

Abstract
The SCID-hu mouse implanted with human fetal tissue is a novel model for investigating human viral pathogenesis. Infection of human skin implants was used to investigate the basis for the clinical attenuation of the varicella-zoster virus (VZV) strain, V-Oka, from which the newly licensed vaccine is made. The pathogenicity of V-Oka was compared with that of its parent, P-Oka, another low-passage clinical isolate, strain Schenke (VZV-S), and VZV-Ellen, a standard laboratory strain. The role of glycoprotein C (gC) in infectivity for human skin was assessed by using gC-negative mutants of V-Oka and VZV-Ellen. Whereas all of these VZV strains replicated well in tissue culture, only low-passage clinical isolates were fully virulent in skin, as shown by infectious virus yields and analysis of implant tissues for VZV DNA and viral protein synthesis. The infectivity of V-Oka in skin was impaired compared to that of P-Oka, providing the first evidence of a virologic basis for the clinical attenuation of V-Oka. The infectivity of V-Oka was further diminished in the absence of gC expression. All strains except gC-Ellen retained some capacity to replicate in human skin, but cell-free virus was recovered only from implants infected with P-Oka or VZV-S. Although VZV is closely related to herpes simplex virus type 1 (HSV-1) genetically, experiments in the SCID-hu model revealed differences in tropism for human cells that correlated with differences in VZV and HSV-1 disease. VZV caused extensive infection of epidermal and dermal skin cells, while HSV-1 produced small, superficial lesions restricted to the epidermis. As in VZV, gC expression was a determinant for viral replication in skin. VZV infects human CD4+ and CD8+ T cells in thymus/liver implants, but HSV-1 was detected only in epithelial cells, with no evidence of lymphotropism. These SCID-hu mouse experiments show that the clinical attenuation of the varicella vaccine can be attributed to decreased replication of V-Oka in skin and that tissue culture passage alone reduces the ability of VZV to infect human skin in vivo. Furthermore, gC, which is dispensable for replication in tissue culture, plays a critical role in the virulence of the human alphaherpesviruses VZV and HSV-1 for human skin.
Varicella-zoster virus (VZV) is a human alphaherpesvirus that causes varicella, or chickenpox, as the primary infection in susceptible individuals (2). The critical events in the pathogenesis of primary VZV infection include inoculation of respiratory mucosa, the occurrence of cell-associated viremia, and the transfer of infectious virus to skin, resulting in the characteristic vesicular exanthem (27, 28, 36, 39). Like other alphaherpesviruses, VZV establishes latency in sensory ganglia (12, 32). VZV reactivation from the latent state causes herpes zoster, manifesting as a localized rash in a unilateral, dermatomal distribution that is often associated with severe neuropathic pain (2, 44).
A live attenuated varicella vaccine was developed to reduce the morbidity due to VZV infection and is the first herpesvirus vaccine licensed for use in humans (29). The varicella vaccine is derived from the Oka strain, a Japanese clinical isolate, which was attenuated by passage in semipermissive guinea pig embryo fibroblasts (42). While most healthy children and adults who are given the varicella vaccine develop immunity without experiencing any signs of disease, the virologic basis for this clinical attenuation of the vaccine Oka strain (V-Oka) is not known. Further evidence for the attenuation of V-Oka was seen in children vaccinated by intranasal inoculation, the presumed route of natural infection, who developed immune responses without signs of disease (6). The pattern of replication of V-Oka in tissue culture cells resembles that of other VZV strains, and restriction endonuclease analysis of genomic DNA does not reveal obvious differences between V-Oka and other geographically related VZV isolates (17, 30). In fact, it is not necessary to assume that the absence of clinical symptoms after immunization is due to an intrinsic altered virulence of V-Oka. Alternatively, early immunologic responses that are induced by subcutaneous administration of the vaccine virus, which is not the natural route of VZV inoculation, or by the noninfectious, viral protein content of the vaccine could modulate the course of V-Oka replication in vaccine recipients (5).
The SCID-hu mouse model provides a unique opportunity to examine the attenuation of V-Oka and other aspects of VZV pathogenesis (35). Virus-cell interactions can be assessed in intact human tissues independently of the effects of the host immune response on viral replication (1, 4, 23, 34). The inoculation of human skin implants in SCID-hu mice with a low-passage clinical isolate of VZV induced histopathologic changes typical of VZV skin lesions observed in patients with varicella or herpes zoster (9, 35). VZV replicated in CD4+ and CD8+ human T cells in thymus/liver (thy/liv) implants in SCID-hu mice, demonstrating that it must be classified as a lymphotropic as well as a neurotropic herpesvirus (12, 13, 32, 35). Although reduced infectivity of V-Oka for T cells could have accounted for its attenuation, V-Oka was as infectious for human T cells as the low-passage clinical isolate of VZV. Therefore, the first objective of these experiments was to determine whether a change in virulence for human skin could provide a virologic explanation for the clinical attenuation of V-Oka.
Diminished expression of glycoprotein C (gC) has been proposed as a genetic factor that may be related to the clinical attenuation of V-Oka (24). gC is one of six known VZV envelope glycoproteins and has 34% amino acid homology to herpes simplex virus type 1 (HSV-1) gC (25). HSV-1 gC binds to heparan sulfate on the cell surface and to the C3b component of complement (16, 21). While HSV-1 gC is dispensable for infection in vitro, it mediates binding to the apical surface of polarized MDCK cells (8, 40). Similarly, VZV gC is not required for replication, and its synthesis is variable when VZV isolates are grown in tissue culture (11, 24). VZV variants that do not produce any detectable gC arise with a low frequency in vitro and can be isolated by repeated plaque purification. Whether this phenomenon has implications for VZV virulence in vivo is not known. Therefore, our second objective was to compare the infectivity for human skin of gC-negative strains of VZV, including a derivative of V-Oka and of a standard laboratory virus, the Ellen strain, with that of their respective parent strains, in vivo. Third, to define similarities or differences between VZV and HSV-1, which is the prototype of the alphaherpesviruses, we examined the tropism of these viruses for human cells in skin and thy/liv implants in the SCID-hu model.
The comparative analysis of V-Oka and the parent Oka strain (P-Oka) along with other VZV strains in the SCID-hu mouse model established that V-Oka has a diminished capacity to replicate in human skin, in the absence of any modulation by host responses. The clinical attenuation of V-Oka can be explained by this decreased virulence for human skin, a characteristic which is associated with prolonged passage in tissue culture cells and is independent of a requirement for passage in nonhuman cells. Although the attenuation of V-Oka was not attributable to decreased gC expression, gC was a specific virulence determinant in VZV infection of human skin cells, as well as for the epidermal cell tropism of HSV-1. The finding that VZV gC is required for effective viral replication in skin is the first evidence of an essential role for any VZV gene product in the pathogenesis of human infection. Since all of the VZV strains that we evaluated were indistinguishable in their patterns of replication in tissue culture cells, these experiments also demonstrated that whether virus strains differ in pathogenicity and whether particular genes are critical for virus-cell interactions must be determined in intact tissue in vivo.
MATERIALS AND METHODS
SCID-hu mice.
Male homozygous C.B-17 scid/scid mice were bred and maintained at SyStemix, Inc, Palo Alto, Calif. When the mice were 8 weeks old, human skin from 18- to 23-week fetuses was introduced subcutaneously as full-thickness dermal grafts. The tissue was allowed to engraft for 3 to 5 weeks before use. Human fetal tissues were obtained with informed consent according to federal and state regulations and were screened for human immunodeficiency virus.
The general care of the experimental animals used for this study was done in accordance with National Institutes of Health guidelines for laboratory animals and in compliance with the Animal Welfare Act (Public Law 94-279). This specific project was reviewed and approved by the Stanford University Administrative Panel on Laboratory Animal Care.
Viral strains and culture conditions.
The VZV strains included a low-passage clinical isolate, strain Schenke (VZV-S), P-Oka, V-Oka, a gC-minus Oka variant (gC−-Oka), VZV Ellen strain (VZV-Ellen), and a gC-minus Ellen variant (gC−-Ellen). VZV-S was recovered from a cutaneous lesion and passaged twice in human foreskin fibroblasts and four times in MRC-5 cells. P-Oka was isolated from a child with varicella, passaged six times in human foreskin fibroblasts, and stored at −70°C (42). V-Oka is the varicella vaccine strain manufactured by Merck & Co., Inc.; it was derived from P-Oka by growth at low temperature (32°C) and passaged 11 times in human embryonic lung cells, 12 times in guinea pig embryo fibroblasts, once in WI-38 cells, and 9 times in MRC-5 cells. gC−-Oka is a naturally occurring variant which was plaque purified from V-Oka and was kindly provided by Lawrence Gelb, Washington University, St. Louis, Mo. VZV-Ellen is a standard laboratory strain passaged more than 100 times since its isolation in 1964 (7, 41); gC−-Ellen is a plaque-purified variant of VZV-Ellen, previously designated L-N strain (24). Before inoculation into skin implants, all VZV strains were passed three times in MRC-5 cells in minimal essential medium (Mediatech, Washington, D.C.) supplemented with 50 IU of penicillin, 50 mg of streptomycin (Pen/Strep; ICN Biomedicals, Inc., Costa Mesa, Calif.), and 0.5 mg of amphotericin B (Fungizone; Flow Laboratories, McLean, Va.) with 10% fetal calf serum (FCS; Tissue Culture Biologicals, Tulare, Calif.) (tissue culture medium [TCM]). The monolayer was trypsinized; the cells were counted, centrifuged, resuspended, and briefly stored on ice before injection into the SCID-hu mouse implants; mock-infected implants were injected with an equal number of uninfected MRC-5 cells. The titer of each inoculum was determined by infectious focus assay and was approximately 2 × 105 to 4 × 105 PFU/ml for each VZV isolate.
HSV-1 strains included KOS, a gC-minus deletion mutant designated ΔgC2-3, and the rescued virus ΔgC2-3rev, constructed by Herold and colleagues (21), which were kindly provided by Curtis R. Brandt, University of Wisconsin, Madison. HSV-1 strains were passaged once in MRC-5 cells before injection into SCID-hu mouse skin or thy/liv implants; plaque titrations were done in Vero cells at the time of inoculation and resulted in approximately 6.0 × 105 to 6.2 × 105 PFU/ml for each HSV strain.
Inoculation of skin implants.
Mice were anesthetized with a solution of 5% (wt/vol) ketamine (Aveco Co., Inc., Fort Dodge, Iowa) and 2.5% (wt/vol) xylazine (LyphoMed, Inc., Rosemont, Ill.) in phosphate-buffered saline (PBS; 140 mM NaCl, 2.7 mM KCl, 15 mM Na2HPO4, 1.5 mM KH2PO4 [pH 7.6]) by intraperitoneal injection. Bilateral skin implants were exposed through a 1-cm dorsal, midsagittal incision. The inoculum (approximately 10 ml) was injected into the graft by using a 27-gauge needle. At 14, 21, and 28 days after inoculation, the implants were dissected from the murine skin and divided; one 2.0-mm central slice was fixed in 4% paraformaldehyde for histology and in situ hybridization, approximately one half was frozen in PBS at −20°C for Western blot analysis, and the other half was placed in SPGA buffer (218 mM sucrose, 3.8 mM KH2PO4, 7.2 mM K2HPO4, 4.9 mM sodium glutamate, 1% bovine albumin, 10% FCS) for virus isolation. Each implant was minced and vortexed thoroughly in 1.0 ml of SPGA buffer; an aliquot of the suspension was titered directly, and a second aliquot was filtered through a 0.45-μm-pore-size membrane before titration to detect cell-free virus. In some experiments, tissue was placed in 2.5% glutaraldehyde for electron microscopy analysis.
HSV-1-infected skin implants were harvested 6 days after inoculation. One half of each implant was minced, resuspended in 1.0 ml of TCM, frozen in a dry ice-ethanol bath, thawed, and sonicated for 30 s. The cell debris was removed by centrifugation, and the supernatants were titered in a plaque assay on Vero cells. The other half of the tissue block was fixed for histology and in situ hybridization as described above. Thy/liv implants were inoculated with HSV-1 as previously described for VZV inoculation (35). Infected implants were harvested at 2, 4, 6, and 7 days after inoculation and then prepared for titration and histology. Apoptotic T cells in HSV-infected thy/liv implants were detected by using an In Situ Cell Death Detection Kit, AP (Boehringer Mannheim, Indianapolis, Ind.) according the instructions provided.
Infectious focus assay.
Virus titrations of infected MRC-5 cell inocula, skin implant suspensions, or cell-free filtrates of skin implant suspensions were done with specimens serially diluted 10-fold in TCM with 5% FCS. A 0.1-ml cell suspension, mixed with 1.5 × 105 Vero cells in 0.9 ml of TCM, was added to 24-well plates in triplicate. The plates were incubated for 6 days at 37°C in 5% CO2; 1.0 ml of fresh TCM with 5% FCS was added on day 3. Following aspiration of the supernatant, the wells were flooded with crystal violet stain (5% ethanol, 5% formaldehyde, and 0.13% crystal violet in PBS) for 2 to 5 min. The stain was aspirated, the wells were air dried, and plaques were counted in an inverted light microscope (magnification, ×40). The level of detection of the infectious focus assay was 10 PFU per specimen.
Western blot analysis.
One half of each infected skin implant was minced to a paste and sonicated for 1 min in detergent extract buffer containing protease inhibitors (10 mM Tris [pH 7.4], 150 mM NaCl, 0.5% Triton X-100, 4 mM Pefabloc SC [Boehringer Mannheim], and 0.2 U of aprotinin per ml). Following standard techniques, the skin extract supernatants were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) in 7.5% gels, transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore, Bedford, Mass.), and stained with amido black (1% amido black [naphthol blue black], 45% methanol, 10% acetic acid) to reveal total protein before Western blot analysis was performed. The amount of total protein in 15 μl of each sample was equivalent and was verified by staining the membranes with amido black. For detection of gC, infected cell lysates were analyzed by SDS-PAGE and transferred to membranes in a similar manner.
VZV proteins were detected with a high-titer polyclonal human immune serum, gC was detected in infected cell lysates with a high-titer polyclonal human monospecific serum (gift of P. Kinchington), and a secondary goat anti-human immunoglobulin G-horseradish peroxidase conjugate was used for enhanced chemiluminscence (ECL) detection of bound antibodies. ECL reagents (Amersham, Buckinghamshire, England) were added, and the blots were immediately exposed to a phosphorimager screen for exactly 1 h. The screen was scanned with a Bio-Rad (Hercules, Calif.) GS-505 Molecular Imager System and analyzed with Molecular Analyst software (Bio-Rad). The intensity of bands was quantitated in density units for each test sample, and the statistical significance of differences from controls was determined with Statview II software (Abacus Concepts, Inc., Berkeley, Calif.).
Histology and in situ hybridization.
Skin implants were fixed in 4% paraformaldehyde overnight at 4°C, embedded in paraffin, cut into 3-μm sections, and stained with hematoxylin and eosin. Unstained sections were deparaffinated in xylene and rehydrated in graded ethanols before use. In situ hybridization was done as described previously (35). Briefly, sections were probed with a 12.9-kb biotinylated plasmid, pVZV-C, that consists of a pBR322 vector carrying the HindIII fragment C of VZV genomic DNA. A negative control probe consisting of pBR322 vector alone was used at the same concentration as the VZV-specific probe. The probe used for in situ hybridization in HSV experiments was a biotinylated plasmid containing the EcoRI A fragment of HSV-1 genome cloned in pACYC184. Hybridization was detected with a streptavidin-alkaline phosphatase conjugate and visualized with nitroblue tetrazolium salt and 5-bromo-4-chloro-3-indolylphosphate p-toluidine salt. The tissue sections were counterstained with hematoxylin and examined by light microscopy.
Electron microscopy.
Skin implants infected with VZV-S were recovered 21 days after inoculation, and a 2- by 5-mm piece was placed in 2.5% glutaraldehyde in 0.1 M sodium phosphate (pH 7.2) for 24 h. Postfixation transmission electron microscopy procedures were performed as previously described (20).
Northern blots.
VZV-S, P-Oka, V-Oka, and gC−-Oka, VZV-Ellen, and gC−-Ellen were evaluated for transcription of glycoprotein genes by using a standard Northern blot procedure. RNA was prepared and analyzed as described previously (26), with some modifications. Briefly, 80% confluent monolayers of MeWo cells on 75-cm2 flats were infected with different VZV strains by overlay of infected cells at 1 infected cell per 20 uninfected cells and incubated at 35°C for 56 h. Total cell RNA was prepared by the guanidinium isothiocyanate-phenol method (10). From the extracted RNA, mRNA was prepared by using a MicroFastTrack kit (InVitrogen Corp., Inc., Carlsbad, Calif.). For Northern blot analyses, 1.5 mg of each RNA was electrophoresed on formaldehyde-denaturing agarose gels and transferred to a GeneScreen membrane (NEN Dupont Nemours, Inc., Boston, Mass.) by capillary action. RNA was fixed to the membranes by UV cross-linking, using the automatic setting on a UV Stratalinker (Stratagene Inc., La Jolla, Calif.).
RNA blots were prehybridized and hybridized to probes as described previously and exposed to a phosphorimager screen for exactly 24 h (26). The screen was scanned with a Bio-Rad GS-505 Molecular Imager System and analyzed with Molecular Analyst software (Bio-Rad). The intensity of bands was quantitated in density units for each test sample, and ratios of glycoprotein RNA transcripts were determined. All probes were double-stranded DNA fragments labeled to high specific activity by using oligonucleotide-primed repair synthesis and [α-32P]dCTP (3,000 Ci/mmol). Each specific probe was generated from previously derived DNA clones from VZV strain Scott, a low-passage clinical isolate (25). VZV gC-specific probes were generated by using a BstN1-AhdI fragment, representing bp 19480 to 21116. VZV gE-specific probes were generated from a SmaI-BglI fragment, representing bp 117870 to 115712 (approximately 100 bp of this probe overlaps with the sequences encoding the putative open reading frame [ORF] 69). The VZV gB-specific probe was generated from a mixture of a BamHI and an NsiI-BamHI fragment representing bp 56994 to 59326. The VZV gH-specific probe was generated from a PstI-SalI fragment representing bp 66583 to 69349 (approximately 520 bp of this fragment overlaps with the coding region of ORF 38). The VZV ORF 13-specific probe was generated from a Bst107I fragment representing positions 17547 to 19040, and the ORF 15 probe was generated from a BamHI-NdeI fragment representing bp 21264 to 23062.
Restriction enzyme analysis.
Nucleocapsids were purified from VZV-infected cells, resuspended in STE buffer (1.0 M Tris-Cl, 0.5 M EDTA, 2% SDS), proteinase K (EM Science) at 1.0 mg/ml was added, and the solution was incubated for 30 min at 50°C (41). VZV DNA was extracted once with Tris-saturated phenol, once with 1:1 phenol-chloroform containing 2% isoamyl alcohol, and once with chloroform alone. The DNA was ethanol precipitated and resuspended in Tris-EDTA buffer. Genomic VZV DNA samples were cleaved with restriction endonucleases HpaI, BglII, EcoRI, and BamHI (New England Biolabs, Beverly, Mass.) and subjected to electrophoresis in 0.8% agarose gels. DNA bands were visualized with ethidium bromide.
Sequencing of gC promoter region.
VZV genomic DNA was used for PCR amplification of the gC promoter region. A 504-bp fragment spanning the intergenic region between VZV ORFs 14 and 15 was amplified by using primers 5′-GGGTGTGGGTTGAGATTC-3′ and 5′-CAGGGTTTTGCCGTTTTA-3′, which map to codons 21039 and 21543 of the VZV genomic sequence (14). Both strands of amplified product from VZV-S, P-Oka, V-Oka, gC−-Oka, VZV-Ellen, and gC−-Ellen were sequenced by using an Applied Biosystems automated sequencing apparatus (model 373A, version 2.0.1S).
RESULTS
Unique characteristics of VZV infection of human skin.
Our previous experiments in the SCID-hu mouse model demonstrated that infection of human skin with VZV-S, a low-passage clinical isolate, results in the formation of lesions typical of varicella (35). P-Oka, the parent of V-Oka, shares this virulence phenotype (Fig. 1). After 21 days, infection with P-Oka had spread deep into the dermis, balloon cells were prevalent, and acellular material from degenerated cells was enclosed by a keratinized surface layer. In situ hybridization of these lesions revealed VZV DNA within glandular cells and fibroblasts of the dermis (Fig. 1a). Further analysis of skin implants infected with VZV-S, representing wild-type virus, by electron microscopy showed that virions were carried to the cell surface in a cytoplasmic vacuole, egressed through the cell membrane, and dispersed from the surface of the cell (Fig. 2), indicating that the highly cell-associated pattern of VZV replication in tissue culture does not reflect virus-cell interactions in vivo (17). Remnants of collapsed transport vacuoles were clearly visible beneath virions on the cell surface. Many mature virions produced in skin were enclosed in a membrane bilayer and were intact morphologically, in contrast to the predominance of defective VZV particles observed in vitro. The infectivity of cell-free virions produced in skin was confirmed by infectious focus assay.
FIG. 1.
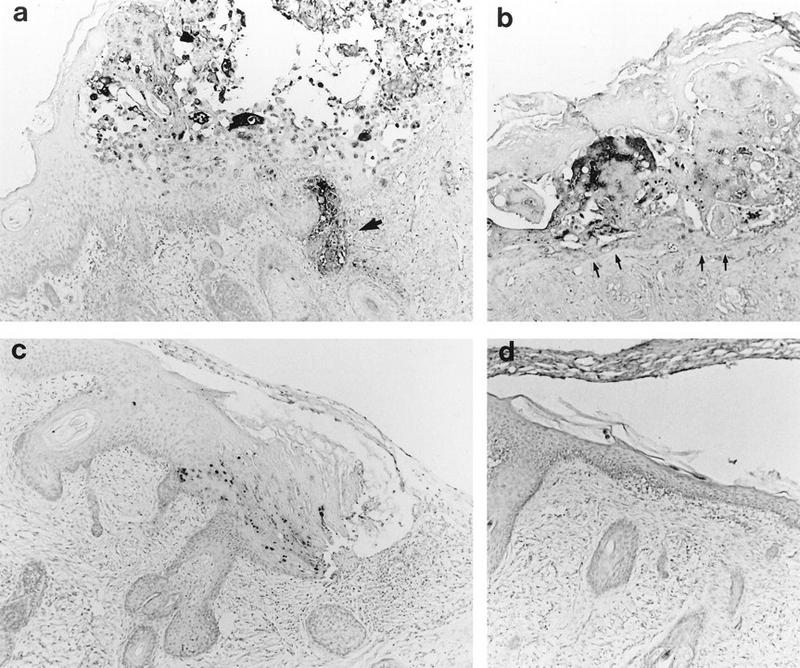
Histological analysis of VZV-infected skin implants. Subcutaneous skin implants infected with P-Oka (a), V-Oka (b), or gC−-Oka (c) or mock infected (d) were fixed in paraformaldehyde, paraffin embedded, and cut into 3-μm sections before in situ hybridization. Darkly stained cells indicate VZV DNA; tissue was counterstained lightly with hematoxylin. At day 21 postinfection, gC−-Oka lesions appeared in the epidermis (c). V-Oka lesions were larger; however, the basement membrane remained intact (arrows), and virus did not penetrate into the dermis (b). P-Oka skin lesions were largest (a), and VZV DNA was detected deep within dermal fibroblasts (arrow). Mock-infected skin had a normal appearance (d), and no DNA was detected in the implant. Histology shown is representative of four implants. Magnification, ×83.
FIG. 2.
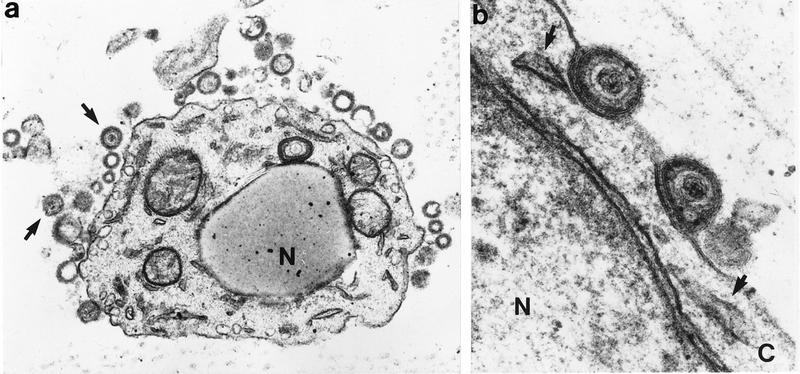
VZV release from skin cells, determined by transmission electron microscopy of skin cells 21 days after inoculation with VZV-S. (a) Enveloped virons containing dense cores have dispersed from the cell (arrows). Vacuoles from which particles egressed are visible adjacent to the cell membrane. Nucleocapsids are not visible in the nucleus (N) by this staining method. Magnification, ×24,000. (b) Higher-magnification view of virions with intact lipid bilayer on cell surface. Remnants of cytoplasmic vacuoles which carried virions to plasma membrane are indicated by arrows. N, nucleus; C, cytoplasm. Magnification, ×108,000.
Tissue culture passage of VZV reduces the synthesis and release of infectious virus in human skin in vivo. P-Oka and VZV-S, representing low-passage clinical isolates, were compared with V-Oka and VZV-Ellen to determine whether growth in tissue culture altered the virulence of VZV in vivo. V-Oka was less infective for human skin than P-Oka or VZV-S. When the infectious virus yields from skin implants were compared at 14, 21, and 28 days, the titer of V-Oka in unfiltered cell suspensions was approximately two-thirds less than the titer of P-Oka at each time point (Fig. 3). By day 21, the mean titer in six skin specimens infected with V-Oka was 1,600 PFU (range, 0 to 2,600 PFU), whereas the mean titer was 6,200 PFU (range, 4,300 to 9,700 PFU) in six implants infected with P-Oka. The difference in mean titer was statistically significant (P = 0.002, Student’s t test). In addition to decreased production of intracellular virus, V-Oka did not replicate in two of six skin implants.
FIG. 3.

Infectious virus in VZV-infected skin implants. Implants were inoculated with P-Oka, V-Oka, or gC−-Oka and harvested 14, 21, or 28 days postinfection. Cell-associated virus was measured in an infectious focus assay, and PFU per implant was calculated. Error bars indicate the standard error of the mean. On day 21, all differences between strains were statistically significant (P ≤ 0.02, Student’s t test).
The patterns of V-Oka and P-Oka replication in human skin also differed with respect to the release of virus from infected cells. Cell-free virus was detected in filtered suspensions of skin infected with P-Oka at 14, 21, and 28 days, whereas no cell-free virus could be detected in implants infected with V-Oka at any time point. The maximum release of virus was observed on day 21, when five of six implants inoculated with P-Oka had concentrations of cell-free virus ranging from 67 to 2,200 PFU; titers declined by day 28 as the infection progressed to cause extensive necrosis of the implant.
VZV-Ellen, a standard laboratory strain which has been passaged extensively in human tissue culture cell lines, was also significantly less virulent in skin implants than the low-passage clinical isolates, P-Oka and VZV-S. Cell-associated infectious virus was recovered from only one of eight skin implants infected with VZV-Ellen. VZV-Ellen was also less infective than V-Oka even though V-Oka was specifically passaged in nonhuman cells to derive a vaccine strain.
Cell-associated infectious virus recovered from implants infected with V-Oka or VZV-Ellen was reinoculated directly into fresh skin implants to determine whether a single passage in vivo restored wild-type levels of infectivity. After 21 days, protein synthesis was detected in one of four implants inoculated with V-Oka although none of the implants yielded infectious virus (data not shown). No viral protein synthesis was detected by Western blotting, and no infectious virus was cultured from any of four implants inoculated with VZV-Ellen that had undergone one passage in skin implants.
Tissue culture passage is associated with decreased viral protein synthesis by VZV in human skin in vivo. Synthesis of viral proteins in the range of 70 to 120 kDa was quantitated by phosphorimager analysis of Western blots prepared from extracts of infected skin implants and incubated with polyclonal antiserum to VZV. Inoculation of skin implants with P-Oka or VZV-S resulted in equivalent synthesis of viral proteins at day 21 (Fig. 4). Based on the mean of values measured in 10 implants inoculated with each strain, both P-Oka and VZV-S produced levels of VZV protein of approximately 2,000 density units (P = 0.19, Student’s t test). In contrast, inoculation with V-Oka resulted in protein levels of approximately 1,000 density units in 10 implants, which was significantly less than for P-Oka or VZV-S (P = 0.01). In five experiments, protein synthesis by V-Oka was undetectable by Western blotting in 4 of 14 of skin implants derived from the same donor, infected with an equivalent inoculum, and harvested 21 days after injection. Two of eight implants injected with VZV-Ellen showed trace amounts of viral protein by Western blotting. The mean density units numbered 11, which is just above the level of detection of the assay and was significantly less than for V-Oka (P = 0.03) as well as P-Oka and VZV-S (P < 0.01). No VZV protein synthesis was detectable in experiments using standard autoradiography of Western blots prepared from skin extracts inoculated with VZV-Ellen.
FIG. 4.

VZV protein synthesis in skin implants. Implants were inoculated with VZV and harvested on day 21 postinfection. Protein was extracted from infected implants, separated by SDS-PAGE, and transferred to nylon membranes. Western blot analysis was performed; VZV proteins were detected with a high-titer human polyclonal serum and ECL on a phosphorimager. The concentration of VZV protein in the range of 70 to 120 kDa was measured digitally in density units. The means and standard error are shown for each VZV strain. All paired VZV strains are statistically different from each other (P ≤ 0.04, Student’s t test) except (i) VZV-S and P-Oka and (ii) VZV-Ellen and gC−-Ellen.
Effect of gC expression on VZV replication in skin.
The infectivities for skin of gC−-Oka and gC−-Ellen strains, which are two naturally occurring variants of VZV that fail to synthesize gC, were compared with each parent strain. Before inoculation of skin implants, transcription and translation of gC by each strain were evaluated by Northern and Western blot analyses. A marked decrease in the transcription of gC mRNA by gC−-Oka and gC−-Ellen was documented in comparison with VZV-S, P-Oka, V-Oka, and VZV-Ellen; transcription of gC in P-Oka, VZV-S, V-Oka, and VZV-Ellen from the 1.9-kb transcript, the predominant coding transcript, was equivalent, with small differences due to variability of cytopathic effect (CPE) at time of harvest (Fig. 5A). The low level of transcription of the 2.5-kb mRNA by P-Oka did not affect synthesis of gC (Fig. 6). CPE and yields of cell-associated infectious virus in vitro were indistinguishable for all strains. Transcription of gE, gB, and gH was evaluated and compared to that of gC to ensure that differences in infectivity of the gC− derivatives for skin were not associated with disrupted mRNA transcription of other major viral glycoprotein genes; no alterations were observed (Fig. 5B). To confirm that the absence of gC transcription by gC−-Oka and gC−-Ellen was not a global defect in transcription of that region of the genome, Northern blot analyses detecting ORFs 13 and 15 were performed. In agreement with a detailed transcriptional analysis of this region described by others (31), all strains tested synthesized equivalent amounts of ORF 13 and ORF 15 mRNAs (data not shown). By Western blotting, P-Oka, VZV-S, V-Oka, and VZV-Ellen produced the characteristic 105-kDa gC protein in MRC-5 cells, with some variability in quantity and size which is typical of this glycoprotein (11, 24). In contrast, gC expression could not be detected in cells infected with gC−-Oka or gC−-Ellen in vitro (Fig. 6). Restriction enzyme digest patterns were identical when DNA preparations of P-Oka, VZV-S, V-Oka, VZV-Ellen, gC−-Ellen, and gC−-Oka DNA were analyzed by using HpaI, BglII, EcoRI, and BamHI (data not shown).
FIG. 5.
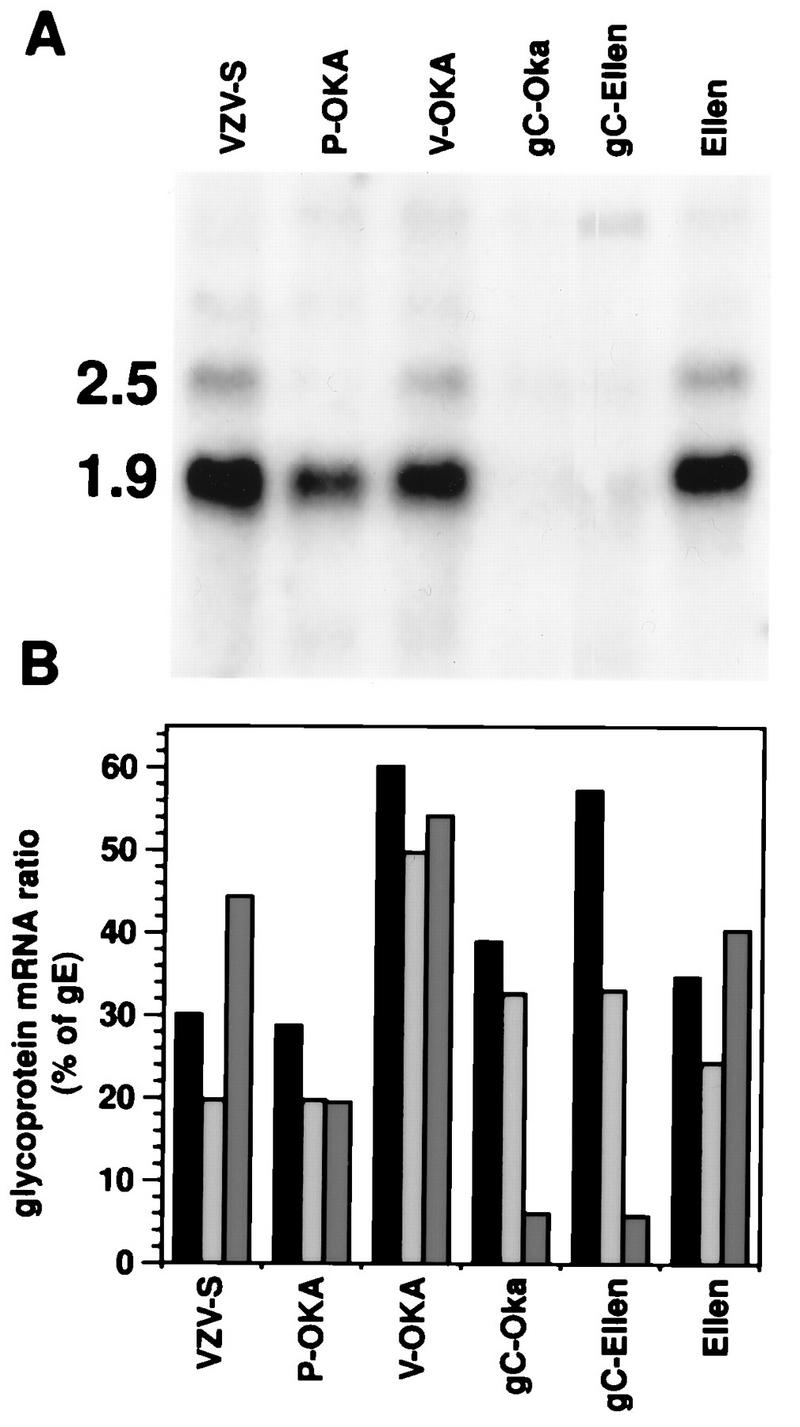
VZV glycoprotein mRNA ratios and expression of gC mRNA in vitro. VZV-S, P-Oka, V-Oka, gC−-Oka, gC−-Ellen, and VZV-Ellen were grown to equivalent CPE in MeWo cells, and total cell mRNA was prepared. RNA was electrophoresed, transferred to membranes, and hybridized with 32P-labeled probes specific for gB, gC, gE, and gH transcripts. Northern blots were visualized, and band intensities were quantitated by phosphorimager analysis. (A) Northern blot showing the 2.5- and 1.9-kb gC mRNA transcripts. Transcription of gC from gC−-Oka and gC−-Ellen was markedly decreased. (B) gB/gE (black bars), gH/gE (gray bars), and gC/gE (dark gray bars) ratios were calculated as a percentage of the gE transcript for each strain. Transcription of gC was deficient in gC−-Oka and gC−-Ellen, whereas gB and gH transcription was not altered. VZV-S, P-Oka, V-Oka, and VZV-Ellen had normal glycoprotein profiles.
FIG. 6.

Western blot analysis of gC expression in vitro. Protein was extracted from VZV-infected or uninfected MRC-5 cells, separated by SDS-PAGE, and transferred to a polyvinylidene difluoride membrane. On duplicate membranes, VZV proteins were detected with a polyclonal human immune serum (lower panel) and gC was detected with a monospecific human serum directed against a vaccinia virus-gC recombinant (upper panel). Bound antibodies were detected with horseradish peroxidase conjugated anti-human immunoglobulin G and visualized by ECL. Typical broad bands of VZV proteins were detected in all lanes of infected MRC-5 cells and not in uninfected cells. The approximately 105-kDa gC band, indicated by the arrow, can be seen on the blot probed with anti-VZV serum in all strains except gC−-Oka and gC−-Ellen, and the corresponding bands are missing on the blot probed with anti-gC serum. Molecular masses (in kilodaltons) are shown on the left.
Sequencing of the gC promoter regions of P-Oka, V-Oka, gC−-Oka, VZV-Ellen, and gC−-Ellen strains showed no differences that could account for the reduced transcription of gC mRNA in gC−-Oka and gC−-Ellen. The putative 145-bp gC promoter regions of all strains tested were identical to the consensus VZV genomic DNA sequence (Dumas strain) (14); this homology extended into the coding regions of ORFs 14 and 15 (data not shown).
The absence of gC expression was associated with a further decrease in the already diminished capacity of V-Oka to replicate in human skin. Some cell-associated infectious virus was recovered after inoculation of skin implants with gC−-Oka, but the concentration of virus was significantly lower than yields from skin infected with P-Oka or V-Oka, from which it was derived (Fig. 3). At day 21, a mean of 143 PFU of cell-associated virus was cultured from six implants infected with gC−-Oka, compared to 6,200 PFU of P-Oka (P = 0.0005) and 1,600 PFU of V-Oka (P = 0.02) (Fig. 3). Infectious virus was recovered in unfiltered cell suspensions from five of the six implants inoculated with gC−-Oka, but no cell-free virus was detected. The level of VZV protein expression in skin implants infected with gC−-Oka was 308 density units, which was significantly less than protein synthesis detected after inoculation with P-Oka or V-Oka (P ≤ 0.02) (Fig. 4).
The decreased replication and VZV protein expression in implants infected with gC−-Oka was confirmed by histology and in situ hybridization (Fig. 1). Sections of infected skin stained with hematoxylin and eosin showed that gC−-Oka produced small lesions in the epidermis, compared to the large vesicular areas of necrosis extending deep into the dermis in skin infected with V-Oka and P-Oka. Mock-infected skin had a normal appearance. Viral DNA was detected by in situ hybridization only in the superficial keratinocyte layer of the epidermis in gC−-Oka-infected skin (Fig. 1c), while V-Oka and P-Oka DNA was present in lower layers of the dermis that consist primarily of fibroblasts (Fig. 1a and b). VZV DNA was not detected in the mock-infected control (Fig. 1d).
Given the marked inhibition of the virulence of VZV-Ellen, the failure to express gC by gC−-Ellen could not be implicated as the cause of any further decrease in infectivity for human skin. In two experiments, infectious virus was not recovered from any of nine implants inoculated with gC−-Ellen, and VZV protein synthesis was not detected by Western blot in these implants by standard autoradiography. The enhanced sensitivity of phosphorimager analysis revealed VZV proteins in eight implants inoculated with gC−-Ellen and harvested at day 21, at an average of 14 density units, which was similar to the minimal synthesis of VZV proteins in implants infected with the parent strain, VZV-Ellen, in vivo (Fig. 4).
Comparison of VZV and HSV-1 infectivity in thy/liv implants and effect of gC expression on HSV-1 replication in human skin.
Thy/liv implants were inoculated with HSV-1 KOS, the gC deletion strain ΔgC2-3, and its gC-positive revertant ΔgC2-3rev. Replication peaked on day 4, and there was no difference in virus yields between the three strains (Table 1). In situ hybridization showed HSV-1 DNA in large cells scattered throughout the thy/liv implant (Fig. 7). Combined in situ hybridization and immunohistochemistry using an antikeratin monoclonal antibody showed that HSV DNA was present in cortical epithelial cells (data not shown). The presence or absence of gC did not affect HSV infectivity for cortical epithelial cells. Colocalization experiments using a monoclonal antibody to T-cell or macrophage markers showed no HSV DNA in these cell types. T cells were depleted in regions of most pronounced cytopathology, and many had undergone apoptosis, demonstrated by enzymatic in situ labeling of apoptosis-induced DNA strand breaks (data not shown).
TABLE 1.
Replication of HSV-1 KOS and the HSV-1 gC deletion mutant in thy/liv and skin implants
| Implant tissue | Mean log PFU/implant ± SE (no. of implants that yielded virus/no. analyzed)a
|
||
|---|---|---|---|
| KOS | ΔgC2-3rev | ΔgC2-3 | |
| Thy/liv | 6.6 ± 1.2 (2/2) | 5.4 ± 1.2 (2/2) | 6.5 ± 0.4 (3/3) |
| Skin | 6.7 ± 0.3 (6/8) | 7.6 ± 0.8 (8/8) | 6.1 ± 0.5 (3/10) |
Implants were inoculated with HSV-1 strains and harvested on day 4 (thy/liv) or day 6 (skin) after infection, and titers in cell lysates were determined.
FIG. 7.

Histological analysis of an HSV-infected thy/liv implant inoculated with HSV-1 KOS and harvested on day 2 after infection. The implant was fixed in paraformaldehyde, paraffin embedded, and cut into 3-μm sections before in situ hybridization was performed. Darkly stained cells indicate where HSV-1 DNA was detected in cortical epithelial cells. Uninfected T cells were lightly counterstained with hematoxylin. Magnification, ×131.
ΔgC2-3, the gC deletion strain of HSV-1 KOS, was tested in skin implants to determine whether this HSV protein, which is homologous to VZV gC, was a virulence factor in human skin cells (Fig. 8). Skin implants were infected with the gC deletion strain, the gC-expressing revertant strain ΔgC2-3rev, or the parent strain KOS in duplicate experiments. The implants were harvested on day 6 after inoculation and examined for infectious virus yields, histopathologic changes, and localization of viral DNA by in situ hybridization. Virus was recovered from six of eight implants infected with HSV-1 KOS, with a mean titer of 5.1 × 106 PFU, and from eight of eight implants infected with the revertant, with a mean titer of 3.6 × 107 PFU. HSV-1 ΔgC2-3, the gC deletion strain, was recovered from 3 of 10 infected implants, with a mean titer of 1.3 × 106 PFU, while no infectious virus was detected in the remaining seven implants (Table 1).
FIG. 8.
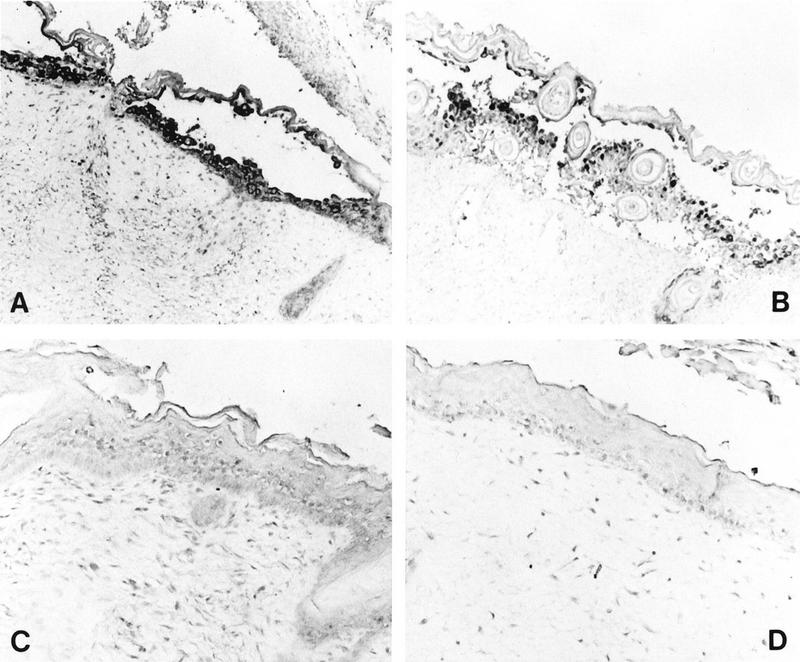
Histological analysis of HSV-infected skin implants. Subcutaneous skin implants were inoculated with HSV-1 KOS (A), HSV-1 ΔgC2-3rev (B), or HSV-1 ΔgC2-3 (C) or mock infected (D) and harvested on day 6 postinfection. Implants were fixed in paraformaldehyde, paraffin embedded, and cut into 3-μm sections before in situ hybridization was performed. HSV-1 DNA was detected in the equivalent epidermal lesions produced by KOS and ΔgC2-3rev (A and B). No HSV-1 DNA was detected in the epidermis of ΔgC2-3- or mock-infected implants (C and D). Hematoxylin counterstain revealed tissue histology. Magnifications: A and B, ×61; C and D, ×122.
When skin implants were inoculated with HSV-1 KOS or the revertant ΔgC2-3rev, the virus caused superficial lesions restricted to the epidermis whereas VZV DNA was detected in dermal layers (Fig. 1a and 8A and B). No lesion formation or other pathologic changes were observed, and no HSV DNA was detected in skin implants inoculated with the HSV-1 gC deletion strain. With respect to histologic appearance and in situ hybridization analysis, the implants were indistinguishable from mock-infected controls (Fig. 8C and D). The skin implant shown in Fig. 8C was one of the three implants from which virus was recovered, yet no evidence for viral replication was seen in the tissue. The cellular site of replication of the gC deletion strain in the three skin implants from which HSV-1 ΔgC2-3 was recovered was likely to have been the membrane of murine origin which forms around the subcutaneous skin implants.
DISCUSSION
These experiments using human skin implants in the SCID-hu mouse model have provided the first opportunity to document differences in the pathogenicity of VZV strains that are indistinguishable by the characteristics of their replication in tissue culture cells in vitro. The most important observation is that V-Oka, the varicella vaccine strain, is attenuated in its infectivity for human skin by virologic measures, including reduced yields of infectious virus and decreased viral protein synthesis. The diminished virulence of V-Oka for human skin as demonstrated in the SCID-hu mouse model explains several aspects of the clinical experience with this newly licensed herpesvirus vaccine. Vaccine-related rashes are rare in healthy children, but immunocompromised children with leukemia who are immunized with V-Oka often have scattered, vesicular skin lesions from which the vaccine virus can be recovered, indicating that viremia has occurred (18). Our experiments in the SCID-hu model show that V-Oka is not altered in its infectivity for CD4+ or CD8+ T cells (35). As a consequence, V-Oka can cause cell-associated viremia in the absence of the immediate host response which is elicited by the immunization of healthy children and adults (29). The occurrence of viremia in immunocompromised children indicates that the attenuation of V-Oka for skin is not sufficient to restrict its replication to the cutaneous site of inoculation if the host response is delayed or diminished.
In our experiments, the impaired infectivity of V-Oka for human skin contrasted with the extensive replication of low-passage clinical isolates, including P-Oka and VZV-S. The virulence of these wild-type strains as demonstrated in the SCID-hu model is consistent with early reports about VZV pathogenesis showing that healthy children developed typical varicella when they were inoculated with unpassaged virus recovered directly from VZV vesicles (43). In contrast to V-Oka, wild-type VZV is likely to replicate in skin and infect T cells before virus-specific immunity is induced even in healthy individuals. Our observation that the attenuation phenotype of V-Oka was not reversed by a single passage in human skin confirms clinical evidence suggesting that the reduced virulence of V-Oka is maintained after one phase of replication in vaccine recipients. Although V-Oka was transmitted to some susceptible healthy siblings of children with leukemia, the symptoms of varicella caused by V-Oka were mild (18).
V-Oka was derived from P-Oka by using a traditional approach for creating a live attenuated vaccine in which the wild-type virus is passaged in a nonhuman cell line. Guinea pig embryo fibroblasts were used because VZV replication occurs in guinea pigs despite the otherwise narrow host range of the virus. This strategy resulted in the attenuation of P-Oka, but our experiments in the SCID-hu model also demonstrated that prolonged passage in human cells alone was sufficient to reduce the virulence of VZV for human skin. VZV-Ellen, which has been passaged more than 100 times since its isolation in 1964, replicated poorly in skin even though no specific laboratory procedures were undertaken to attenuate the strain (7). Attenuation by passage in human cells has been observed in the effort to develop a live attenuated vaccine for human cytomegalovirus, another human herpesvirus, in which the Towne strain became avirulent after 129 passages in WI-38 cells (37).
One of the distinguishing characteristics of VZV is the highly cell-associated nature of its replication in vitro (2, 20). In contrast, infectious virus was released from skin infected with P-Oka and VZV-S in vivo. Similarly, vesicular fluid from skin lesions of patients with varicella and herpes zoster contains intact, cell-free virus (17). By electron microscopy, the morphology of VZV virions produced in skin implants in the SCID-hu model differed markedly from the pattern of defective particles and virion degradation observed in tissue culture cells. V-Oka, like other VZV strains, produces a mixture of viral particles in embryonic lung fibroblasts, many of which have aberrant capsids, are not enveloped, and are therefore not infectious (19, 20). Nevertheless, despite adjusting the inoculum to equivalent titers of infectious virus, V-Oka was less infective for human skin than its parent strain. Prolonged passage in tissue culture cells appears to induce virologic changes that have a significant impact on VZV virulence for human epidermal and dermal cells in vivo since only the low-passage clinical isolates, P-Oka and VZV-S, were fully virulent in skin implants. These changes occurred without any major genomic alterations detectable by restriction enzyme analysis using HpaI, BglII, EcoRI, and BamHI.
The tropism of VZV for human T cells and its infectivity for skin are both essential elements of its pathogenicity in human disease. These experiments in the SCID-hu mouse model demonstrated that these tropisms are mediated by different virulence determinants. V-Oka was indistinguishable from low-passage VZV in its peak titers for CD4+ and CD8+ T cells, whereas its pathogenic potential in human skin was reduced substantially, as assessed by the extent of the cutaneous lesions, viral protein synthesis, infectious virus yields, and release of infectious virus. This altered infectivity of V-Oka for human skin in vivo was not predictive of a corresponding attenuation of the same VZV strain with respect to its T-cell tropism.
The comparison of V-Oka and its derivative strain, gC−-Oka, demonstrated that gC is a specific determinant for VZV virulence in skin, although the reduced infectivity of V-Oka relative to P-Oka indicates that gC expression is not the only factor. The analysis of viral localization by in situ hybridization showed that lack of gC expression was associated with a failure of VZV to penetrate through the epidermal cell layer into dermal fibroblasts. Although T-cell tropism was preserved, deletion of gC may have other attenuating effects on VZV, in addition to reduced skin infectivity. As is true of other alphaherpesviruses, VZV gC is not required for replication in tissue culture. In HSV-1, gC binds to a proteoglycan receptor on the apical surface of polarized MDCK cells but is not required for HSV-1 entry via the basolateral surface (40). VZV spreads by cell fusion in nonpolarized tissue culture cells, which probably accounts for the fact that VZV gC is dispensable and its expression is variable in vitro. In contrast, the requirement for VZV gC expression for infectivity in skin implants in SCID-hu mice suggests that gC is involved in viral interactions with polarized epidermal cells in intact skin in vivo. That gC expression is a virulence determinant for alphaherpesvirus replication in human skin was further substantiated by the failure of the HSV-1 gC deletion mutant ΔgC2-3 to replicate in epidermal cells in skin implants. In vivo, both VZV gC and HSV-1 gC could affect viral transport across the basal lamina and into the tightly joined cells of the epidermis. Enhanced infectivity for polarized cells is likely to be the important common pathogenic function of gC in VZV and HSV-1 since other activities of the alphaherpesvirus gC homologs, such as increasing infectivity by binding to heparan sulfate on the cell surface or binding to the C3b component of complement, do not appear to be shared by VZV (11, 15, 16, 21, 22, 23a, 33, 38, 46). Of note, the transcriptional inactivation of VZV gC in the gC−-Oka and gC−-Ellen variants and their altered virulence in vivo occurred despite the presence of an intact gC promoter sequence. Despite this extensive analysis of gC transcription and the ORF 14 regulatory region, additional mutations that arose elsewhere in the genomes of the gC−-Oka and gC−-Ellen strains cannot be excluded as contributing factors in these strains’ loss of infectivity for skin. A possible explanation for the absence of ORF 14 transcription is that one or more transactivating proteins are involved in regulation of its promoter (31). A detailed analysis of ORF 14 transcription by Ling et al. showed that the 1.9-kb transcript is the predominant coding transcript and is sufficient to code for the entire gC protein (31). Levels of the 2.5-kb transcript, a minor species polyadenylated at an alternative site within ORF 13, may vary at the late stages of infection when transcriptional control starts to erode. This could account for the absence of generation of the 2.5-kb transcript by P-Oka.
The comparative analysis of VZV and HSV-1 replication in the SCID-hu model revealed significant differences in cell tropisms that correlate with clinical observations about their pathologic effects. Although T cells within thy/liv implants are highly permissive for VZV replication, HSV-1 was detected only in nonlymphoid, cortical epithelial cells. Varicella is characterized by the appearance of scattered cutaneous vesicles, while primary HSV-1 infection is associated with localized, mucocutaneous lesions (2). This widespread rash is a consequence of the lymphocyte-associated viremia caused by VZV during primary infection, whereas HSV viremia is an unusual complication (3, 35). Immunocompromised patients are also susceptible to cell-associated viremia during VZV reactivation, which is a rare occurrence during HSV reactivation (45). HSV-1 can infect activated T-cells in vitro, and abortive replication in T cells was not not excluded in the thy/liv implant experiments. In SCID-hu mice, VZV caused extensive necrosis in deeper dermal layers, but HSV-1 necrosis was confined to the epidermis of skin implants. Similarly, the initial cutaneous lesions produced by primary VZV infection extend into the dermis and often cause scarring before an immune response develops, while HSV-1 lesions remain superficial.
In general, the documentation of virulence phenotypes among VZV strains that are indistinguishable in vitro, including the differentiation of V-Oka from its parent, and the differences in cell tropisms of VZV and HSV-1 underscore the relevance of the SCID-hu model for studies of viral pathogenesis and the development of live attenuated viral vaccines.
ACKNOWLEDGMENTS
J.F.M. was supported by a training grant from the Division of Developmental and Neonatal Biology (HD07249) and NRSA AI09195. The work was supported by Public Health Service grants AI20459, AI36884, and P01-CA49605 to A.M.A. and RO1-EY09397 and P30-EY08098 to P.R.K.
We thank Marvin Sommer, Judie Boisvert, and Jean-Paul Vergnes for technical assistance.
REFERENCES
- 1.Aldrovandi G M, Feuer G, Gao L, Jamieson B, Kristeva M, Chen I S Y, Zack J A. The SCID-hu mouse as a model for HIV-1 infection. Nature. 1993;363:732–736. doi: 10.1038/363732a0. [DOI] [PubMed] [Google Scholar]
- 2.Arvin A M. Varicella-zoster virus. In: Fields B N, Knipe D N, Howley P M, editors. Virology. Philadelphia, Pa: Lippincott-Raven Publishers; 1996. pp. 2547–2585. [Google Scholar]
- 3.Arvin A M, Koropchak C M, Williams B R G, Grumet F C, Foung S K. Early immune response in healthy and immunocompromised subjects with primary varicella-zoster virus infection. J Infect Dis. 1986;154:422–429. doi: 10.1093/infdis/154.3.422. [DOI] [PubMed] [Google Scholar]
- 4.Auwaerter P G, Kaneshima H, McCune J M, Wiegand G, Griffin D E. Measles virus infection of thymic epithelium in the SCID-hu mouse leads to thymocyte apoptosis. J Virol. 1996;70:3734–3740. doi: 10.1128/jvi.70.6.3734-3740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bergen R E, Diaz P S, Arvin A M. The immunogenicity of the Oka/Merck varicella vaccine in relation to infectious varicella-zoster virus and relative viral antigen content. J Infect Dis. 1990;162:1049–1054. doi: 10.1093/infdis/162.5.1049. [DOI] [PubMed] [Google Scholar]
- 6.Bogger-Goren S, Baba K, Hurley P, Yabuuchi H, Takahashi M, Ogra P L. Antibody response to varicella-zoster virus after natural or vaccine-induced infection. J Infect Dis. 1982;146:260–265. doi: 10.1093/infdis/146.2.260. [DOI] [PubMed] [Google Scholar]
- 7.Brunell P A, Casey H L. Crude tissue culture antigen for determination of varicella-zoster complement fixing antibody. Public Health Rep. 1964;79:839–842. [PMC free article] [PubMed] [Google Scholar]
- 8.Centifanto-Fitzgerald Y M, Yamaguchi T, Kaufman H E, Tognon M, Roizman B. Ocular disease pattern induced by herpes simplex virus is genetically determined by a specific region of viral DNA. J Exp Med. 1982;155:475–489. doi: 10.1084/jem.155.2.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheatham W J, Weller T H, Dolan T F J, Dower J C. Varicella: report of two fatal cases with necropsy, virus isolation, and serologic studies. Am J Pathol. 1956;32:1015–1035. [PMC free article] [PubMed] [Google Scholar]
- 10.Chomczynski P, Sacchi N. Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 11.Cohen J I, Seidel K E. Absence of varicella-zoster virus (VZV) glycoprotein V does not alter growth of VZV in vitro or sensitivity to heparin. J Gen Virol. 1994;75:3087–3093. doi: 10.1099/0022-1317-75-11-3087. [DOI] [PubMed] [Google Scholar]
- 12.Croen K, Ostrove J, Dragovic L, Straus S. Patterns of gene expression and sites of latency in human nerve ganglia are different for varicella-zoster and herpes simplex viruses. Proc Natl Acad Sci USA. 1988;85:9773–9777. doi: 10.1073/pnas.85.24.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Croen K, Straus S. Varicella-zoster virus latency. Annu Rev Microbiol. 1991;4:2391–99. doi: 10.1146/annurev.mi.45.100191.001405. [DOI] [PubMed] [Google Scholar]
- 14.Davison A J, Scott J E. The complete DNA sequence of varicella-zoster virus. J Gen Virol. 1986;67:1759–1816. doi: 10.1099/0022-1317-67-9-1759. [DOI] [PubMed] [Google Scholar]
- 15.Flynn S J, Ryan P. The receptor-binding domain of pseudorabies virus glycoprotein gC is composed of multiple discrete units that are functionally redundant. J Virol. 1996;70:1355–1364. doi: 10.1128/jvi.70.3.1355-1364.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman H M, Cohen G H, Eisenberg R J, Seidel C A, Cines D B. Glycoprotein C of HSV-1 functions as a C3b receptor on infected endothelial cells. Nature. 1984;309:633–635. doi: 10.1038/309633a0. [DOI] [PubMed] [Google Scholar]
- 17.Gelb L D. Varicella-zoster virus: molecular biology. In: Roizman B, Whitley R J, Lopez C, editors. The human herpes viruses. New York, N.Y: Raven Press; 1993. pp. 257–279. [Google Scholar]
- 18.Gershon, A. A., P. LaRussa, I. Hardy, S. Steinberg, and S. Silverstein. 1992. Varicella vaccine: the American experience. J. Infect. Dis. 166(Suppl. 1):S63–S68. [DOI] [PubMed]
- 19.Grose C. Pathogenesis of infection with varicella vaccine. In: Ellis R W, White C J, editors. Infectious disease clinics of North America. W. B. Philadelphia, Pa: Saunders Co.; 1996. pp. 489–505. [DOI] [PubMed] [Google Scholar]
- 20.Harson R, Grose C. Egress of varicella-zoster virus from the melanoma cell: a tropism for the melanocyte. J Virol. 1995;69:4994–5010. doi: 10.1128/jvi.69.8.4994-5010.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herold B C, WuDunn D, Soltys N, Spear P G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J Virol. 1991;65:1090–1098. doi: 10.1128/jvi.65.3.1090-1098.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huemer H P, Larcher C, Coe N E. Pseudorabies virus glycoprotein-III derived from virions and infected cells binds to the 3rd component of complement. Virus Res. 1992;23:271–280. doi: 10.1016/0168-1702(92)90113-n. [DOI] [PubMed] [Google Scholar]
- 23.Kaneshima H, Shih C-C, Namikawa R, Rabin L, Outzen G, Machado S G, McCune J M. Human immunodeficiency virus infection of human lumph nodes in the SCID-hu mouse. Proc Natl Acad Sci USA. 1991;88:4523–4527. doi: 10.1073/pnas.88.10.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23a.Kinchington, P., and G. Cohen. Personal communication.
- 24.Kinchington P R, Ling P, Pensiero M, Gershon A, Hay J, Ruyechan W T. A Possible role for glycoprotein gpV in the pathogenesis of varicella-zoster virus. In: Lopez C, editor. Immunobiology and prophylaxis of human herpesvirus infections. New York, N.Y: Plenum Press; 1990. pp. 83–91. [DOI] [PubMed] [Google Scholar]
- 25.Kinchington P R, Remenick J, Ostrove J M, Straus S E, Ruyechan W T, Hay J. Putative glycoprotein gene of varicella-zoster virus with variable copy numbers of a 42-base-pair repeat sequence has homology to herpes simplex virus glycoprotein C. J Virol. 1986;59:660–668. doi: 10.1128/jvi.59.3.660-668.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinchington P R, Vergnes J-P, Defechereux P, Piette J, Turse S E. Transcriptional mapping of the varicella-zoster virus regulatory genes encoding open reading frames 4 and 63. J Virol. 1994;68:3570–3581. doi: 10.1128/jvi.68.6.3570-3581.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koropchak C M, Diaz P S, Arvin A M. Investigation of varicella-zoster virus infection of lymphocytes by in situ hybridization. J Virol. 1989;63:2392–2395. doi: 10.1128/jvi.63.5.2392-2395.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koropchak C M, Graham G, Palmer J, Winsberg M, Ting S F, Wallace M, Prober C G, Arvin A M. Investigation of varicella-zoster virus infection by polymerase chain reaction in the immunocompetent host with acute varicella. J Infect Dis. 1991;163:1016–1022. doi: 10.1093/infdis/163.5.1016. [DOI] [PubMed] [Google Scholar]
- 29.Krause P R, Klinman D M. Efficacy, immunogenicity, safety, and use of live attenuated chickenpox vaccine. J Pediatr. 1995;127:518–525. doi: 10.1016/s0022-3476(95)70106-0. [DOI] [PubMed] [Google Scholar]
- 30.LaRussa P, Lungu O, Hardy I, Gershon A, Steinberg S P, Silverstein S. Restriction fragment length polymorphism of polymerase chain reaction products from vaccine and wild-type varicella-zoster virus isolates. J Virol. 1992;66:1016–1020. doi: 10.1128/jvi.66.2.1016-1020.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ling P, Kinchington P R, Ruychan W T, Hay J. A detailed analysis of transcripts mapping to varicella zoster virus gene 14 (glycoprotein V) Virology. 1991;184:625–635. doi: 10.1016/0042-6822(91)90432-b. [DOI] [PubMed] [Google Scholar]
- 32.Mahalingam R, Wellish M, Wolf W, Dueland A N, Cohrs R, Vafai A, Gilden D. Latent VZV DNA in human trigeminal and thoracic ganglia. N Engl J Med. 1990;323:627–631. doi: 10.1056/NEJM199009063231002. [DOI] [PubMed] [Google Scholar]
- 33.Mettenleiter T C, Zsak L, Zuckermann F, Sugg N, Kern H, Ben-Porat T. Interaction of glycoprotein gIII with a cellular heparinlike substance mediates adsorption of pseudorabies virus. J Virol. 1990;64:278–286. doi: 10.1128/jvi.64.1.278-286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mocarski E S, Bonyhadi M, Salimi S, McCune J M, Kaneshima H. Human cytomegalovirus in a SCID-hu mouse: thymic epithelial cells are prominent targets of viral replication. Proc Natl Acad Sci USA. 1993;90:104–108. doi: 10.1073/pnas.90.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moffat J F, Stein M D, Kaneshima H, Arvin A M. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J Virol. 1995;69:5236–5242. doi: 10.1128/jvi.69.9.5236-5242.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozaki T, Ichikawa T, Matsui Y, Kondo H, Nagai T, Asano Y, Yamanishi K, Takahashi M. Lymphocyte-associated viremia in varicella. J Med Virol. 1986;19:249–253. doi: 10.1002/jmv.1890190307. [DOI] [PubMed] [Google Scholar]
- 37.Plotkin S A, Starr S E, Friedman H M, Gonczol E, Weibel R E. Protective effects of Towne cytomegalovirus vaccine against low-passage cytomegalovirus administered as a challenge. J Infect Dis. 1989;159:860–865. doi: 10.1093/infdis/159.5.860. [DOI] [PubMed] [Google Scholar]
- 38.Robbins A K, Whealy M E, Watson R J, Enquist L W. Pseudorabies virus gene encoding glycoprotein gIII is not essential for growth in tissue culture. J Virol. 1986;59:635–645. doi: 10.1128/jvi.59.3.635-645.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawyer M H, Wu Y N, Chamberlin C J, Burgos C, Brodine S K, Bowler W A, LaRocco A, Oldfield III E C, Wallace M R. Detection of varicella-zoster virus DNA in the oropharynx and blood of patients with varicella. J Infect Dis. 1992;166:886–888. doi: 10.1093/infdis/166.4.885. [DOI] [PubMed] [Google Scholar]
- 40.Sears A E, McGwire B S, Roizman B. Infection of polarized MDCK cells with herpes simplex virus 1: two asymmetrically distributed cell receptors interact with different viral proteins. Proc Natl Acad Sci USA. 1991;88:5087–5091. doi: 10.1073/pnas.88.12.5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Straus S, Aulakh H S, Ruychan W T, Hay J, Casey T A, Vande Woude G F, Owens J, Smith H A. Structure of varicella-zoster virus DNA. J Virol. 1981;40:516–525. doi: 10.1128/jvi.40.2.516-525.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi M, Otsuka T, Okuno Y, Asano Y, Yazaki T, Isomura S. Live vaccine used to prevent the spread of varicalla in children in hospital. Lancet. 1974;ii:1288–1290. doi: 10.1016/s0140-6736(74)90144-5. [DOI] [PubMed] [Google Scholar]
- 43.von Bokay J. Das Auftreten der Schafblattern uter besonderen Umstanden. Ungerer’s Arch Med. 1892;1:159. [Google Scholar]
- 44.Whitley R J. In: Antiviral agents and viral diseases of man. Galasso G, Whitley R, Merigan T C, editors. New York, N.Y: Raven Press; 1990. pp. 235–264. [Google Scholar]
- 45.Wilson A, Sharp M, Koropchak C M, Ting S F, Arvin A M. Subclinical varicella-zoster virus viremia, herpes zoster and recovery of T-lymphocyte responses to varicella-zoster viral antigens after allogeneic and autologous bone marrow transplantation. J Infect Dis. 1992;165:119–126. doi: 10.1093/infdis/165.1.119. [DOI] [PubMed] [Google Scholar]
- 46.Zhu Z, Gershon M D, Ambron R, Gabel C, Gershon A A. Infection of cells by varicella zoster virus: inhibition of viral entry by mannose 6-phosphate and heparin. Proc Natl Acad Sci USA. 1995;92:3546–3550. doi: 10.1073/pnas.92.8.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]