

Abstract
Expression of the coronavirus gene 1-encoded polyproteins, pp1a and pp1ab, is linked to a series of proteolytic events involving virus-encoded proteinases. In this study, we used transfection and immunoprecipitation assays to show that the human coronavirus 229E-encoded papain-like cysteine proteinase, PCP1, is responsible for the release of an amino-terminal protein, p9, from the gene 1-encoded polyproteins. The same protein, p9, has also been identified in virus-infected cells. Furthermore, using an in vitro trans-cleavage assay, we defined the proteolytic cleavage site at the carboxyl terminus of p9 as pp1a-pp1ab amino acids Gly-111 and Asn-112. These results and a comparative sequence analysis suggest that substrate positions P1 and P5 seem to be the major determinants of the PCP1 cleavage site and that the latter can occupy a variable position at the amino terminus of the coronavirus pp1a and pp1ab polyproteins. By combining the trans-cleavage assay with deletion mutagenesis, we were also able to locate the boundaries of the active PCP1 domain between pp1a-pp1ab amino acids Gly-861–Glu-975 and Asn-1209–Gln-1285. Finally, codon mutagenesis was used to show that Cys-1054 and His-1205 are essential for PCP1 proteolytic activity, suggesting that these amino acids most likely have a catalytic function.
The coronaviruses are a group of enveloped, positive-stranded RNA viruses that are associated predominantly with respiratory and gastrointestinal diseases in their natural hosts (28). The human coronaviruses (HCV), which are represented by the prototypes HCV 229E and HCV OC43, are responsible for 5 to 30% of all upper respiratory tract infections in humans, and their involvement in lower respiratory tract illness and gastroenteritis has also been documented (18, 25, 30).
The HCV 229E genome is comprised of approximately 27,000 nucleotides. Gene 1, which is located at the 5′ end of the genome, encodes the viral RNA replicase and encompasses two large, overlapping open reading frames (ORFs), ORF1a and ORF1b (14). ORF1a encodes a polyprotein, pp1a, with a calculated molecular weight of 454,000. The downstream ORF, ORF1b, is expressed by ribosomal frameshifting as a fusion protein with pp1a (12), and the predicted gene product, pp1ab, has a calculated molecular weight of 754,000.
Proteolytic processing, and in particular the processing of replicase polyproteins, is a crucial step in the life cycle of many positive-stranded RNA viruses (7, 20). Generally, these processing events are carried out by virus-encoded proteinases. Coronaviruses are no exception, and sequence motifs characteristic of both papain-like cysteine proteinases and a chymotrypsin-like enzyme, the 3C-like proteinase, have been identified in the regions of pp1a and pp1ab encoded by ORF1a (8, 9, 14, 22). Recent studies have confirmed that these activities are indeed responsible for the proteolytic processing of replicase polyproteins and can be implicated in the generation of a functional replication complex (see, for example, references 4, 10, 15, 16, 23, and 31).
Sequence analysis of four different coronaviruses, HCV 229E (14), infectious bronchitis virus (5), murine hepatitis virus (MHV) (2, 22), and transmissible gastroenteritis virus (TGEV) (8), has suggested that either one infectious bronchitis virus or two (HCV 229E, MHV, and TGEV) papain-like proteinase activities are encoded in the amino-proximal region of pp1a and pp1ab. To date, no experimental evidence has demonstrated that the carboxyl-proximal domain, PCP2, is functional. In contrast, both in vivo and in vitro data have shown that the amino-proximal domain, PCP1, is active. Thus, the MHV PCP1 domain has been shown to be responsible for the release of two proteins from nascent replicase polyproteins in vitro, the amino-terminal protein p28 and the adjacent protein, p65 (1, 4). Deletion mutagenesis studies have identified the boundaries of the active MHV PCP1 proteinase, and codon mutagenesis has shown that the catalytic residues most likely are Cys-1137 and His-1288 (1). Finally, the MHV pp1a-pp1ab amino acids Gly-247 and Val-248 have been identified as the cleavage site for the release of p28 by MHV PCP1, and amino acids Gly-247 and Arg-246 have been identified as the major determinants for cleavage site recognition (6, 17).
In this paper, we report an analysis of the HCV 229E PCP1 activity. Our results show that the location and catalytic properties of the HCV 229E enzyme are similar to those described for MHV but that there are some peculiarities in the position and structure of the cleavage site used to release the amino-terminal protein, p9, of pp1a and pp1ab. These differences could not be predicted by previous sequence comparisons (8a, 17).
MATERIALS AND METHODS
Virus and cells.
The methods for HCV 229E propagation in MRC-5 cells and for concentration of virus by use of polyethylene glycol have been described elsewhere (26, 32). HeLa cells (ATCC CCL2) were grown in monolayers in minimal essential medium with Earle’s salts and containing 10% heat-inactivated fetal bovine serum, 25 mM HEPES, GLUTAMAX 1 (l-alanyl-l-glutamine), antibiotics, and nonessential amino acids. The recombinant vaccinia virus MVA-T7, which expresses the bacteriophage T7 RNA polymerase, was propagated in chicken embryo fibroblasts as described previously (29).
Preparation of antigen and antiserum.
A 632-bp SphI/KpnI cDNA fragment corresponding to nucleotides 412 to 1043 of the genomic RNA of HCV 229E was excised from plasmid pJ12E6 (14) and ligated with SphI/KpnI-digested pQE30 DNA (Diagen, Hilden, Germany). The ligated DNA was transformed into competent Escherichia coli JM109, and individual clones were analyzed by restriction enzyme digestion and sequencing. The correct construct was designated pI1a.1.
Expression of the recombinant protein encoded by pI1a.1 was induced by isopropyl-β-d-thiogalactopyranoside in E. coli M15/pRep4. The recombinant protein comprises 12 amino acids at the amino terminus that are encoded by the expression vector, including 6 consecutive histidines; 210 amino acids encoded by the HCV 229E replicase gene (corresponding to amino acids 41 to 250 of ORF1a); and 2 vector-derived amino acids at the carboxyl terminus. Purification of the fusion protein and immunization of rabbits have been described elsewhere (32). The resulting pI1a.1-encoded protein-specific antiserum was designated IS1720.
Construction of DNAs encoding carboxyl-terminally extended pp1a and pp1ab proteins.
Polyadenylated RNA was isolated from HCV 229E-infected MRC-5 cells and reverse transcribed (oligonucleotide 1; Table 1) (13). Then, 2 μl of the reaction mixture was used as a template in a PCR (oligonucleotides 2 and 3; Table 1) to amplify a DNA that corresponds to nucleotides 387 to 12850 of the HCV 229E genomic RNA. Elongase polymerase mixture (Life Technologies, Eggenstein, Germany) was used for all PCR amplifications with the recommended buffer conditions. The cycle conditions were as follows: initial denaturation, 94°C for 30 s; 12 cycles at 94°C for 30 s, 50°C for 30 s, and 68°C for 12 min; 18 cycles at 94°C for 30 s, 50°C for 30 s, and 68°C for 12 min, with 15 s for extension per cycle; and final elongation, 72°C for 10 min.
TABLE 1.
Oligonucleotides used in this study
| Oligo- nucleotide | Sequencea | Methodb | Usec |
|---|---|---|---|
| 1 | 5′-AAT GAG GAC ATA CAC CGT GTG-3′ | cDNA synthesis | |
| 2 | 5′-GCG AGG CCG CTA GCA ATG G-3′ | RT-PCR | Produce pp1a-pp1ab DNA template |
| 3 | 5′-ATC TTA CTA AAT ACA CCA TGA-3′ | RT-PCR | Produce pp1a-pp1ab DNA template |
| 4 | 5′-GGC GAA TTG GAG CTC CAC-3′ | PCR | |
| 5 | 5′-CGG TAA GAG AGG TGG TGG-3′ | PCR | Produce pp1a-pp1ab 1–111 |
| 6 | 5′-CAC AAG TCA CAG TGG TTG G-3′ | PCR | Produce pp1a-pp1ab 1–613 |
| 7 | 5′-GTG CTG ATT GAA TAG TCT TAC-3′ | PCR | Produce pp1a-pp1ab 1–956 |
| 8 | 5′-GTA GTT GTA CCA GTG GTA GG-3′ | PCR | Produce pp1a-pp1ab 1–1132 |
| 9 | 5′-GTT AGT CTG GTA ATG ACC AC′-3′ | PCR | Produce pp1a-pp1ab 1–1209 |
| 10 | 5′-GAC AAT GTA GCT TTC TAC CAA G-3′ | PCR | Produce pp1a-pp1ab 1–1285 |
| 11 | 5′-GCA AGG TTC TCA TTA GCA-3′ | PCR | Produce pp1a-pp1ab 1–1309 |
| 12 | 5′-GGA TGA CCG TGG TTT AGC TCT AG-3′ | PCR | Produce pp1a-pp1ab 1–1500 |
| 13 | 5′-CCA ATG GCA AGA TGT AGT CAG CTG G-3′ | PCR | Produce pp1a-pp1ab 1–1589 |
| 14 | 5′-CAT AAC CAC CAA CCA TAA CAA CTG ATG TG-3′ | PCR | Produce pp1a-pp1ab 1–1900 |
| 15 | 5′-CTA AAT TGA CTT AAC TCT TGG-3′ | PCR | Produce pp1a-pp1ab 1–2058 |
| 16 | 5′-GTA ACT GTT CAC CAT GGC CTG CAA CCG TGT GAC A-3′ | rec-PCR | Introduce NcoI site to pJ12E6 |
| 17 | 5′-TGC AGG CCA TGG TGA ACA GTT ACT GGT TTC CAC A-3′ | rec-PCR | Introduce NcoI site to pJ12E6 |
| 18 | 5′-TTG TTT CTV GCT ATG TAA ATC TAC CTA CTT-3′ | rec-PCR–codon mut. | Change pp1a-pp1ab Cys-962 to Gly |
| 19 | 5′-TAC ATA GCB AGA AAC AAC AGA AAG TGC TGA-3′ | rec-PCR–codon mut. | Change pp1a-pp1ab Cys-962 to Gly |
| 20 | 5′-ATA ACA ACV GCT GGG TTA ACT CAG TTA TGT-3′ | rec-PCR–codon mut. | Change pp1a-pp1ab Cys-1054 to Arg, Gly, or Ser |
| 21 | 5′-AAC CCA GCB GTT GTT ATC CAA TTG TTT GAG-3′ | rec-PCR–codon mut. | Change pp1a-pp1ab Cys-1054 to Arg, Gly, or Ser |
| 22 | 5′-CTT GTG GTG BGT ACC AGA CTA ACA TCT ATT CA-3′ | rec-PCR–codon mut. | Change pp1a-pp1ab His-1205 to Arg or Gly |
| 23 | 5′-GTC TGG TAC VCA CCA CAA GAC ACA GCA CCA CG-3′ | rec-PCR–codon mut. | Change pp1a-pp1ab His-1205 to Arg or Gly |
| 24 | 5′-TCC TCG TTG BGG ACA ATG TAG CTT TCT ACC AA-3′ | rec-PCR–codon mut. | Change pp1a-pp1ab His-1278 to Arg, Gly, or Val |
| 25 | 5′-ACA TTG TCC VCA ACG AGG AAG GCG TTC AAC TT-3′ | rec-PCR–codon mut. | Change pp1a-pp1ab His-1278 to Arg, Gly, or Val |
| 26 | 5′-CAC ACG GTT GCA GGC CAT GGT ATT-3′ | del mut. | |
| 27 | 5′-ACT GCC ATG GCC AGT CCA AAT AGT GTG-3′ | del mut. | Produce pp1a-pp1ab 1 + 578–1315 |
| 28 | 5′-ACT GCC ATG GGT GGT ATT TTG GCA GTA ATA-3′ | del mut. | Produce pp1a-pp1ab 1 + 861–1315 |
| 29 | 5′-ACT GCC ATG GGC GGT AAT GAC TTG AGT TTG-3′ | del mut. | Produce pp1a-pp1ab 1 + 976–1315 |
| 30 | 5′-ACT GCC ATG GGA TTT GAA GAG TTA AAT GGT TTA-3′ | del mut. | Produce pp1a-pp1ab 1 + 1037–1315 |
V = GAC; B = GTC.
rec-PCR, recombination PCR; codon mut., codon mutagenesis; del mut., deletion mutagenesis.
Ranges indicate amino acids. For oligonucleotides 27 to 30, 1 indicates the initiating methionine.
The reverse transcription (RT)-PCR product and pT7-IRES-1a/N (see below) were digested with SapI and ligated with T4 DNA ligase. Approximately 1 ng of the ligation product was then used as a template for 11 different PCRs. In each case, oligonucleotide 4 (Table 1) was the upstream primer, and oligonucleotides 5 to 15 (Table 1) were used as downstream primers. The cycle conditions were as follows: initial denaturation, 94°C for 30 s; 30 cycles at 94°C for 30 s, 50°C for 30 s, and 68°C for 1.25 min/1 kb to be amplified; and final elongation, 72°C for 10 min. The DNAs generated are designated DNA 4/5 (primers 4 and 5), DNA 4/6 (primers 4 and 6), and so on, to DNA 4/15 (primers 4 and 15). They encode a series of carboxyl-terminally extended pp1a and pp1ab proteins that terminate between amino acids 111 and 2058 (Table 1).
Construction of pT7-IRES-Pap.
The construction of plasmid pT7-IRES-Pap is complex and is illustrated in Fig. 1. The starting plasmids, pPap, pBluescript II KS+, pTM3, and pJ12E6, have all been described elsewhere (13, 14, 24) (Stratagene, Heidelberg, Germany). Briefly, a DNA fragment containing the T7 RNA polymerase promoter and the encephalomyocarditis virus internal ribosomal entry site (IRES) element, derived from pTM3, was cloned into pBluescript II KS+; subsequently, most of the multiple cloning site was removed, resulting in plasmid pT7-IRES(CX). An NcoI site was introduced into pJ12E6 by in vivo recombination PCR (oligonucleotides 16 and 17; Table 1) (12), and HCV 229E ORF1a nucleotides 1 to 1207 were cloned behind the T7-IRES element of pT7-IRES(CX) to produce plasmid pT7-IRES-1a/N. Finally, the small NotI/SpeI fragment of pT7-IRES-1a/N was used to replace the small ApaI/SpeI fragment of pPap to produce plasmid pT7-IRES-Pap. The nucleotide sequence of pT7-IRES-Pap was determined, and two PCR-derived nucleotide misincorporations that led to changes in the deduced amino acid sequence compared to the published sequence (14) (GAG [Glu-1023] → GGG [Gly]; AAA [Lys-1316] → TAA [*]) were identified. Thus, pT7-IRES-Pap encodes a protein corresponding to the amino-terminal 1,315 amino acids of pp1a and pp1ab.
FIG. 1.

Construction of pT7-IRES-Pap. The diagram illustrates the original plasmids and strategy used to construct plasmid pT7-IRES-Pap. Relevant restriction sites, the major steps in the cloning procedure, important functional elements in the RNA, and proteins encoded by the various plasmids are indicated. UTR, untranslated region; nts, nucleotides.
Codon and deletion mutagenesis of pT7-IRES-Pap.
Codon mutations were introduced into pT7-IRES-Pap by in vivo recombination PCR (oligonucleotides 18 to 25; Table 1) (12). Deletions were made in the HCV 229E ORF1a coding region of pT7-IRES-Pap by PCR. Oligonucleotide 26 was used as a downstream primer, and oligonucleotide 27, 28, 29, or 30 was used as an upstream primer (Table 1). The resulting PCR products were digested with NcoI, religated with T4 DNA ligase, and transformed into E. coli Top 10F′ bacteria (Invitrogen, Leck, The Netherlands). The nucleotide sequences of the resulting plasmids were determined to exclude PCR-derived nucleotide misincorporations. The proteins encoded by this plasmid series correspond to the initiating methionine of HCV 229E ORF1a followed by pp1a-pp1ab amino acids 578 to 1315 (pT7-IRES-Papdel2-577), 861 to 1315 (pT7-IRES-Papdel2-860), 976 to 1315 (pT7-IRES-Papdel2-975), and 1037 to 1315 (pT7-IRES-Papdel2-1036).
Metabolic labeling, cell lysis, and immunoprecipitation.
Metabolic labeling of virus-specific polypeptides was done essentially as described previously (32). Briefly, 2 × 106 HeLa cells in 56-cm2 dishes were mock infected or infected with HCV 229E at a multiplicity of 10 PFU per cell. After 1 h, the supernatant was replaced with 10 ml of fresh medium. Radioactive labeling of newly synthesized proteins was done for 3 h at 33°C, between either 4 to 7 h postinfection or 7 to 10 h postinfection. Before labeling, the cells were washed twice with methionine- and cysteine-free Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 2% dialyzed fetal bovine serum. Pro-Mix l-35S in vitro cell-labeling mixture (SJQ 0079; Amersham, Braunschweig, Germany) was added to the cells to yield concentrations of 100 μCi of l-[35S]methionine and 42 μCi of l-[35S]cysteine per ml of medium. After labeling, the cells were lysed, and immunoprecipitation was done with IS1720 or preimmune serum essentially as described by Ziebuhr et al. (32). Proteins were analyzed by electrophoresis in sodium dodecyl sulfate (SDS)–10 to 17.5% polyacrylamide gradient gels.
T7 RNA polymerase-mediated transient expression.
Proteins encoded by circular DNA (plasmids) or linear DNA (PCR products) were expressed in HeLa cells with recombinant vaccinia virus MVA-T7 as a source of bacteriophage T7 RNA polymerase (29). To do this, 2 × 105 HeLa cells in 10-cm2 dishes were washed twice with OptiMEM (Life Technologies) and transfected with 5 μg of DNA by use of 12.5 μl of Lipofectin (Life Technologies) according to the manufacturer’s protocol. After 2 h, the transfection mixture was removed, and the cells were washed twice with medium and infected with MVA-T7 at a multiplicity of 5 PFU per cell. When linear DNA was transfected, the intracellular proteins were metabolically labeled for 6 h starting at 2 h postinfection. When circular DNA was used for transfection, the intracellular proteins were labeled for 2 h starting at 4 h postinfection. Cell labeling, lysis, and immunoprecipitation were done as described above for HCV 229E-infected cells.
In vitro trans-cleavage assay.
RNA was synthesized in vitro by use of a MEGAscript T7 kit (Ambion, Austin, Tex.). For the preparation of a labeled substrate, RNA transcribed from the PCR product encoding pp1a-pp1ab amino acids 1 to 956 was translated (100 ng/μl of reaction mixture) in a reticulocyte lysate (Promega, Heidelberg, Germany) in the presence of [35S]methionine as described previously (13). Proteins with putative proteolytic activity were translated in a separate reaction in which [35S]methionine was replaced with [32S]methionine at a concentration of 50 μM. Both the substrate and the enzyme translation reactions were stopped after 1 h of incubation at 30°C by the addition of 0.1 volume of TL-stop mix (10 μg of cycloheximide per μl, 100 ng of RNase A per μl, 5 mM [35S]methionine). Then, 1 volume of substrate and 2.5 volumes of enzyme reaction mixtures were mixed and incubated for 3 h at 30°C. Immunoprecipitation with IS1720 and protein analysis in SDS-polyacrylamide gels was done as described above.
Amino-terminal protein sequence analysis.
trans-Cleavage assays with 100 μl of substrate and 250 μl of enzyme reaction mixtures were done as described above, except that the in vitro-synthesized substrates were radiolabeled separately with either [35S]methionine or [35S]cysteine. The products of the cleavage reactions were immunoprecipitated with IS1720, separated by electrophoresis in SDS–10% polyacrylamide gels, and transferred electrophoretically to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Munich, Germany) (32). The areas of the membranes containing cleavage products were identified by autoradiography and isolated. The bound proteins were then subjected to 20 cycles of Edman degradation by use of a pulsed-liquid protein sequencer (ABI 467A; Applied Biosystems, Weiterstadt, Germany). The eluate from each cycle was mixed with scintillation cocktail, and the radioactivity was measured.
RESULTS
Identification of four proteins derived from the amino-terminal region of the HCV 229E pp1a and pp1ab polyproteins in vivo.
To facilitate the analysis of proteolytic processing events at the amino terminus of the HCV 229E replicase polyproteins, we first generated a polyclonal rabbit antiserum containing antibodies specific for a region of pp1a/pp1ab corresponding to amino acids 41 to 250 (Fig. 2A). This serum, IS1720, reacted strongly with the bacterial fusion protein used for immunization but not with other bacterial proteins (data not shown).
FIG. 2.
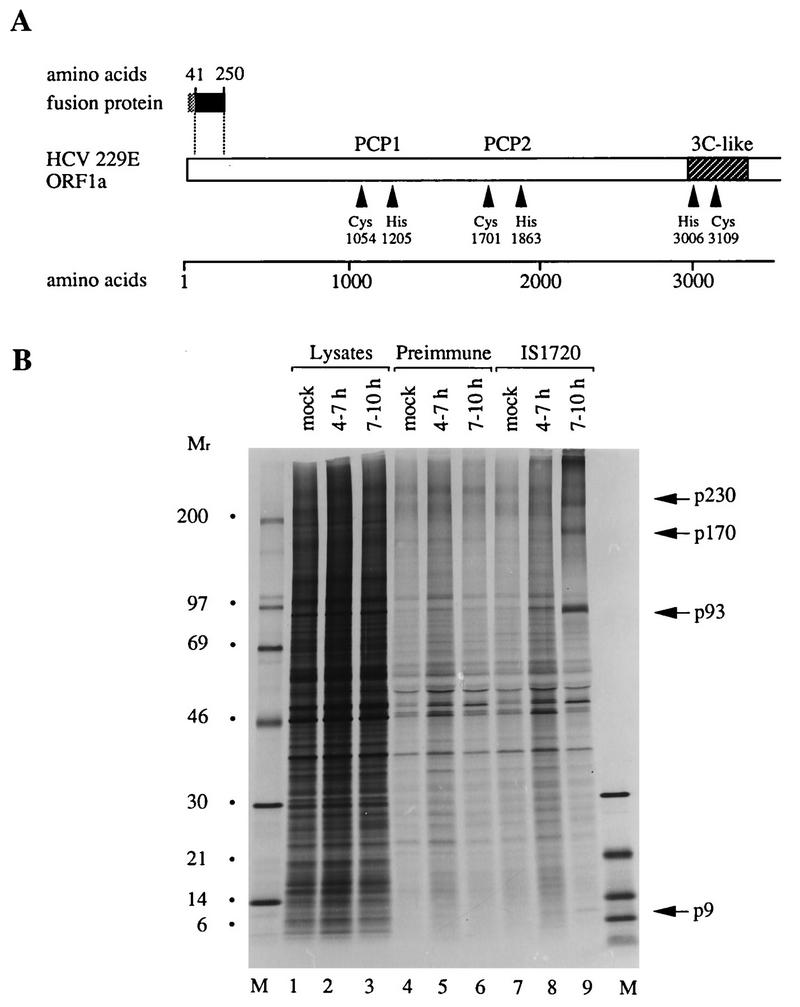
Detection of p9 in HCV 229E-infected cells. (A) Schematic representation of HCV 229E ORF1a; the relative position of the bacterially expressed fusion protein is shown. The putative catalytic amino acids of PCP1, PCP2, and the 3C-like proteinase are indicated, as are the boundaries of the domain of the 3C-like proteinase. (B) Metabolically labeled lysates from mock- or HCV 229E-infected HeLa cells were analyzed by electrophoresis with an SDS-containing 10 to 17.5% polyacrylamide gradient gel before or after immunoprecipitation with preimmune serum or IS1720 immune serum. The cells were labeled from 4 to 7 h or 7 to 10 h postinfection. Either 1-μl lysates from mock- or HCV 229E-infected cells were analyzed directly (lanes 1 to 3) or 140-μl lysates were analyzed after immunoprecipitation with preimmune serum (lanes 4 to 6) or IS1720 immune serum (lanes 7 to 9). Protein molecular weight markers (in thousands; lanes M) (CFA 626; Amersham), p9, and infection-specific higher-molecular-weight polypeptides are indicated.
HeLa cells were infected with HCV 229E at a multiplicity of 10 PFU per cell and metabolically labeled from 4 to 7 h postinfection or 7 to 10 h postinfection. Cell protein lysates were then immunoprecipitated with IS1720 or the corresponding preimmune serum, and the precipitated proteins were analyzed by gel electrophoresis and autoradiography. This experiment revealed four proteins that had apparent molecular weights of 9,000 (p9), 93,000 (p93), 170,000 (p170), and approximately 230,000 (p230) and that were specifically precipitated by immune serum from lysates of HCV 229E-infected cells (Fig. 2B, lanes 8 and 9). These proteins did not react with preimmune serum and were not present in mock-infected cells (Fig. 2B, lanes 4, 5, 6, and 7).
Taking into account the specificity of IS1720, the simplest interpretation of the data shown in Fig. 2B is that p9 represents the amino-terminal cleavage product of pp1a and pp1ab and that the larger proteins represent either precursors or cleavage products. It should be noted that we cannot exclude the possibility that a very small polypeptide is cleaved from the amino terminus of pp1a and pp1ab but, at the moment, there is no indication that this is the case. Furthermore, it is obvious that, in this experiment, it is difficult to detect p9. We do not believe that this is due to poor labeling of p9 (which is predicted to contain 9 radiolabeled residues compared to, for example, 35 in p93) but consider it more likely to be a reflection of processing kinetics, differential protein stability, or even the particular properties of the antiserum used.
HCV 229E PCP1 is likely to be responsible for the generation of the p9 protein.
It has been shown that MHV PCP1 is a papain-like proteinase responsible for the cleavage of amino-terminal p28 from pp1a and pp1ab (1, 17). Therefore, it seemed likely that the cleavage of p9 would also be mediated by a homologous enzyme, HCV 229E PCP1. To test this hypothesis, we produced a series of PCR products that contained a T7 RNA polymerase promoter, an EMCV IRES element at the 5′ end, and different 3′ extensions representing HCV 229E ORF1a from codons 1 to 111 (DNA 4/5) to codons 1 to 2058 (DNA 4/15) (Fig. 3A). These PCR DNA templates could be synthesized in quantitative amounts and were sufficiently homogeneous to be used in transfection experiments without further purification (data not shown). Seven of these DNAs were transfected into HeLa cells. Subsequently, the cells were infected with recombinant vaccinia virus MVA-T7. Newly synthesized proteins were metabolically labeled from 2 to 8 h postinfection, and cell protein lysates were then immunoprecipitated with IS1720 serum. The precipitated proteins were analyzed by gel electrophoresis and autoradiography.
FIG. 3.
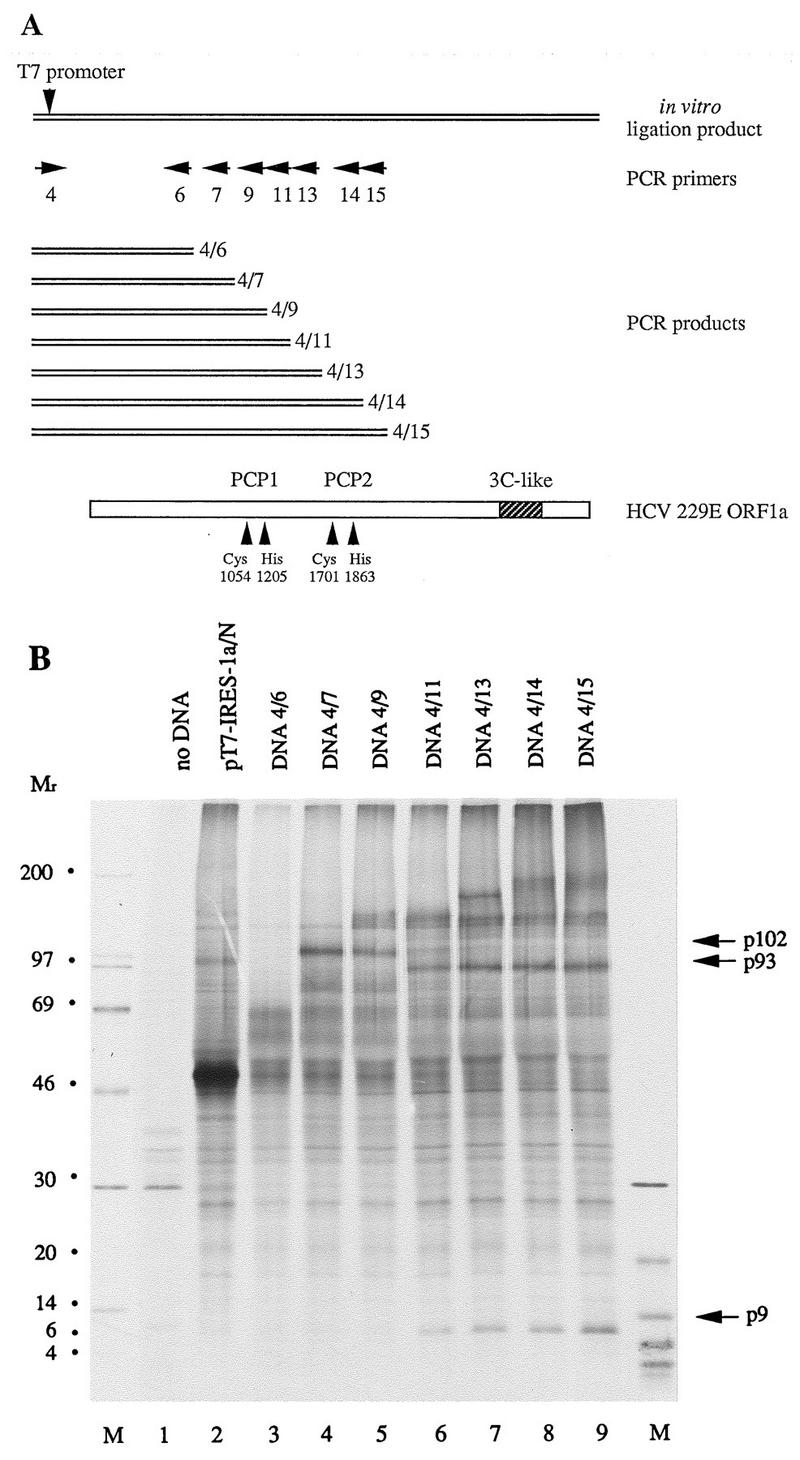
Demonstration of HCV 229E PCP1 activity in transfected cells. (A) Schematic representation of HCV 229E ORF1a, the in vitro ligation product, and the positions of the PCR primers used to generate PCR products. The putative catalytic amino acids of PCP1 and PCP2 are indicated, as are the boundaries of the domain of the 3C-like proteinase. (B) Metabolically labeled lysates from mock-infected (lane 1) or DNA-transfected (lanes 2 to 9) HeLa cells that had been coinfected with MVA-T7 were immunoprecipitated with IS1720 antiserum. The immunoprecipitated proteins were analyzed by electrophoresis in an SDS-containing 10 to 17.5% polyacrylamide gradient gel. Protein molecular weight markers (in thousands; lanes M), p9, p93, and p102 are indicated.
The results of this experiment are shown in Fig. 3B. First, and most importantly, it is clear that p9 was generated only when the primary translation product included the predicted PCP1 domain (Fig. 3B, lanes 6 to 9, DNA 4/11, DNA 4/13, DNA 4/14, and DNA 4/15) (14). This result strongly suggests that HCV 229E PCP1 is responsible for the cleavage of p9 from the replicase polyproteins. Second, the data suggested that p9 was derived from a precursor, p102, which was cleaved to generate p9 and p93. This precursor was most clearly seen in the translation products of RNA derived from DNA 4/9 (Fig. 3B, lane 5), and it is evident that PCP1 activity was not required for the cleavage of p102 from larger precursors. Third, the cleavage of p102 to p9 and p93 appeared to be more effective than the cleavage of p102 from its precursor(s).
Obviously, there are other, more complex interpretations of the data shown in Fig. 3B. For example, p102 could represent a premature termination product of translation rather than a proteolytic product. Also, at least in vivo, the cleavage of p9 from its precursor could precede the generation of p93. Further experiments will be needed to resolve these questions.
Codon mutagenesis of HCV 229E PCP1.
The results shown above suggest that HCV 229E PCP1 is responsible for the cleavage of p9 from the replicase polyproteins. To strengthen this conclusion and to provide experimental data to support the prediction of Cys-1054 and His-1205 as the catalytic residues of this proteolytic activity (14), we carried out codon mutagenesis of the HCV 229E PCP1 domain.
A recombinant plasmid, pT7-IRES-Pap, containing a T7 RNA polymerase promoter and an EMCV IRES element followed by the coding sequence of the amino-terminal 1315 amino acids of pp1a/pp1ab was constructed. Derivatives of this plasmid were then generated by in vivo recombination mutagenesis. In these plasmids, the codons for the cysteine residues Cys-962 and Cys-1054 and the histidine residues His-1205 and His-1278 of pp1a and pp1ab have been changed. The resulting plasmids were transfected into HeLa cells, and transcripts were synthesized after infection with vaccinia virus MVA-T7. Newly synthesized proteins were metabolically labeled from 4 to 6 h postinfection, and cell protein lysates were immunoprecipitated with IS1720 serum.
In cells transfected with pT7-IRES-Pap DNA, p9 and a processed form of the full-length translation product (p137) could be easily identified (Fig. 4, lane 2). When Cys-962 and His-1278 were changed to either Gly (pT7-IRES-PapC962G [Fig. 4, lane 3]) or Gly, Val, and Ala (pT7-IRES-PapH1278G [lane 9], pT7-IRES-PapH1278A [lane 10], and pT7-IRES-PapH1278V [lane 11], respectively), proteolytic processing remained unaffected. In contrast, changes in the predicted catalytic amino acids Cys-1054 to Arg, Gly, and Ser (pT7-IRES-PapC1054R [Fig. 4, lane 4], pT7-IRES-PapC1054G [lane 5], and pT7-IRES-PapC1054S [lane 6], respectively) and His-1205 to Ala and Gly (pT7-IRES-PapH1205A [lane 7] and pT7-IRES-PapH1205G [lane 8], respectively) completely abolished the generation of p9.
FIG. 4.

Codon mutagenesis of HCV 229E PCP1. HeLa cells were mock transfected (lane 1) or DNA transfected (lanes 2 to 11) and then infected with MVA-T7. The cells were metabolically labeled, and cell protein lysates were immunoprecipitated with IS1720 antiserum. The immunoprecipitated proteins were analyzed by electrophoresis with an SDS-containing 10 to 17.5% polyacrylamide gradient gel. Protein molecular weight markers (in thousands; lanes M) and p9, p93, p102, p137, and p146 are indicated.
In cells transfected with plasmids coding for changes in the predicted catalytic residues, a full-length translation product with a molecular weight of 146,000 was detected. In cells transfected with plasmids coding for changes in noncatalytic residues, the size of this translation product was reduced to 137,000. Proteins that corresponded in size to p102 (inactive proteinase) and p93 (active proteinase) were also detected in this experiment, but they were no more prominent than numerous other proteins that were probably the result of premature termination events during transcription or translation.
Mapping of the PCP1 domain.
In order to identify the amino-terminal and carboxyl-terminal borders of the active HCV 229E PCP1 proteinase, we used an in vitro trans-cleavage assay in combination with deletion mutagenesis. This approach allows for the modification of the enzyme without introducing changes in the substrate molecule and significantly simplifies the interpretation of the results.
As a substrate in these experiments, we used an in vitro translation product representing the amino-terminal 956 amino acids of pp1a and pp1ab (encoded by DNA 4/7). To produce carboxyl-terminally truncated proteins with putative enzymatic activity, we used a series of PCR DNA templates (DNA 4/5 to DNA 4/12) that encode HCV 229E pp1a-pp1ab amino acids 1 to 111 through 1 to 1500 (Fig. 5A). To produce amino-terminally truncated proteins with putative enzymatic activity, we used derivatives of plasmid pT7-IRES-Pap with deletions affecting codons 2 to 577 (pT7-IRES-Papdel2-577), 2 to 860 (pT7-IRES-Papdel2-860), 2 to 975 (pT7-IRES-Papdel2-975), and 2 to 1036 (pT7-IRES-Papdel2-1036) of HCV 229E ORF1a (Fig. 5A).
FIG. 5.
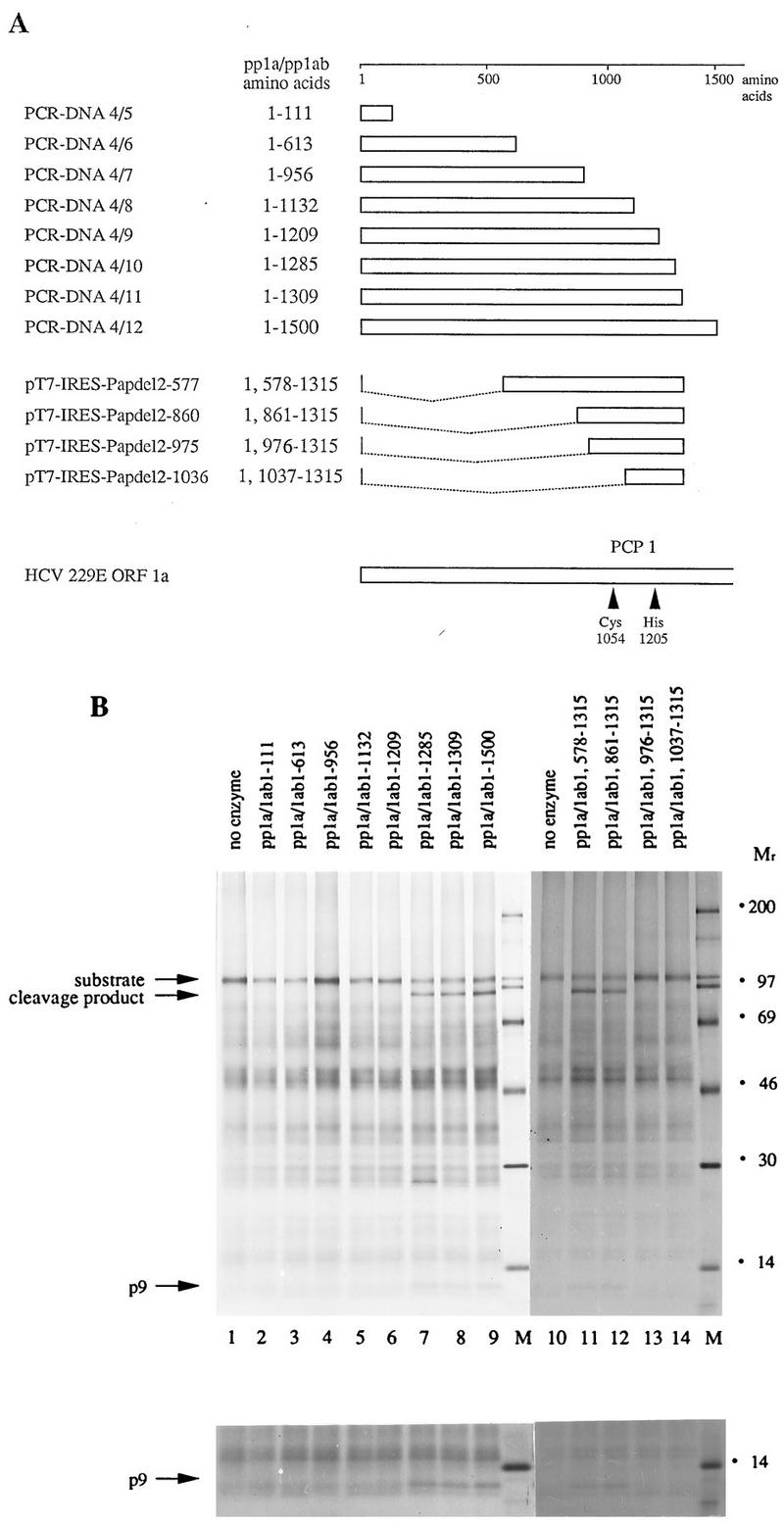
Mapping the borders of HCV 229E PCP1. (A) Schematic representation of HCV 229E ORF1a and the polypeptides tested for proteolytic activity. The putative catalytic amino acids of PCP1 are also indicated. (B) trans-Cleavage assay with amino-terminally and carboxyl-terminally truncated pp1a and pp1ab proteins. To produce a substrate for the trans-cleavage assay, the amino-terminal 956 amino acids of pp1a and pp1ab were translated in vitro in a reticulocyte lysate in the presence of [35S]methionine. The polypeptides to be tested for proteolytic activity were translated in vitro in a reticulocyte lysate in the presence of [32S]methionine. After the termination of translation, 1 volume of substrate reaction mixture was incubated without (lanes 1 and 10) or with (lanes 2 to 9 and 11 to 14) 2.5 volumes of enzyme reaction mixture for 3 h at 30°C. The cleavage reaction products were then immunoprecipitated with IS1720 antiserum, and the proteins were analyzed by electrophoresis in an SDS-containing 10 to 17.5% polyacrylamide gradient gel. Protein molecular weight markers (in thousands; lanes M), the uncleaved substrate, and the two cleavage products, p9 and a larger protein, are indicated. The lower panel shows a longer exposure of the low-molecular-weight region of the gel.
The results of this experiment are shown in Fig. 5B. With respect to the carboxyl-terminal truncations, pp1a and pp1ab proteins extending to amino acid 1285 or beyond (Fig. 5B, lanes 7, 8, and 9) had proteolytic activity, as evidenced by the generation of a larger cleavage product (molecular weight, approximately 90,000) and p9. Proteins ending at amino acid 1209 or earlier (Fig. 5B, lanes 2 to 6) did not have this enzymatic activity. Thus, the carboxyl-terminal border of HCV 229E PCP1 must lie between amino acids 1209 and 1285. With respect to the amino-terminal deletions, the data showed that proteins lacking pp1a-pp1ab amino acids 2 to 578 or 2 to 861 retained proteinase activity (Fig. 5B, lanes 12 and 13), while a deletion of amino acids 2 to 975 or 2 to 1037 rendered the proteins inactive (lanes 14 and 15). Thus, the amino-terminal border of HCV 229E PCP1 must lie between amino acids 861 and 975. As expected, the probable catalytic residues, Cys-1054 and His-1205, lie within these boundaries.
PCP1 cleaves the Gly-111–Asn-112 peptide bond.
The in vitro trans-cleavage assay described above also allowed us to determine the HCV 229E PCP1 cleavage site used for the generation of p9. Thus, the amino-terminal 956 amino acids of pp1a and pp1ab were translated in vitro with either [35S]cysteine or [35S]methionine as the radiolabel. These substrates were incubated together with in vitro-synthesized enzyme and, after transfer to PVDF membranes, the position of the carboxyl-terminal proteolytic product was determined by autoradiography. This area of the membrane was then isolated, and the bound protein was subjected to 20 cycles of Edman degradation. The results are shown in Fig. 6. Peaks of radioactivity were found at position 10 when the substrate was labeled with [35S]cysteine and at position 18 when the substrate was labeled with [35S]methionine.
FIG. 6.

Identification of the PCP1 cleavage site. Preparative-scale trans-cleavage reactions were carried out with [35S]methionine- or [35S]cysteine-labeled substrate. After immunoprecipitation, SDS-polyacrylamide gel electrophoresis, and electrophoretic transfer to PVDF membranes, the position of the carboxyl-terminal cleavage product was determined by autoradiography, the isolated proteins were subjected to 20 cycles of Edman degradation, and the distribution of radiolabeled amino acids was determined. The amino acid sequence of pp1a and pp1ab from positions 112 to 131 is shown. The amino acids Cys-121 and Met-129 are underlined.
The amino acid sequence of the amino-terminal 956 amino acids of pp1a and pp1ab was examined for the pattern Cys-X7-Met, and only the sequence Cys-Gly-Ala-Asp-Gly-Lys-Pro-Val-Met at positions 121 to 129 was found. Thus, the amino terminus of the carboxyl-terminal cleavage product can be identified as pp1a-pp1ab amino acids NH2-111AsnValThr113, and the HCV 229E PCP1 cleavage site used to generate p9 can be deduced to be NH2-107LysArgGlyGlyGly|AsnValThrTyrThr116-COOH, with cleavage occurring between residues Gly-111 and Asn-112.
DISCUSSION
The data presented in this paper represent the first characterization of the HCV 229E PCP1 proteinase. In many respects, the results that we obtained parallel those of earlier studies on the MHV PCP1 proteinase (1, 6, 17); in other respects, they reveal some intriguing and significant differences.
First, in keeping with previous predictions (14), mapping of the HCV 229E PCP1 domain indicates that the HCV 229E proteinase shares a common location with its counterpart from MHV, approximately 900 to 1300 amino acids from the amino terminus of the replicase polyproteins pp1a and pp1ab. However, despite this overall congruity, a closer analysis of the data reveals some specific differences. For example, our results located the amino-terminal border of the active HCV 229E PCP1 domain between pp1a-pp1ab amino acids 861 and 975. Bonilla et al. (3) reported that the active MHV PCP1 domain lies between pp1a-pp1ab amino acids 1084 and 1316. When the HCV 229E and MHV pp1a and pp1ab sequences are optimally aligned in this region (8a), the MHV pp1a-pp1ab amino acid Ala-1084 is found to correspond to the HCV 229E pp1a-pp1ab amino acid Thr-1025. Thus, the HCV 229E PCP1 proteinase domain seems to be extended at the amino terminus relative to the MHV PCP1 proteinase domain. Further detailed experiments will be required to assess the significance of this observation.
Second, as indicated by a sequence alignment analysis (14), our results, combined with those of Baker et al. (1), suggest that homologous residues (Cys-1054 and His-1205 for HCV 229E and Cys-1121 and His-1272 for MHV) probably act as the catalytic amino acids for both enzymes. Also, on the basis of our results, it is most likely that coronavirus PCP1 enzymes are able to function in trans, not only in vitro (4) but also in transfected cells.
Third, it is striking that the cleavage sites used by the HCV 229E and MHV PCP1 proteinases at the amino terminus of the replicase polyproteins are different in both position and sequence. Thus, the MHV activity cleaves a protein, p28, from the amino terminus of pp1a and pp1ab, and the recognition site is NH2-243ArgGlyTyrArgGlyValLysProIleLeu252-COOH, with cleavage between Gly-247 and Val-248 (6, 17). The HCV 229E PCP1 proteinase cleaves a protein, p9, from the amino terminus of pp1a and pp1ab, and the recognition site is NH2-107LysArgGlyGlyGlyAsnValThrTyrThr116-COOH, with cleavage between Gly-111 and Asn-112. Thus, not only the position and recognition sequence but also the scissile bond of PCP1-mediated processing are clearly different for these two viruses.
The coronavirus PCP1 substrates described in this paper and by others (4, 6, 17) obey a general pattern which includes cleavage between small uncharged residues: a basic amino acid at the P5 position and relative flexibility at the P2, P3, and P4 positions. Within this framework, the viruses differ by using, for instance, different small residues in the P1 and P1′ positions. The MHV p28-p65 cleavage site, the HCV 229E PCP1 cleavage site, a homologous sequence from TGEV, and their neighboring sequences can be aligned as two ungapped blocks, AI and AII (Fig. 7A). Block AI comprises the aligned cleavage sites but is not statistically significant and cannot be selected without prior knowledge of the locations of (putative) functionally equivalent residues which are conserved in the essential P1 and P5 positions of the HCV 229E and MHV PCP1 cleavage sites. According to the alignment of Fig. 7A, a region around the PCP1 cleavage site might have evolved by accepting replacements as well as insertions or deletions immediately downstream of this site in the three coronavirus lineages. This alignment also predicts that Arg-106 and Gly-110 will occupy, respectively, the P5 and P1 positions of a putative PCP1 cleavage site in the TGEV pp1a and pp1ab polyproteins.
FIG. 7.

Multiple amino acid alignments of three coronavirus pp1a and pp1ab sequences around the PCP1 cleavage site. The gap-free alignments (blocks) shown in uppercase letters were generated by use of the MACAW program (27) with a Gibbs sampler (21) and the Blossum62 scoring table (11). The statistical significance (19) of the alignments was assessed with a searching space comprised of the MHV A59, HCV 229E, and TGEV pp1a sequences. Fragments of the sequences not aligned by the program are shown in lowercase type. Roman bold type indicates residues conserved in any two sequences; italic bold type indicates a positive residue occupying the P5 position relative to the (putative) PCP1 cleavage site, whose position is indicated in the MHV A59 and HCV 229E sequences with ><. The positions of the fragments aligned within the pp1a and pp1ab sequences and sequence database accession numbers are indicated on the left and right sides of the alignments, respectively (GB, GenBank; SP, Swissprot). (A) Alignment constrained by the need to have the P5 and P1 residues of the PCP1 cleavage sites of MHV A59 and HCV 229E in equivalent positions. The statistical significances of the similarities in block AI and block AII were 1.0e+0 (statistically nonsignificant) and 1.2e−3, respectively. (B) Alignment including the statistically most favorable block encompassing the PCP1 cleavage sites in MHV A59 and HCV 229E. The statistical significances of the similarities in block BI and block BII were 1.0e+0 (statistically nonsignificant) and 4.4e−5, respectively.
It is important to note, however, that an alternative alignment of the region encompassing the PCP1 cleavage site can be deduced by a comparative sequence analysis (Fig. 7B). This alignment includes a block-ungapped BII, the only statistically significant block identified within the amino-terminal region of the pp1a and pp1ab polyproteins upstream of the PCP1 domain. Most notably, in the analysis shown in Fig. 7B, the MHV and HCV 229E cleavage sites are shifted by two residues relative to one another. This alignment suggests that no insertions or deletions have been accepted in a region delimited between the cleavage site and the downstream conserved region (tripeptide Asp-Gln-Tyr) in the three coronavirus lineages. If this suggestion is correct, then the position of the PCP1 cleavage site has migrated in these polyproteins over the course of evolution. Also, for TGEV, cleavage at either Gly-110–Ala-111 or Thr-107–Gly-108, both of which conform to PCP1 site rules (see above), would be compatible with this model.
Irrespective of which analysis correctly reflects the ancestral relationships among the proteins of the three coronaviruses, both alternatives can be reconciled if it is assumed that the PCP1 cleavage site region is multifunctional and under complex selective pressure driven by both divergent and convergent evolution.
It is also worth noting that, in contrast to the situation with MHV (4), our data provide no indication of further HCV 229E PCP1-mediated cleavages in the replicase polyproteins, at least within the first 1,500 amino acids.
Very little is known about the function of HCV 229E protein p9 or, indeed, of any of the proteins derived from coronavirus pp1a and pp1ab proteins by PCP1 activity. This lack of knowledge is partly due to the fact that there are no obvious sequences from which a putative function can be deduced. Immunofluorescence assays of HCV 229E-infected cells with IS1720 serum showed a punctate pattern of staining in the perinuclear region, like that found with antisera specific for the HCV 229E 3C-like proteinase, antisera specific for the putative metal-binding and helicase protein (p71) (16), and a monoclonal antibody specific for p41, a 3C-like proteinase-mediated processing product encoded by ORF1b (15). This result suggests that at least one of the proteins reacting with IS1720 serum remains associated with the viral replication complex and therefore may have a role in RNA replication and transcription.
Now that we have identified the trans-active domain of HCV 229E PCP1 and a corresponding substrate recognition sequence, we will try to (over)express a biologically active form of the PCP1 protein in bacteria or eucaryotic cells. This approach has been very successful for HCV 229E 3C-like proteinase (31, 32) and would allow for detailed biochemical and structural studies on the papain-like proteinases of coronaviruses. Structural studies on a purified form of HCV 229E PCP1 would be very desirable because this enzyme is an obvious target for the design of synthetic inhibitors to control coronavirus infections.
ACKNOWLEDGMENTS
We thank A. Weidmann for the preparation of MVA-T7 stocks and J. Hoppe and V. Hoppe for protein sequencing data.
This work was supported by a grant from the DFG (SFB 165/B1). During this work, A.E.G. was an SFB Visiting Professor at the Institute of Virology, Würzburg, Germany. A.E.G. was supported by The Netherlands Organization for Scientific Research (NWO) and the Russian Fund for Basic Research (grant 96-04-49562).
REFERENCES
- 1.Baker S C, Yokomori K, Dong S, Carlisle R, Gorbalenya A E, Koonin E V, Lai M M C. Identification of the catalytic sites of a papain-like cysteine proteinase of murine coronavirus. J Virol. 1993;67:6056–6063. doi: 10.1128/jvi.67.10.6056-6063.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonilla P J, Gorbalenya A E, Weiss S R. Mouse hepatitis virus strain A59 RNA polymerase ORF 1a: heterogeneity among MHV strains. Virology. 1994;198:736–740. doi: 10.1006/viro.1994.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonilla P J, Hughes S A, Pinon J D, Weiss S R. Characterization of the leader papain-like proteinase of MHV-A59: identification of a new in vitro cleavage site. Virology. 1995;209:489–497. doi: 10.1006/viro.1995.1281. [DOI] [PubMed] [Google Scholar]
- 4.Bonilla P J, Hughes S A, Weiss S R. Characterization of a second cleavage site and demonstration of activity in trans by the papain-like proteinase of the murine coronavirus mouse hepatitis virus strain A59. J Virol. 1997;71:900–909. doi: 10.1128/jvi.71.2.900-909.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boursnell M E, Brown T D K, Foulds I J, Green P F, Tomley F M, Binns M M. Completion of the sequence of the genome of the coronavirus avian infectious bronchitis virus. J Gen Virol. 1987;68:57–67. doi: 10.1099/0022-1317-68-1-57. [DOI] [PubMed] [Google Scholar]
- 6.Dong S, Baker S C. Determinants of the p28 cleavage site recognized by the first papain-like cysteine-proteinase of murine coronavirus. Virology. 1994;204:541–549. doi: 10.1006/viro.1994.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dougherty W G, Semler B L. Expression of virus-encoded proteinases: functional and structural similarities with cellular enzymes. Microbiol Rev. 1993;57:781–822. doi: 10.1128/mr.57.4.781-822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eleouet J-F, Rasschaert D, Lambert P, Levy L, Vende P, Laude H. Complete sequence (20 kilobases) of the polyprotein-encoding gene 1 of transmissible gastroenteritis virus. Virology. 1995;206:817–822. doi: 10.1006/viro.1995.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a.Gorbalenya, A. E. Unpublished data.
- 9.Gorbalenya A E, Koonin E V, Donchenko A P, Blinov V M. Coronavirus genome: prediction of putative functional domains in the nonstructural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grötzinger C, Heusipp G, Ziebuhr J, Harms U, Süss J, Siddell S G. Characterization of a 105-kDa polypeptide encoded in gene 1 of the human coronavirus HCV 229E. Virology. 1996;222:227–235. doi: 10.1006/viro.1996.0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henikoff S, Henikoff J G. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herold J, Siddell S G. An elaborated pseudoknot is required for high frequency frameshifting during translation of HCV 229E polymerase mRNA. Nucleic Acids Res. 1993;21:5838–5842. doi: 10.1093/nar/21.25.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herold J, Siddell S G, Ziebuhr J. Characterization of coronavirus RNA polymerase gene products. Methods Enzymol. 1996;275:68–69. doi: 10.1016/S0076-6879(96)75007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herold J, Raabe T, Schelle-Prinz B, Siddell S G. Nucleotide sequence of the human coronavirus 229E RNA polymerase locus. Virology. 1993;195:680–691. doi: 10.1006/viro.1993.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heusipp, G., C. Grötzinger, J. Herold, S. G. Siddell, and J. Ziebuhr. Identification and subcellular localization of a 41 kDa, polyprotein lab processing product in human coronavirus 229E-infected cells. J. Gen. Virol., in press. [DOI] [PubMed]
- 16.Heusipp G, Harms U, Siddell S G, Ziebuhr J. Identification of an ATPase activity associated with a 71-kilodalton polypeptide encoded by gene 1 of the human coronavirus 229E. J Virol. 1997;71:5631–5634. doi: 10.1128/jvi.71.7.5631-5634.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes S A, Bonilla P J, Weiss S R. Identification of the murine coronavirus p28 cleavage site. J Virol. 1995;69:809–813. doi: 10.1128/jvi.69.2.809-813.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnston S, Holgate S. Epidemiology of viral respiratory tract infections. In: Myint S, Taylor D, editors. Viral and other infections of the human respiratory tract. London, United Kingdom: Chapman & Hall, Ltd.; 1996. pp. 1–38. [Google Scholar]
- 19.Karlin S, Altschul S F. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc Natl Acad Sci USA. 1990;87:2264–2268. doi: 10.1073/pnas.87.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kräusslich H-G, Wimmer E. Viral proteinases. Annu Rev Biochem. 1988;57:701–754. doi: 10.1146/annurev.bi.57.070188.003413. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence C E, Altschul S F, Boguski M S, Liu J S, Neuwald A F, Wootton J C. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- 22.Lee H J, Shieh C K, Gorbalenya A E, Koonin E V, la Monica N, Tuler J, Bagdzhadzhyan A, Lai M M C. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology. 1991;180:567–582. doi: 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu D X, Brown T D K. Characterization and mutational analysis of an ORF 1a-encoding proteinase domain responsible for proteolytic processing of the infectious bronchitis virus 1a/1b polyprotein. Virology. 1995;209:420–427. doi: 10.1006/viro.1995.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moss B, Elroy-Stein O, Mitsukami T, Alexander W A, Fuerst T R. New mammalian expression vectors. Nature. 1990;348:91–92. doi: 10.1038/348091a0. [DOI] [PubMed] [Google Scholar]
- 25.Myint S H. Human coronavirus infections. In: Siddell S G, editor. The coronaviridae. New York, N.Y: Plenum Press; 1995. pp. 389–401. [Google Scholar]
- 26.Raabe T, Schelle-Prinz B, Siddell S G. Nucleotide sequence of the gene encoding the spike glycoprotein of human coronavirus HCV 229E. J Gen Virol. 1990;71:1065–1073. doi: 10.1099/0022-1317-71-5-1065. [DOI] [PubMed] [Google Scholar]
- 27.Schuler G D, Altschul S F, Lipman D J. A workbench for multiple alignment construction and analysis. Proteins. 1991;9:180–190. doi: 10.1002/prot.340090304. [DOI] [PubMed] [Google Scholar]
- 28.Siddell S G. The coronaviridae—an introduction. In: Siddell S G, editor. The coronaviridae. New York, N.Y: Plenum Press; 1995. pp. 1–10. [Google Scholar]
- 29.Sutter G, Ohlmann M, Erfle V. Nonreplicating vaccinia vector efficiently expresses bacteriophage-T7 RNA-polymerase. FEBS Lett. 1995;371:9–12. doi: 10.1016/0014-5793(95)00843-x. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X M, Herbst W, Kousoulas K G, Storz J. Biological and genetic characterization of hemagglutinating coronavirus isolated from a diarrhoeic child. J Med Virol. 1994;44:152–161. doi: 10.1002/jmv.1890440207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ziebuhr J, Heusipp G, Siddell S G. Biosynthesis, purification, and characterization of the human coronavirus 229E 3C-like proteinase. J Virol. 1997;71:3992–3997. doi: 10.1128/jvi.71.5.3992-3997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziebuhr J, Herold J, Siddell S G. Characterization of a human coronavirus (strain 229E) 3C-like proteinase activity. J Virol. 1995;69:4331–4338. doi: 10.1128/jvi.69.7.4331-4338.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]