

Abstract
To better understand the early biochemical events that occur in human rhinovirus (HRV) infections, we examined the kinetics and mechanisms of interleukin-8 (IL-8) and IL-6 production from infected epithelial cells. Several HRV strains caused IL-8 and IL-6 production, but HRV-16 induced maximal IL-8 and IL-6 mRNA expression and protein production more rapidly than did HRV-14, despite similar rates of replication of the two viral strains. Viral induction of cytokine mRNA does not require new protein synthesis, since it was unaffected by cycloheximide treatment. The potent glucocorticoid budesonide did not affect viral replication or cytokine mRNA induction but modestly inhibited cytokine protein production. Interestingly, the nitric oxide donor 3-(2-hydroxy-2-nitroso-1-propylhydrazino)-1-propanamine (NONOate) inhibited both rhinovirus replication and cytokine production in a dose-dependent fashion without reducing levels of cytokine mRNA. The NONOate effects were due to release of nitric oxide, because NONOate that had been depleted of its nitric oxide content had no effect. Thus, nitric oxide may play an important anti-inflammatory and antiviral role in colds and nitric oxide donors may represent a novel therapeutic approach.
Rhinovirus infections are the predominant cause of the common cold (18), the most frequently experienced acute respiratory illness in humans. Recent evidence also implicates rhinovirus infections as important precipitating factors for the exacerbation of asthma (21, 22, 37), chronic bronchitis (35), sinusitis (19, 50), and otitis media (3). Despite the high health care costs associated with rhinovirus infections, the underlying process by which viral infection leads to symptomatology is poorly understood.
The epithelial cell is the primary site of rhinovirus infection (6, 51). In contrast to other respiratory viruses, such as influenza, cytotoxic damage of infected epithelial cells does not appear to play a role in the pathogenesis of rhinovirus infections, since cytotoxicity is not observed either in infected human epithelial cell cultures (49) or in the nasal mucosa of infected individuals (53, 54). In light of this, emphasis has been focused on the concept that symptoms may result from the actions of proinflammatory mediators that are generated as a consequence of rhinovirus infection. Support for this hypothesis has come from two lines of evidence: (i) studies of subjects with experimentally induced or naturally acquired colds have demonstrated increased levels of several mediators, including kinins (36, 41), interleukin-1 (IL-1) (40), and IL-6 (55), in nasal secretions during symptomatic rhinovirus infections; and (ii) infection of purified human respiratory epithelial cell populations with rhinovirus has been shown to induce production of proinflammatory cytokines, including IL-8, IL-6, and granulocyte-macrophage colony-stimulating factor (49, 55), that could contribute to disease pathogenesis. To date, however, the specific biochemical events involved in the production of each of these cytokines by rhinoviruses are incompletely understood and the role of specific cytokines, and other mediators, in the pathogenesis of colds remains to be established.
The present studies were undertaken to further delineate the kinetics and mechanisms of rhinovirus-induced cytokine generation by epithelial cells and to evaluate the effects of potential therapeutic interventions on these pathways. We have focused on viral production of IL-8 and IL-6, because these cytokines are produced in relatively large amounts upon rhinovirus infection and because they have biological properties that are of interest with respect to the pathogenesis of colds. IL-8 is a potent chemoattractant for, and activator of, neutrophils (5) and also has chemotactic activity for lymphocytes (28), the two predominant cell types in the nasal mucosa during rhinovirus infections (29, 54). IL-6 is not only capable of stimulating T-cell activation and inducing B-cell differentiation and antibody production (1); it can also stimulate mucosal immunoglobulin A immune responses (42). Of the potential interventions, we have used two approaches. Based upon the wide-ranging immunomodulatory and anti-inflammatory effects of glucocorticoids (44), including their ability to inhibit the production of several cytokines in a variety of cell types (45), we examined the effects of the potent glucocorticoid budesonide on rhinovirus infection in epithelial cells. As a novel alternative approach, we also investigated the ability of a nitric oxide (NO) donor to inhibit viral replication and virally induced cytokine production in epithelial cells. Studies have demonstrated that the vasodilator NO can exert modulatory effects on inflammation (39), and nitric oxide has been shown to have antiviral effects in some animal models (7, 12, 20, 24, 32), but this property has not been examined in human respiratory epithelial cells. Our studies show that budesonide modestly inhibits rhinovirus-induced cytokine generation without affecting viral replication. By contrast, NO markedly inhibits rhinovirus-induced cytokine generation as well as viral replication and may play a therapeutic role in rhinovirus infections.
MATERIALS AND METHODS
Materials.
The following reagents were purchased from the indicated suppliers: Dulbecco’s minimal essential medium, Eagle’s minimal essential medium (EMEM), Ham’s F-12 medium, Hanks balanced salt solution (HBSS), l-glutamine, penicillin-streptomycin-amphotericin B (Fungizone), trace elements, and retinoic acid (Biofluids, Rockville, Md.); hydrocortisone, epithelial cell growth factor, and endothelial cell growth supplement (Collaborative Research, Bedford, Mass.); fetal bovine serum (Gemini Bio Products, Inc., Calabasa, Calif.); transferrin and insulin (GIBCO BRL, Grand Island, N.Y.); 3-(2-hydroxy-2-nitroso-1-propylhydrazino)-1-propanamine (NONOate) (Cayman Chemical Company, Ann Arbor, Mich.); RNAzol B (Tel-Test, Inc., Friendswood, Tex.); agarose (FMC Bioproducts, Rockland, Maine); MOPS (morpholinepropanesulfonic acid) (Boehringer Mannheim, Indianapolis, Ind.); and [α-32P]dCTP (Amersham, Arlington Heights, Ill.). All other chemicals were purchased from Sigma Chemical Company (St. Louis, Mo.). Budesonide was generously provided by Per Andersson and Ralph Brattsand (Astra Pharmaceuticals, Lund, Sweden).
The following stock buffers were employed: 10× MOPS (0.2 M MOPS, 0.05 M sodium acetate, 0.01 M EDTA), 50× Denhardt’s solution (1% Ficoll, 1% polyvinylpyrrolidone, 1% bovine serum albumin), and 20× SSPE (175.3 g of NaCl, 27.6 g of NaH2PO4 · H2O, 7.4 g of EDTA in 1 liter of H2O [pH 7.4]).
Viruses and cell lines.
Human rhinovirus type 14 (HRV-14), HRV-16, HRV-39, and HRV-1A, WI-38 cells, and HeLa cells were purchased from the American Type Culture Collection (Rockville, Md.). Additional viral stocks for HRV-14 and HRV-16 were generated by passage in HeLa or WI-38 cells, respectively, as previously described (49). It was not possible to generate equivalent stocks of these two viral strains with the same host cell line, since the two strains displayed marked preferences in terms of their capacities to infect and replicate in these cell lines. This variable sensitivity of host cells to different strains of rhinovirus has been documented previously (11). For some experiments, HRV-16 was purified to remove ribosomes and soluble factors of WI-38 origin by centrifugation through sucrose, according to published methods (16). Inactivation of HRV-16 was performed by UV exposure for 30 min as previously described (49). For experiments using HRV-39 and -1A, viral stocks were used directly as obtained from the supplier. The HRV-39 stock had been prepared in WI-38 cells, while the HRV-1A stock was generated in HeLa cells. The BEAS-2B cell line (43) was generously provided by Curtis Harris (National Cancer Institute, Bethesda, Md.).
Epithelial cell culture.
Primary human tracheal epithelial cells were obtained by protease digestion of human tissue as previously described (10). Both primary cells and BEAS-2B cells were grown in culture medium consisting of Ham’s F-12 nutrient medium with penicillin (100 U/ml), streptomycin (100 U/ml), amphotericin B (250 ng/ml), l-glutamine (2 mM), phosphoethanolamine-ethanolamine (0.5 mM), transferrin (10 μg/ml), endothelial cell growth supplement (3.75 μg/ml), epidermal growth factor (12.5 ng/ml), insulin (5 μg/ml), hydrocortisone (10−7 M), cholera toxin (10 ng/ml), 3,3′,5-triiodothyronine (3 × 10−9 M), retinoic acid (0.1 ng/ml), and trace elements. This medium is hereafter referred to as F12/10×. The cells were incubated at 37°C in 95% air and 5% CO2. For the experiments, cells between passages 35 and 50 were plated on 6-well plates or in 75-cm2 flasks (Costar, Cambridge, Mass.) at a density of 2.5 × 104/cm2.
Viral infection of BEAS-2B cells.
Monolayers of BEAS-2B cells (70 to 80% confluent) were washed three times with HBSS. HRV-14, -16, -39, or -1A was added to the cells at a concentration of 104 50% tissue culture infective dose (TCID50) units/ml of HBSS. This equates to an infectious dose of 0.01 TCID50 units/BEAS-2B cell, although it is unclear what this represents in terms of multiplicity of infection (infectious units per cell) for BEAS-2B cells, since the capacity of rhinovirus to infect different host cells is quite variable (see above). The cells were incubated with the virus at 34°C for 1 h and washed three times with F12/10×, and then fresh F12/10× medium was added to the cells. Supernatants were removed from the cells at various times after infection and stored at −80°C for later analysis of cytokine protein production and viral content. In some experiments, total cellular RNA was extracted from the cells at various times after infection and stored at −80°C for later analysis.
Titration of viruses.
Supernatants from infected cultures of BEAS-2B cells were collected at various times postinfection and assessed for viral content by cytotoxicity assays. For detection of HRV-14, confluent monolayers of HeLa cells in 96-well plates were exposed to serial dilutions of the supernatants as described previously (49). In a similar assay, HRV-16 was detected by incubating confluent monolayers of WI-38 cells in 96-well plates with serial dilutions of the virus-containing supernatants in EMEM with 10% fetal bovine serum. The plates were incubated at 34°C for 5 days. The medium was removed, and the cells were washed with HBSS and then fixed with methanol for 1 min. The methanol was removed, and the cells were incubated with 0.1% crystal violet in distilled water for 20 min. The plates were washed, and the absorbance at 570 nm was determined with a plate reader. The dilution of supernatant required to infect 50% of the monolayers was determined and expressed as log TCID50 units.
Quantification of IL-8 and IL-6.
Levels of cytokines in cell supernatants were determined by specific enzyme-linked immunosorbent assays (ELISAs). Measurements of IL-8 were performed by a previously described ELISA sensitive to 30 pg of cytokine/ml (49), while levels of IL-6 were assayed with a commercial kit sensitive to 15 pg of IL-6/ml (Biosource International, Camarillo, Calif.). Neither the culture medium nor vehicles for drugs used in our experiments caused any nonspecific interference effects in either assay.
Probes for Northern blotting.
A full-length cDNA for IL-8 was obtained by reverse transcription-PCR with RNA extracted from the human BEAS-2B cell line. The full-length cDNA was cloned into a pCR II vector (Invitrogen Corp., San Diego, Calif.) between two EcoRI sites and grown in competent Escherichia coli XL1-Blue cells (Stratagene, La Jolla, Calif.). The sequence of the cDNA probe used for Northern analysis was identical to the published sequence of IL-8 (31, 34), as determined by dideoxy sequencing. A full-length cDNA for IL-6 was kindly provided by Steven Gillis (Immunex, Seattle, Wash.). The full-length cDNA for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Clontech (Palo Alto, Calif.). Probes for IL-8, IL-6, and GAPDH were labeled to a high specific activity by the random-primer method (15) with [α32P]dCTP and a random-primer DNA labeling kit (Boehringer Mannheim). Unincorporated nucleotides were separated with Nuctrap push columns (Stratagene).
RNA extraction and Northern analysis.
Total cellular RNA was extracted from BEAS-2B cells with RNAzol B (1 ml/10 cm2) in a modification of the method of Chomczynski and Sacchi (9). Briefly, cell monolayers were lysed with RNAzol B and transferred to a 13-ml polypropylene tube to which chloroform (0.1 ml/1 ml RNAzol) was added. After being chilled on ice for 5 min, the samples were centrifuged at 7,900 × g for 30 min at 4°C. The aqueous phase was precipitated with an equal volume of ice-cold 95% ethanol at −20°C overnight. After a second centrifugation, the RNA pellet was washed twice in 75% ethanol, dried, and dissolved in 50 μl of 0.2% diethylpyrocarbonate-treated water. The integrity of each RNA was assessed by electrophoresis of an aliquot (0.5 μg) on a 1% agarose gel with 0.5 μg of ethidium bromide/ml of buffer. RNA was stored at −80°C.
For Northern analysis, equal amounts (15 to 20 μg) of RNA from each experimental condition were electrophoresed on a 1% agarose–2.2 M formaldehyde gel in a MOPS buffer system. The RNA was transferred to a nylon membrane (GeneScreen Plus; New England Nuclear Research Products, Wilmington, Del.). The membranes were cross-linked by exposure to UV light and then prehybridized in 10 ml of buffer containing 4.5 ml of formamide, 2.5 ml of 10× Denhardt’s solution, 2 ml of 20× SSPE, 1 ml of 20% sodium dodecyl sulfate (SDS), and 100 μg of denatured salmon sperm DNA/ml in a hybridization oven for 2 h at 42°C. Immediately following the prehybridization, the appropriate α-32P-labeled cDNA probe was added to the prehybridization solution, and the mixture was rotated for an additional 18 h at 42°C. The blots were washed to a final stringency of 0.2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–0.2% SDS at 60°C and exposed to film (Biomax MS; Kodak, New Haven, Conn.) with two Lightening Plus screens at −70°C. Films were routinely developed for various times to ensure that band intensities assessed by densitometry were within the linear range for the film. Densitometry was performed with a scanning densitometer (UVP [San Gabriel, Calif.] gel documentation system), and densitometric analysis was performed with National Institutes of Health Image software.
Effect of cycloheximide on HRV-16-induced IL-8 and IL-6 mRNA expression.
BEAS-2B cells were treated with cycloheximide (10 μg/ml) or the medium control for 1 h before viral infection. The drug was also present during and after infection with HRV-16. At 1 h postinfection, RNA was harvested for Northern analysis. This concentration of cycloheximide was used because it has previously been shown to inhibit tumor necrosis factor alpha (TNF-α)- and gamma interferon-induced expression of RANTES mRNA in this cell line (48).
Effect of budesonide on cytokine production and viral replication.
Budesonide was prepared as a 10−2 M stock solution in dimethyl sulfoxide. Since the BEAS-2B cells are usually maintained in growth medium containing low levels of hydrocortisone, the cells for these experiments were placed in medium without hydrocortisone for 24 h prior to treatment with the glucocorticoid. The cells were then treated with 10−7 M budesonide or appropriately diluted vehicle control for 24 h prior to viral infection. Budesonide was again included in the medium after viral infection. The concentration of budesonide used was selected because it has previously been shown to maximally inhibit TNF-α-induced RANTES production from BEAS-2B cells (48). Supernatants from cells with and without budesonide were removed at various times after viral infection and stored at −70°C for determination of IL-8 and IL-6 protein and viral content. In some experiments, Northern analysis was used to compare RNA extracted from the budesonide-treated cells 1 h after infection with that extracted from control infected cells.
Effect of NONOate on cytokine production and viral replication.
NONOate was prepared in alkaline solution (0.01 M NaOH) as a 100 mM stock solution, which was kept at 4°C until use. New stock solutions of NONOate were prepared for each experiment and used within 1 h of preparation. The defined half-life of NO release from NONOate is 76 min at pH 7.4 and 22°C (Cayman Chemical Co.). Under alkaline conditions, the NONOate does not release nitric oxide. Aliquots of the alkaline stock solution were added directly to the BEAS-2B culture medium (pH 7.4) in a final concentration range of 100 to 1,000 μM. For most experiments, the NONOate was present both during and following the virus exposure. In some experiments, the NONOate was added only during or only after exposure to virus. Supernatants from BEAS-2B cells incubated with or without NONOate were removed at various times after viral infection and stored at −70°C for later determination of IL-8 and IL-6 protein and viral content. In some cases, RNAs extracted at various times after infection from the NONOate-treated cells and from control infected cells were compared by Northern analysis. To control for nonspecific effects of the NONOate compound, experiments were performed in which cells were treated with an inactive solution of NONOate. Inactivation was accomplished by placing a 1,000 μM solution of NONOate in medium at pH 7.4 at room temperature for 24 h to allow the NONOate to release all of the available NO prior to adding it to the cell cultures.
Statistical analysis.
Data are expressed as the mean ± standard error of the mean (SEM). Comparisons of the kinetics of RNA expression, protein secretion, and viral titers were performed by a one-way analysis of variance (ANOVA). The effects of cycloheximide and glucocorticoid on RNA expression and protein secretion were compared by Student’s t test for paired samples. Comparisons of the effects of NONOate on cytokine production, viral titers, and RNA expression were made by two-way ANOVA with one repeated measure, except for the experiments comparing active and inactive NONOate, which were analyzed by a one-way ANOVA. When significant variance ratios were obtained, pairwise comparisons of the means were performed with the least significant difference multiple-range test (47). Differences were considered significant for values of P of <0.05.
RESULTS
Effects of cell passage.
Preliminary studies indicated that there was a marked effect of repeated cell passage on cytokine production from BEAS-2B cells. Although the data obtained were always qualitatively identical for each passage, there was a progressive effect of cell passage on absolute levels of cytokines produced. For example, in four experiments performed with identical protocols with consecutive cell passages, production of IL-8 decreased from 7,690 to 3,090 pg/ml. For this reason, each type of experiment described below was always carried out in matched experiments with the same cell passages.
Comparison of effects of several rhinovirus strains on cytokine production from BEAS-2B cells.
The effects on cytokine production of equal infective doses of four different strains of rhinovirus were compared in cultures of BEAS-2B cells. Three of the strains used, types 14, 16, and 39, are members of the major group of rhinoviruses, which use intercellular adhesion molecule-1 (ICAM-1) as their receptor, while type 1A is a member of the ICAM-1-independent minor group. All of the strains induced IL-8 and IL-6 production measured at 24 h following infection (Fig. 1). With all viral strains, generated levels of IL-8 were approximately 10-fold higher than those of IL-6.
FIG. 1.

Cytokine production from BEAS-2B cells 24 h after infection with each of several strains of HRV. Data represent the mean plus SEM from three experiments. (A) Production of IL-8; (B) production of IL-6.
Kinetics of cytokine mRNA expression and protein secretion.
Figure 2 demonstrates that mRNAs for IL-8 and IL-6 were significantly elevated within 1 h post-HRV-14 infection. Maximal expression occurred by 3 h postinfection, but mRNA levels were still higher than those in noninfected controls at 24 h postinfection. Induction of mRNA was followed by significant elevations in IL-8 and IL-6 proteins in the supernatants. Increased cytokine production occurred by 3 h postinfection and reached maximal concentrations by 24 h. Interestingly, the time course of IL-8 and IL-6 production after HRV-16 infection was more rapid than that observed for HRV-14 (Fig. 3). Maximal mRNA expression occurred within 1 h postinfection, and maximal protein production occurred within 7 h. Consistent with the data shown in Fig. 1, the magnitude of cytokine generation was about fourfold greater following HRV-16 infection than the response following HRV-14 infection (Fig. 3 versus 2). Because HRV-16 produced a more rapid and robust production of cytokines, and because it is the strain that will be used for later in vivo studies, subsequent experiments on the mechanism of virus-induced cytokine generation were performed with HRV-16. To confirm the specificity of HRV-16 effects, three matched experiments were performed comparing IL-8 generation by active and UV-inactivated viral preparations. Active virus generated 3,307 ± 1,156 pg of IL-8/ml, while UV-inactivated virus produced only 520 ± 90 pg/ml (control, noninfected cells produced 345 ± 110 pg/ml). Specificity was further confirmed by demonstrating that similar amounts of IL-8 were generated, in matched experiments, when cells were infected with our standard viral preparation or with an equal infective dose of the same stock of HRV-16 purified by sucrose density centrifugation (2,080 ± 340 and 1,750 ± 500 pg/ml, respectively; n = 3). The time course of viral induction of IL-8 and IL-6 was also identical for the standard and purified preparations of HRV-16 (data not shown).
FIG. 2.

Time course of induction of steady-state mRNA levels and proteins for IL-8 (left) and IL-6 (right) from HRV-14-infected BEAS-2B cells. The upper panels show Northern blots for each cytokine and for the housekeeping gene product, GAPDH. The center panels show densitometric ratios, while the lower panels show protein levels produced at each of the indicated time points. Data are from a representative experiment (n = 3).
FIG. 3.

Time course of induction of steady-state mRNA levels and protein for IL-8 (left) and IL-6 (right) from HRV-16-infected BEAS-2B cells. The upper panels show representative Northern blots for each cytokine and for the housekeeping gene product, GAPDH. The center panels show the means plus SEM of densitometric ratios for four experiments. The lower panels show the means plus SEM of amounts of protein produced for four experiments at each time point. Asterisks indicate significant increases in each parameter relative to the zero time control (P < 0.05 in each case).
Viral titers post-HRV-16 infection.
Supernatants were collected at various times postinfection and assessed for viral titers in the WI-38 cell cytotoxicity assay for HRV-16. Virus was detected in the culture medium beginning approximately 7 h following infection and progressively increased between 7 and 24 h after infection (Table 1). Supernatants collected during a second 24-h period after infection contained levels of virus similar to those seen after 24 h (Table 2). This pattern of viral titers is virtually identical to that previously observed with HRV-14 (49).
TABLE 1.
Viral content of supernatants from BEAS-2B cells at different times after infection with HRV-16
| Time postinfection (h) |
Viral titer (log TCID50 units)a |
|---|---|
| 0 | ND |
| 1 | ND |
| 3 | ND |
| 7 | 1.25 ± 0.25 |
| 16 | 2.25 ± 0.25 |
| 24 | 2.4 ± 0.13 |
Data represent the means ± SEM from four experiments. ND, not detectable.
TABLE 2.
Effect of NONOate on viral titers decreases with time
| Treatment | Viral titer (log TCID50 units)a
|
|
|---|---|---|
| 0–24 h | 24–48 h | |
| HRV-16 | 2.8 ± 0.1 | 2.9 ± 0.1 |
| HRV-16 + 300 μM NONOate | 2.5 ± 0.3 | 2.8 ± 0.1 |
| HRV-16 + 1,000 μM NONOate | 0.5 ± 0.5b | 2.4 ± 0.2 |
Data represent means ± SEM from three experiments.
P < 0.05 versus HRV-16 alone.
Effects of cycloheximide on IL-8 and IL-6 mRNA expression.
Levels of mRNA for IL-8 and IL-6 from HRV-16-infected cells treated with cycloheximide (10 μg/ml) were not different from those of control infected cells (Fig. 4). Comparisons were made at 1 h after viral infection, the time of peak mRNA expression in the kinetics studies described above.
FIG. 4.

Cycloheximide does not alter steady-state mRNA levels for IL-8 (left) and IL-6 (right) measured 1 h after infection with HRV-16. The upper panels show representative Northern blots, while the lower panels show the means plus SEM of densitometric ratios for four experiments.
Effect of glucocorticoid pretreatment on cytokine production and viral titers.
Cells were treated with 10−7 M budesonide or vehicle control for 24 h prior to viral infection. Comparisons of RNA expression and protein production were made at the times of maximal response as determined in the kinetics experiments described above: 1 h for mRNA and 7 h for cytokine protein production. IL-8 and IL-6 mRNA expression in HRV-16-infected BEAS-2B cells was not significantly altered by budesonide (Fig. 5). In every experiment, however, the production of IL-8 and IL-6 proteins from budesonide-treated BEAS-2B cells was lower than from control infected cells (P < 0.05 for paired comparison of normalized data). Viral titers (2.2 ± 0.6 log TCID50 units) were not altered by budesonide exposure.
FIG. 5.
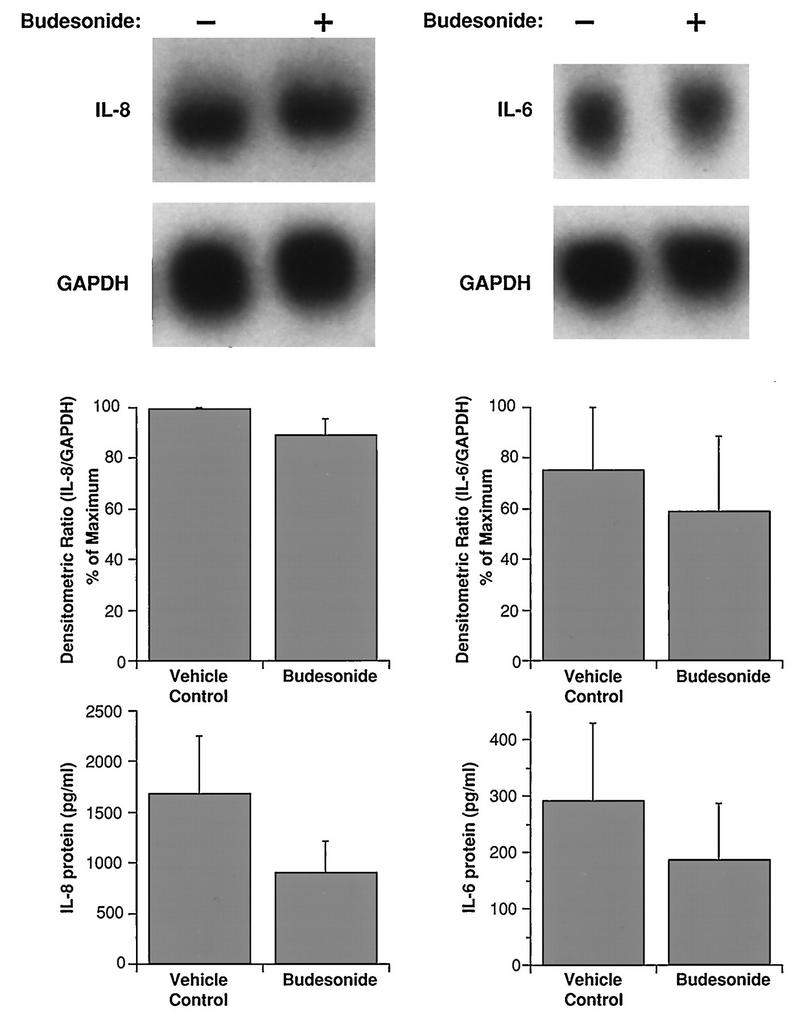
Effects of budesonide (10−7 M) on steady-state mRNA levels and proteins for IL-8 (left) and IL-6 (right) from HRV-16-infected BEAS-2B cells. The upper panels show representative Northern blots with mRNA extracted 1 h after infection. The center panels show the means plus SEM of densitometric ratios from three experiments. The lower panels show the means plus SEM of amounts of protein produced 7 h after infection in three experiments.
Effect of a nitric oxide donor on cytokine production and viral titers.
Supernatants were collected at 4 and 24 h post-HRV-16 infection from BEAS-2B cells incubated in the absence or presence of NONOate and were assayed for viral content and levels of cytokines. NONOate significantly inhibited IL-8 and IL-6 production in a dose-dependent manner (Fig. 6). IL-6 production was significantly inhibited by doses of NONOate as low as 100 μM. The levels of cytokine generated were inhibited more at 4 than at 24 h, presumably due to the waning levels of NO at 24 h. Viral titers were also significantly inhibited by NONOate (Fig. 7). Viral content in the supernatant collected at 24 h was almost completely eliminated by 1,000 μM NONOate. Supernatants from a second 24-h collection, however, contained similar amounts of virus whether the cells had been treated with NONOate or not (Table 2). In parallel studies, the effects of NONOate on epithelial cell viability and cell numbers were assessed. There was no significant effect of NONOate on cell viability at any dose or time. There was a small, but significant, decrease in cell numbers with 1,000 μM NONOate at 24 h only ([1.7 ± 0.4] × 106 cells/well without NONOate versus [1.2 ± 0.4] × 106 cells/well with NONOate [n = 3, P < 0.05]). No such effects were observed at lower NONOate doses. The effects of NONOate were also confirmed with purified HRV-16 (data not shown).
FIG. 6.
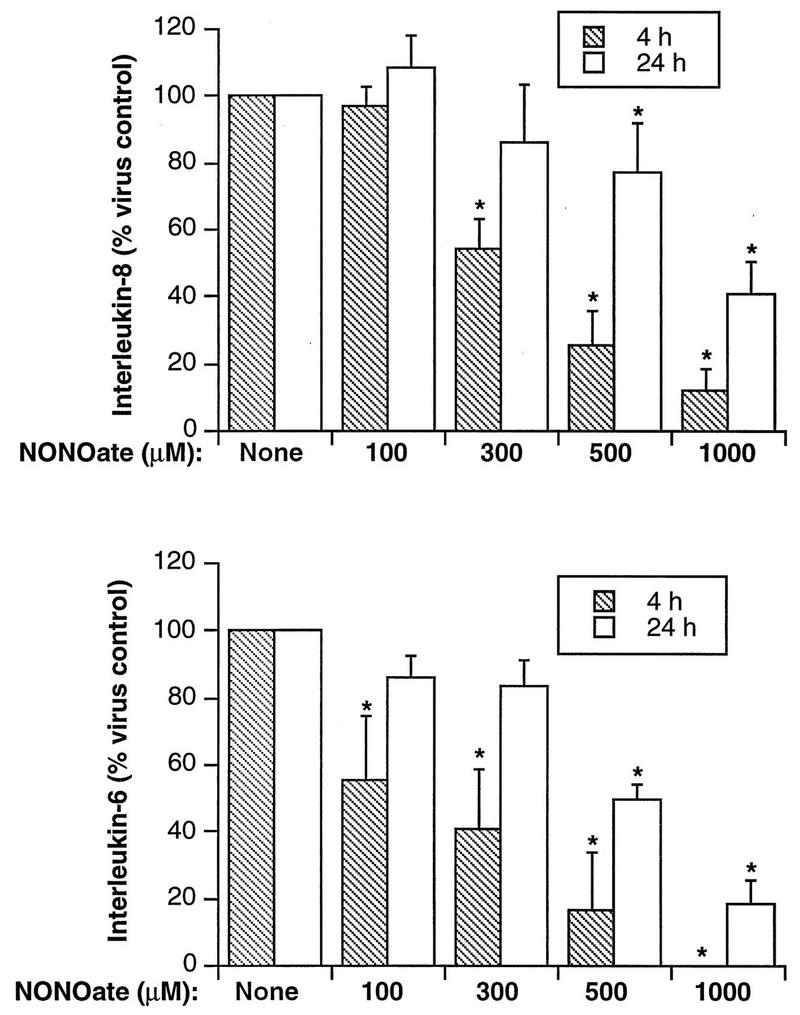
Dose-dependent inhibition of cytokine production from HRV-16-infected BEAS-2B cells by NONOate. The upper panel shows the means plus SEM from four experiments for IL-8 production at 4 and 24 h after HRV-16 infection, while the lower panel shows data for IL-6 production. The asterisks indicate significant inhibition compared to levels produced at the same time after infection in the absence of NONOate (P < 0.05 in each case).
FIG. 7.
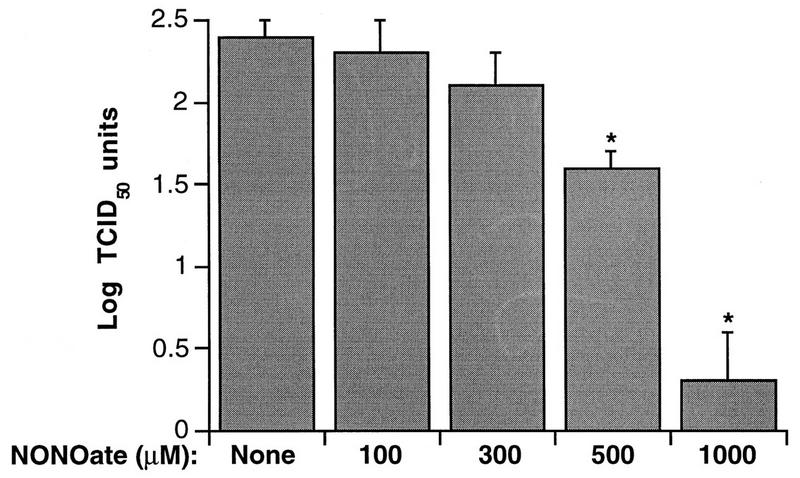
Dose-dependent inhibition by NONOate of HRV-16 titers in BEAS-2B supernatants recovered 24 h after viral exposure. Data represent the means plus SEM of values from four experiments. The asterisks indicate significant inhibition compared to levels produced in the absence of NONOate (P < 0.05 in each case).
The inhibitory effects of NONOate were not limited to HRV-16 infection. Cytokines produced from BEAS-2B cells infected with another major-group strain, HRV-14, or a minor-group strain, HRV-1A, were also significantly inhibited by NONOate. In the presence of 500 μM NONOate, virus-induced IL-8 production in BEAS-2B cells was inhibited by about 60% at 4 h (350 ± 51 to 117 ± 59 pg/ml for HRV-14 and 1,857 ± 58 to 670 ± 64 pg/ml for HRV-1A [n = 3, P < 0.01]). In addition, NONOate inhibited viral titers in supernatants collected from BEAS-2B cells 24 h post-HRV-14 infection (data not shown). The capacity of NONOate to inhibit rhinovirus-induced cytokine production was also observed in primary human cells. In one experiment, 1,000 μM NONOate reduced virally induced levels of IL-8, measured 4 h after infection, from 1,400 to 366 pg/ml, while in a second experiment, IL-8 was reduced from levels of 3,420 pg/ml in virally infected cells to 1,980 and 1,030 pg/ml in cells treated with 500 and 1,000 μM NONOate, respectively.
To further examine NONOate effects, additional experiments were conducted in which NONOate was added only during or after viral infection. Figure 8 demonstrates that NONOate present only during virus exposure, or only following virus infection, inhibited IL-8 and IL-6 production by 50 to 60%. Complete inhibition of protein production was observed if NONOate was present both during and after viral exposure.
FIG. 8.

Comparison of the effects of inactive NONOate and of active NONOate added at different times during the infection procedure on HRV-16-induced cytokine production from BEAS-2B cells. NONOate was used at a final concentration of 1,000 μM, and protein levels were measured 4 h after infection. (A) Means plus SEM of values for IL-8 production from three experiments; (B) data for IL-6. Asterisks indicate significant inhibition compared to levels produced by virus alone (P < 0.05 in each case).
To determine if the observed inhibition was specifically due to nitric oxide, experiments were conducted with active NONOate and with NONOate that had released all the available NO. Figure 8 shows that the inactive compound did not inhibit IL-8 production.
Kinetics and mechanisms of the NONOate inhibition of IL-6 and IL-8 production.
To examine the time course of NONOate inhibition, BEAS-2B cells were studied in the presence (500 μM) and absence of NONOate at various times after HRV-16 infection. The inhibitory effect of NONOate was most pronounced at the earliest time points, with a 60 to 70% reduction in protein levels at 1 h, 50% at 3 h, and 30 to 40% at 7 h (Fig. 9). These results probably reflect the declining concentration of nitric oxide in the medium as the NONOate degraded. Interestingly, the NONOate did not alter levels of cytokine mRNA expression. As shown in Fig. 9, mRNA levels for BEAS-2B cells infected with HRV-16 in the presence or absence of NONOate were not significantly different. There was a tendency for the 3- and 7-h IL-8 mRNA levels to be higher in the NONOate-treated cells. In additional control studies, NONOate alone had no effect on mRNA expression for IL-8 or IL-6, nor did inactive NONOate alter virally induced expression of mRNA for IL-8 or IL-6 in BEAS-2B cells (data not shown).
FIG. 9.

Effects of NONOate (500 μM) on steady-state mRNA levels and proteins for IL-8 (left) and IL-6 (right) at different times after infection with HRV-16. The upper panels show representative Northern blots for each cytokine and for the housekeeping gene product, GAPDH. The center panels show the means plus SEM of densitometric ratios for three experiments. The lower panels show the means plus SEM of amounts of protein produced for three experiments at each time point. The asterisks indicate significant inhibition by NONOate compared to levels produced by virus alone (P < 0.05 in each case).
DISCUSSION
We have previously demonstrated that HRV-14 induces the production of IL-8 and IL-6 from BEAS-2B cells (49), and we now show that other major-group strains (HRV-16 and HRV-39) and HRV-1A of the minor group all share this ability, suggesting that the induction of proinflammatory cytokines may occur with many, if not all, rhinoviruses. We have already shown that cytokine production by HRV-14 can be blocked both by antibodies to ICAM-1 and by UV inactivation of the virus (49). Our present studies showed not only that the effects of HRV-16 can be abrogated by UV inactivation but also that a purified preparation of HRV-16 induced cytokine production. Taken together, these data indicate that cytokine induction is specifically due to virus and not to some contaminant of the viral stock solutions. Moreover, the common nature of this response implies that the induction of epithelial cell cytokine production may play an important role in the pathogenesis of upper respiratory viral infections in humans, a concept that is supported further by the fact that other viruses, such as influenza and respiratory syncytial virus, also induce epithelial cytokine production before they cause overt cytotoxicity (2, 8, 33, 38).
The significance of the differences in levels of cytokine production by each strain is difficult to interpret because the titers of viral strains were determined in several different cell lines and may not be exactly comparable. It is clear, however, that the kinetics of cytokine mRNA expression and protein secretion varied between strains of rhinovirus. Infection with HRV-14 led to a time-dependent accumulation of mRNA for IL-8 and IL-6, with observed levels being maximal at 3 h postinfection and remaining elevated at 24 h after infection. Consistent with our earlier report (49), production of protein for each cytokine increased up to 24 h postinfection but production during a second 24-h period was not different from that of control noninfected cells (data not shown). This time course of protein production was similar to that observed with this viral strain in A549 type II epithelial cells (55), although the time course for mRNA accumulation differs somewhat, presumably reflecting differences of the two cell populations. Interestingly, the time courses of mRNA expression and cytokine production were more rapid, and the magnitude of the response was greater, for cells infected with HRV-16 than for those infected with HRV-14. Not only were maximal mRNA and protein levels achieved more quickly, but they were more transient in nature, being essentially complete within 7 h. As for HRV-14, cytokine production during a second 24-h period after infection with HRV-16 was not different from that for control noninfected cells (not shown). The reasons for the difference in initial rates of IL-8 and IL-6 production by HRV-14 and HRV-16 are unknown but could be related to a difference in recognition, uptake, or uncoating of the two viral strains in BEAS-2B cells. Despite the different rates of cytokine production, no differences in the rates of viral replication were observed between the two strains. In each case, virus was detected in the supernatants of BEAS-2B cells by 7 h after infection and reached maximal levels by 24 h. A second 24-h collection produced titers similar to those of the first 24-h sample, suggesting that viral proliferation and release into the culture medium were occurring at a constant rate. The transient induction of IL-8 and IL-6 by both viral strains in the setting of continued viral replication suggests that an early event in the viral infection, and not viral replication itself, stimulates the production of proinflammatory cytokines. This rapid production of cytokines raises the speculation that this relatively early event in the pathogenesis of colds may be important to initiate rapid inflammatory cell infiltration.
To further elucidate the biochemical mechanisms of virus-induced cytokine generation, we examined the effects of selected drugs on virus-induced expression of mRNA and protein for cytokines. The protein synthesis inhibitor, cycloheximide, did not alter levels of mRNA for IL-8 or IL-6, suggesting that de novo synthesis of proteins was not required for rhinovirus-induced mRNA expression. This is consistent with the recent observations for A549 cells, indicating that induction of IL-6 by HRV-14 occurs via a nuclear factor κB-dependent pathway that is unaffected by cycloheximide (55).
Glucocorticosteroids have been shown to inhibit the production of several cytokines in patients with allergic inflammatory diseases (46, 52) as well as in cell culture systems (45, 48). We evaluated, for the first time, the effects of a potent glucocorticoid on viral replication and on induced IL-8 and IL-6 mRNA expression and protein production in rhinovirus-infected epithelial cells. Budesonide had no effect on mRNA expression for either cytokine but caused a modest inhibition of secreted protein levels. This reduction in protein secretion in the absence of changes in mRNA levels could reflect an ability of glucocorticoids to alter posttranscriptional events involved in cytokine protein production or secretion. It has previously been reported that glucocorticoids at best modestly inhibit IL-8 mRNA and protein production from cultured epithelial cells exposed to cytokines (27, 30), but dexamethasone has been reported to inhibit TNF-α-induced IL-6 mRNA and protein production from BEAS-2B cells (30). The lack of effect of budesonide on viral titers and the modest inhibition of IL-8 and IL-6 secretion are consistent, however, with in vivo studies of experimental rhinovirus infections, in which glucocorticoids had little or no effect on viral shedding and symptoms (14, 17).
It is now clear that NO can perform a broad range of actions, serving as a vasodilator, neurotransmitter, antimicrobial, and immune regulator (39). In recent years, NO has also been shown to have antiviral properties in murine cell lines and in an in vivo mouse model. Replication of several viruses, including vaccinia virus (20), herpes simplex virus type 1 (12, 24), vesicular stomatitis virus (7), coxsackievirus (32), and poliovirus (26), was inhibited by induction of NO synthase, the enzyme that generates NO, or by the addition of the NO donor S-nitroso-1-acetyl penicillamine. Given that levels of NO are increased in exhaled air from human subjects with upper respiratory viral infections (25), we investigated whether NO could inhibit rhinovirus replication and extended these studies to evaluate the effects of NO on rhinovirus-induced production of IL-6 and IL-8. Although normal human respiratory epithelial cells have been shown to express both the constitutive and inducible forms of NO synthase (4), the expression of these enzymes is markedly reduced in the BEAS-2B cell line (data not shown). For this reason, and to ensure a controlled level of NO exposure, we used NONOate, a donor that releases NO with a defined half-life. Our data show, for the first time, that NO can inhibit both rhinovirus replication and rhinovirus-induced production of IL-8 and IL-6 in human respiratory epithelial cells. These effects were dose dependent and occurred in the absence of any effects on epithelial cell viability. Inhibition of cytokine production was more pronounced at 4 than at 24 h postinfection, while viral shedding from epithelial cells also recovered to normal levels during a second 24-h collection period. These data are consistent with the ability of NONOate to cause inhibition only when it is able to release significant amounts of NO, and they indicate that both viral replication and cytokine production resume as the compound degrades. Further support for the key role of NO release is provided by our data showing that inactivated NONOate had no effect on viral titers or cytokine production.
The ability of NONOate to cause partial inhibition of cytokine production even when present only after the viral infection period suggests that NONOate is not inhibiting by directly killing the virus or by preventing the virus from entering the BEAS-2B cells. This is also supported by the ability of viral titers to recover after NONOate degradation. Rather, it seems likely that NO is inhibiting one or more early events in the viral infection process. The failure of NONOate to inhibit cytokine mRNA expression at any time point examined suggests that NO may be functioning by a posttranscriptional mechanism, but further studies are necessary to confirm this. Precedent exists, however, for the capacity of NO to inhibit protein synthesis in other cell types (13, 23).
In summary, we have demonstrated that multiple strains of rhinoviruses induce production of proinflammatory cytokines from human respiratory epithelial cells but that there are variations in the levels and kinetics of cytokine production by different strains. Although glucocorticoids modestly inhibit cytokine secretion induced by rhinovirus infection, they do not alter cytokine mRNA expression or viral replication. In contrast, NO markedly inhibits rhinovirus replication and virally induced cytokine expression without affecting mRNA levels for these cytokines. Although further studies are necessary to elucidate the mechanisms by which NO inhibits viral replication and cytokine production, our data indicate that topical application of NO donors may provide a novel therapeutic approach for the treatment of rhinovirus-induced colds and their complications.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant AI37163 and by a grant from the Center for Indoor Air Research.
REFERENCES
- 1.Akiro S, Hirano T, Taga T, Kishimoto T. Biology of multifunctional cytokines: IL 6 and related molecules (IL 1 and TNF) FASEB J. 1990;4:2860–2867. [PubMed] [Google Scholar]
- 2.Arnold R, Humbert B, Werchau H, Gallati H, König W. Interleukin-8, interleukin-6, and soluble tumour necrosis factor receptor type I release from a human pulmonary epithelial cell line (A549) exposed to respiratory syncytial virus. Immunology. 1994;82:126–133. [PMC free article] [PubMed] [Google Scholar]
- 3.Arola M, Ziegler T, Puhakka H, Lehtonen O P, Ruuskanen O. Rhinovirus in otitis media with effusion. Ann Otol Rhinol Laryngol. 1990;99:451–453. doi: 10.1177/000348949009900607. [DOI] [PubMed] [Google Scholar]
- 4.Asano K, Chee C B E, Gaston B, Lilly C M, Gerard C, Drazen J M, Stamler J S. Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc Natl Acad Sci USA. 1994;91:10089–10093. doi: 10.1073/pnas.91.21.10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baggiolini M, Walz A, Kunkel S L. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J Clin Invest. 1989;84:1045–1049. doi: 10.1172/JCI114265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bardin P G, Johnston S L, Sanderson G, Robinson S, Pickett M A, Fraenkel D J, Holgate S T. Detection of rhinovirus infection of the nasal mucosa by oligonucleotide in situ hybridization. Am J Respir Cell Mol Biol. 1994;10:207–213. doi: 10.1165/ajrcmb.10.2.8110476. [DOI] [PubMed] [Google Scholar]
- 7.Bi Z, Reiss C S. Inhibition of vesicular stomatitis virus infection by nitric oxide. J Virol. 1995;69:2208–2213. doi: 10.1128/jvi.69.4.2208-2213.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi A M K, Jacoby D B. Influenza virus A infection induces interleukin-8 gene expression in human airway epithelial cells. FEBS Lett. 1992;309:327–329. doi: 10.1016/0014-5793(92)80799-m. [DOI] [PubMed] [Google Scholar]
- 9.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 10.Churchill L, Chilton F H, Resau J H, Bascom R, Hubbard W C, Proud D. Cyclooxygenase metabolism of endogenous arachidonic acid by cultured human tracheal epithelial cells. Am Rev Respir Dis. 1989;140:449–459. doi: 10.1164/ajrccm/140.2.449. [DOI] [PubMed] [Google Scholar]
- 11.Couch R B. Rhinoviruses. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 713–734. [Google Scholar]
- 12.Croen K D. Evidence for an antiviral effect of nitric oxide. Inhibition of herpes simplex virus type 1 replication. J Clin Invest. 1993;91:2446–2452. doi: 10.1172/JCI116479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curran R D, Ferrari F K, Kispert P H, Stadler J, Stuehr D J, Simmons R L, Billiar T R. Nitric oxide and nitric oxide-generating compounds inhibit hepatocyte protein synthesis. FASEB J. 1991;5:2085–2092. doi: 10.1096/fasebj.5.7.1707021. [DOI] [PubMed] [Google Scholar]
- 14.Farr B M, Gwaltney J M, Jr, Hendley J O, Hayden F G, Naclerio R M, McBride T, Doyle W J, Sorrentino J V, Riker D K, Proud D. A randomized controlled trial of glucocorticoid prophylaxis against experimental rhinovirus infection. J Infect Dis. 1990;162:1173–1177. doi: 10.1093/infdis/162.5.1173. [DOI] [PubMed] [Google Scholar]
- 15.Feinberg A P, Vogelstein B. A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 16.Gern J E, Vrtis R, Kelly E A B, Dick E C, Busse W W. Rhinovirus produces nonspecific activation of lymphocytes through a monocyte-dependent mechanism. J Immunol. 1996;157:1605–1612. [PubMed] [Google Scholar]
- 17.Gustafson L M, Proud D, Hendley J O, Hayden F G, Gwaltney J M., Jr Oral prednisone therapy in experimental rhinovirus infections. J Allergy Clin Immunol. 1996;97:1009–1014. doi: 10.1016/s0091-6749(96)80077-7. [DOI] [PubMed] [Google Scholar]
- 18.Gwaltney J M, Hendley J O, Simon G, Jordan W S. Rhinovirus infections in an industrial population. I. The occurrence of illness. N Engl J Med. 1966;275:1261–1268. doi: 10.1056/NEJM196612082752301. [DOI] [PubMed] [Google Scholar]
- 19.Gwaltney J M, Jr, Philips C D, Miller R D, Riker D K. Computed tomographic study of the common cold. N Engl J Med. 1994;330:25–30. doi: 10.1056/NEJM199401063300105. [DOI] [PubMed] [Google Scholar]
- 20.Harris N, Buller R M L, Karupiah G. Gamma interferon-induced, nitric oxide-mediated inhibition of vaccinia virus replication. J Virol. 1995;69:910–915. doi: 10.1128/jvi.69.2.910-915.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnston S L, Pattemore P K, Sanderson G, Smith S, Campbell M J, Josephs L K, Cunningham A, Robinson B S, Myint S H, Ward M E, Tyrrell D A J, Holgate S T. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am J Respir Crit Care Med. 1996;154:654–660. doi: 10.1164/ajrccm.154.3.8810601. [DOI] [PubMed] [Google Scholar]
- 22.Johnston S L, Pattemore P K, Sanderson G, Smith S, Lampe F, Josephs L, Sympington P, O’Toole S, Myint S H, Tyrrell D A, Holgate S T. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. Br Med J. 1995;310:1225–1228. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karupiah G, Harris N. Inhibition of viral replication by nitric oxide and its reversal by ferrous sulfate and tricarboxylic acid cycle metabolites. J Exp Med. 1995;181:2171–2179. doi: 10.1084/jem.181.6.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karupiah G, Xie Q-W, Buller R M L, Nathan C, Duarte C, MacMicking J D. Inhibition of viral replication by interferon-γ-induced nitric oxide synthase. Science. 1994;261:1445–1448. doi: 10.1126/science.7690156. [DOI] [PubMed] [Google Scholar]
- 25.Kharitonov S A, Yates D, Barnes P J. Increased nitric oxide in exhaled air of normal human subjects with upper respiratory tract infections. Eur Respir J. 1995;8:295–297. doi: 10.1183/09031936.95.08020295. [DOI] [PubMed] [Google Scholar]
- 26.Komatsu T, Bi Z, Reiss C S. Interferon-γ induced type I nitric oxide synthase activity inhibits viral replication in neurons. J Neuroimmunol. 1996;68:101–108. doi: 10.1016/0165-5728(96)00083-5. [DOI] [PubMed] [Google Scholar]
- 27.Kwon O J, Au B T, Collins P D, Baraniuk J N, Adcock I M, Chung K F, Barnes P J. Inhibition of interleukin-8 by dexamethasone in human cultured airway epithelial cells. Immunology. 1994;81:389–394. [PMC free article] [PubMed] [Google Scholar]
- 28.Larsen C G, Anderson A O, Appella E, Oppenheim J J, Matsushima K. The neutrophil activating protein (NAP-1) is also chemotactic for T lymphocytes. Science. 1989;243:1464–1466. doi: 10.1126/science.2648569. [DOI] [PubMed] [Google Scholar]
- 29.Levandowski R A, Weaver C W, Jackson G G. Nasal secretion leukocyte populations determined by flow cytometry during acute rhinovirus infection. J Med Virol. 1988;25:423–432. doi: 10.1002/jmv.1890250406. [DOI] [PubMed] [Google Scholar]
- 30.Levine S J, Larivée P, Logun C, Angus C W, Shelhamer J H. Corticosteroids differentially regulate secretion of IL-6, IL-8, and G-CSF by a human epithelial cell line. Am J Physiol. 1993;265:L360–L368. doi: 10.1152/ajplung.1993.265.4.L360. [DOI] [PubMed] [Google Scholar]
- 31.Lindley I, Aschauer H, Seifert J M, Lam C, Brunowsky W, Kownatski E, Thelen M, Peveri P, Dewald B, von Tscharner V, Walz A, Baggiolini M. Synthesis and expression in Escherichia coli of the gene encoding monocyte-derived neutrophil-activating factor: biological equivalence between natural and recombinant neutrophil-activating factor. Proc Natl Acad Sci USA. 1988;85:9199–9203. doi: 10.1073/pnas.85.23.9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowenstein C J, Hill S L, Lafond-Walker A, Wu J, Allen G, Landavere M, Rose N R, Herskowitz A. Nitric oxide inhibits viral replication in murine myocarditis. J Clin Invest. 1996;97:1837–1843. doi: 10.1172/JCI118613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsukura S, Kokubo F, Noda H, Tokunaga H, Adachi M. Expression of IL-6, IL-8, and RANTES on human bronchial epithelial cells, NCI-H292, induced by influenza virus A. J Allergy Clin Immunol. 1996;98:1080–1087. doi: 10.1016/s0091-6749(96)80195-3. [DOI] [PubMed] [Google Scholar]
- 34.Matsushima K, Morishita K, Yoshimura T, Lavu S, Kobayashi Y, Lew W, Appella E, Kung H F, Leonard E J, Oppenheim J J. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MDNCF) and the induction of MDNCF by interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167:1883–1893. doi: 10.1084/jem.167.6.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McHardy V U, Inglis J M, Calder M A, Crofton J W, Gregg I, Ryland D A, Taylor P, Chadwick M, Coombs D, Riddell R W. A study of infective and other factors in exacerbations of chronic bronchitis. Br J Dis Chest. 1980;74:228–238. doi: 10.1016/0007-0971(80)90048-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naclerio R M, Proud D, Lichtenstein L M, Kagey-Sobotka A, Hendley J O, Sorrentino J, Gwaltney J M. Kinins are generated during experimental rhinovirus colds. J Infect Dis. 1988;157:133–142. doi: 10.1093/infdis/157.1.133. [DOI] [PubMed] [Google Scholar]
- 37.Nicholson K G, Kent J, Ireland D C. Respiratory viruses and exacerbations of asthma in adults. Br Med J. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noah T L, Becker S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am J Physiol. 1993;265:L472–L478. doi: 10.1152/ajplung.1993.265.5.L472. [DOI] [PubMed] [Google Scholar]
- 39.Nussler A K, Billiar T R. Inflammation, immunoregulation, and inducible nitric oxide synthase. J Leukocyte Biol. 1993;54:171–178. [PubMed] [Google Scholar]
- 40.Proud D, Gwaltney J M, Jr, Hendley J O, Dinarello C A, Gillis S, Schleimer R P. Increased levels of interleukin-1 are detected in nasal secretions of volunteers during experimental rhinovirus colds. J Infect Dis. 1994;169:1007–1013. doi: 10.1093/infdis/169.5.1007. [DOI] [PubMed] [Google Scholar]
- 41.Proud D, Naclerio R M, Gwaltney J M, Hendley J O. Kinins are generated in nasal secretions during natural rhinovirus colds. J Infect Dis. 1990;161:120–123. doi: 10.1093/infdis/161.1.120. [DOI] [PubMed] [Google Scholar]
- 42.Ramsay A J, Husband A J, Ramshaw I A, Bao S, Matthaei K I, Koehler G, Kopf M. The role of interleukin-6 in mucosal IgA antibody responses in vivo. Science. 1994;264:561–563. doi: 10.1126/science.8160012. [DOI] [PubMed] [Google Scholar]
- 43.Reddel R R, Ke Y, Gerwin B I, McMenamin M G, Lechner J F, Su R T, Brash D E, Park J-B, Rhim J S, Harris C C. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- 44.Schleimer R P. Glucocorticosteroids: their mechanism of action and use in allergic diseases. In: Middleton E, Reed C E, Ellis E F, Adkinson N F Jr, Yunginger J W, Busse W W, editors. Allergy: principles and practice. 4th ed. St. Louis, Mo: Mosby; 1993. pp. 893–925. [Google Scholar]
- 45.Schwiebert L A, Beck L A, Stellato C, Bickel C A, Bochner B S, Schleimer R P. Glucocorticosteroid inhibition of cytokine production; relevance to antiallergic actions. J Allergy Clin Immunol. 1996;97:143–152. doi: 10.1016/s0091-6749(96)80214-4. [DOI] [PubMed] [Google Scholar]
- 46.Sim T C, Reece L M, Hilsmeier K A, Grant J A, Alam R. Secretion of chemokines and other cytokines in allergen-induced nasal responses: inhibition by topical steroid treatment. Am J Respir Crit Care Med. 1995;152:927–933. doi: 10.1164/ajrccm.152.3.7545059. [DOI] [PubMed] [Google Scholar]
- 47.Steel R G D, Torrie J H. Principles and procedures of statistics, a biometrical approach. 2nd ed. New York, N.Y: McGraw-Hill; 1980. [Google Scholar]
- 48.Stellato C, Beck L A, Gorgone G A, Proud D, Schall T J, Ono S J, Lichtenstein L M, Schleimer R P. Expression of the chemokine RANTES by a human bronchial epithelial cell line. Modulation by cytokines and glucocorticoids. J Immunol. 1995;155:410–418. [PubMed] [Google Scholar]
- 49.Subauste M C, Jacoby D B, Richards S M, Proud D. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J Clin Invest. 1995;96:549–557. doi: 10.1172/JCI118067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turner B W, Cail W S, Hendley J O, Hayden F G, Doyle W J, Sorrentino J V, Gwaltney J M., Jr Physiologic abnormalities of the paranasal sinuses during experimental rhinovirus colds. J Allergy Clin Immunol. 1992;90:474–478. doi: 10.1016/0091-6749(92)90172-x. [DOI] [PubMed] [Google Scholar]
- 51.Turner R B, Hendley J O, Gwaltney J M., Jr Shedding of infected epithelial cells in rhinovirus colds. J Infect Dis. 1982;145:849–853. doi: 10.1093/infdis/145.6.849. [DOI] [PubMed] [Google Scholar]
- 52.Wang J H, Trigg C J, Devalia J L, Jordan S, Davies R J. Effect of inhaled beclomethasone dipropionate on expression of proinflammatory cytokines and activated eosinophils in the bronchial epithelium of patients with mild asthma. J Allergy Clin Immunol. 1994;94:1025–1034. doi: 10.1016/0091-6749(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 53.Winther B, Brofeldt S, Christensen B, Mygind N. Light and scanning electron microscopy of nasal biopsy material from patients with naturally acquired common colds. Acta Oto-Laryngol. 1984;97:309–318. doi: 10.3109/00016488409130994. [DOI] [PubMed] [Google Scholar]
- 54.Winther B, Farr B, Turner R B, Hendley J O, Gwaltney J M, Jr, Mygind N. Histopathologic examination and enumeration of polymorphonuclear leukocytes in the nasal mucosa during experimental rhinovirus colds. Acta Oto-Laryngol Suppl. 1984;413:19–24. doi: 10.3109/00016488409128537. [DOI] [PubMed] [Google Scholar]
- 55.Zhu Z, Tang W, Ray A, Wu Y, Einarsson O, Landry M L, Gwaltney J M, Jr, Elias J A. Rhinovirus stimulation of interleukin-6 in vivo and in vitro. Evidence for nuclear factor κB-dependent transcriptional activation. J Clin Invest. 1996;97:421–430. doi: 10.1172/JCI118431. [DOI] [PMC free article] [PubMed] [Google Scholar]