014): 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsuboi N., Asano K., Lauterbach M., and Mayadas T. N., “Human Neutrophil Fcgamma Receptors Initiate and Play Specialized Nonredundant Roles in Antibody‐Mediated Inflammatory Diseases,” Immunity 28, no. 6 (2008): 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosenkranz A. R., Knight S., Sethi S., Alexander S. I., Cotran R. S., and Mayadas T. N., “Regulatory Interactions of Alphabeta and Gammadelta T Cells in Glomerulonephritis,” Kidney International 58, no. 3 (2000): 1055–1066. [DOI] [PubMed] [Google Scholar]
- 20. Tang T., Rosenkranz A., Assmann K. J., et al., “A Role for mac‐1 (CDIIb/CD18) in Immune Complex‐Stimulated Neutrophil Function In Vivo: Mac‐1 Deficiency Abrogates Sustained Fcgamma Receptor‐Dependent Neutrophil Adhesion and Complement‐Dependent Proteinuria in Acute Glomerulonephritis,” Journal of Experimental Medicine 186, no. 11 (1997): 1853–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Utomo A., Cullere X., Glogauer M., Swat W., and Mayadas T. N., “Vav Proteins in Neutrophils Are Required for FcgammaR‐Mediated Signaling to Rac GTPases and Nicotinamide Adenine Dinucleotide Phosphate Oxidase Component p40(Phox),” Journal of Immunology 177, no. 9 (2006): 6388–6397. [DOI] [PubMed] [Google Scholar]
- 22. Yoshida Y., Fujigaki H., Kato K., et al., “Selective and Competitive Inhibition of Kynurenine Aminotransferase 2 by Glycyrrhizic Acid and Its Analogues,” Scientific Reports 9, no. 1 (2019): 10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Python Version 3.11.9. Python Software Foundation, https://www.python.org/.
- 24. Zhou J., Abedini A., Balzer M. S., et al., “Unified Mouse and Human Kidney Single‐Cell Expression Atlas Reveal Commonalities and Differences in Disease States,” Journal of the American Society of Nephrology 34, no. 11 (2023): 1843–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Monaco G., Lee B., Xu W., et al., “RNA‐Seq Signatures Normalized by mRNA Abundance Allow Absolute Deconvolution of Human Immune Cell Types,” Cell Reports 26, no. 6 (2019): 1627–1640.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ball H. J., Yuasa H. J., Austin C. J., Weiser S., and Hunt N. H., “Indoleamine 2,3‐Dioxygenase‐2; a New Enzyme in the Kynurenine Pathway,” International Journal of Biochemistry & Cell Biology 41, no. 3 (2009): 467–471. [DOI] [PubMed] [Google Scholar]
- 27. Prendergast G. C., Metz R., Muller A. J., Merlo L. M., and Mandik‐Nayak L., “IDO2 in Immunomodulation and Autoimmune Disease,” Frontiers in Immunology 5 (2014): 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trabanelli S., Očadlíková D., Ciciarello M., et al., “The SOCS3‐Independent Expression of IDO2 Supports the Homeostatic Generation of T Regulatory Cells by Human Dendritic Cells,” Journal of Immunology 192, no. 3 (2014): 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Merlo L. M. F., Pigott E., DuHadaway J. B., et al., “IDO2 Is a Critical Mediator of Autoantibody Production and Inflammatory Pathogenesis in a Mouse Model of Autoimmune Arthritis,” Journal of Immunology 192, no. 5 (2014): 2082–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmiedel B. J., Singh D., Madrigal A., et al., “Impact of Genetic Polymorphisms on Human Immune Cell Gene Expression,” Cell 175, no. 6 (2018): 1701–1715.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hou W., Huang G., Cao X., Zhang Y., Zhang J., and Li Y., “Suppression of Experimental Autoimmune Glomerulonephritis by Tryptophan,” Journal of Nephrology 27, no. 1 (2014): 19–28. [DOI] [PubMed] [Google Scholar]
- 32. Frumento G., Rotondo R., Tonetti M., Damonte G., Benatti U., and Ferrara G. B., “Tryptophan‐Derived Catabolites Are Responsible for Inhibition of T and Natural Killer Cell Proliferation Induced by Indoleamine 2,3‐Dioxygenase,” Journal of Experimental Medicine 196, no. 4 (2002): 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fallarino F., Grohmann U., Vacca C., et al., “T Cell Apoptosis by Tryptophan Catabolism,” Cell Death and Differentiation 9, no. 10 (2002): 1069–1077. [DOI] [PubMed] [Google Scholar]
- 34. Orabona C., Puccetti P., Vacca C., et al., “Toward the Identification of a Tolerogenic Signature in IDO‐Competent Dendritic Cells,” Blood 107, no. 7 (2006): 2846–2854. [DOI] [PubMed] [Google Scholar]
- 35. DiNatale B. C., Murray I. A., Schroeder J. C., et al., “Kynurenic Acid Is a Potent Endogenous Aryl Hydrocarbon Receptor Ligand That Synergistically Induces Interleukin‐6 in the Presence of Inflammatory Signaling,” Toxicological Sciences: An Official Journal of the Society of Toxicology 115, no. 1 (2010): 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mezrich J. D., Fechner J. H., Zhang X., Johnson B. P., Burlingham W. J., and Bradfield C. A., “An Interaction Between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells,” Journal of Immunology 185, no. 6 (2010): 3190–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Opitz C. A., Litzenburger U. M., Sahm F., et al., “An Endogenous Tumour‐Promoting Ligand of the Human Aryl Hydrocarbon Receptor,” Nature 478, no. 7368 (2011): 197–203. [DOI] [PubMed] [Google Scholar]
- 38. Beamer C. A., Seaver B. P., and Shepherd D. M., “Aryl Hydrocarbon Receptor (AhR) Regulates Silica‐Induced Inflammation but Not Fibrosis,” Toxicological Sciences: An Official Journal of the Society of Toxicology 126, no. 2 (2012): 554–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang J., Simonavicius N., Wu X., et al., “Kynurenic Acid as a Ligand for Orphan G Protein‐Coupled Receptor GPR35,” Journal of Biological Chemistry 281, no. 31 (2006): 22021–22028. [DOI] [PubMed] [Google Scholar]
- 40. Tiszlavicz Z., Németh B., Fülöp F., et al., “Different Inhibitory Effects of Kynurenic Acid and a Novel Kynurenic Acid Analogue on Tumour Necrosis Factor‐α (TNF‐α) Production by Mononuclear Cells, HMGB1 Production by Monocytes and HNP1‐3 Secretion by Neutrophils,” Naunyn‐Schmiedeberg's Archives of Pharmacology 383, no. 5 (2011): 447–455. [DOI] [PubMed] [Google Scholar]
- 41. Fallarini S., Magliulo L., Paoletti T., de Lalla C., and Lombardi G., “Expression of Functional GPR35 in Human iNKT Cells,” Biochemical and Biophysical Research Communications 398, no. 3 (2010): 420–425. [DOI] [PubMed] [Google Scholar]
- 42. Wang G., Cao K., Liu K., et al., “Kynurenic Acid, an IDO Metabolite, Controls TSG‐6‐Mediated Immunosuppression of Human Mesenchymal Stem Cells,” Cell Death and Differentiation 25, no. 7 (2018): 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dyer D. P., Thomson J. M., Hermant A., et al., “TSG‐6 Inhibits Neutrophil Migration via Direct Interaction With the Chemokine CXCL8,” Journal of Immunology 192, no. 5 (2014): 2177–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURES S1‐S4: fsb271076‐sup‐0001‐FigureS1‐S4.pdf.
Table S1: fsb271076‐sup‐0002‐TableS1.pdf.
Table S2: fsb271076‐sup‐0003‐TableS2.pdf.
Data Availability Statement
The datasets and analysis code supporting this study are available from the corresponding author upon request. We plan to deposit all relevant materials in a public repository (e.g., Zenodo) and make them publicly accessible by the time of manuscript acceptance.