

Abstract
The development of neoplasia frequently involves inactivation of the p53 and retinoblastoma (Rb) tumor suppressor pathways and disruption of cell cycle checkpoints that monitor the integrity of replication and cell division. The human papillomavirus type 16 (HPV-16) oncoproteins, E6 and E7, have been shown to bind p53 and Rb, respectively. To further delineate the mechanisms by which E6 and E7 affect cell cycle control, we examined various aspects of the cell cycle machinery. The low-risk HPV-6 E6 and E7 proteins did not cause any significant change in the levels of cell cycle proteins analyzed. HPV-16 E6 resulted in very low levels of p53 and p21 and globally elevated cyclin-dependent kinase (CDK) activity. In contrast, HPV-16 E7 had a profound effect on several aspects of the cell cycle machinery. A number of cyclins and CDKs were elevated, and despite the elevation of the levels of at least two CDK inhibitors, p21 and p16, CDK activity was globally increased. Most strikingly, cyclin E expression was deregulated both transcriptionally and posttranscriptionally and persisted at high levels in S and G2/M. Transit through G1 was shortened by the premature activation of cyclin E-associated kinase activity. Elevation of cyclin E levels required both the CR1 and CR2 domains of E7. These data suggest that cyclin E may be a critical target of HPV-16 E7 in the disruption of G1/S cell cycle progression and that the ability of E7 to regulate cyclin E involves activities in addition to the release of E2F.
Decisions to enter S phase and proliferate, arrest in the G0/G1 phase of the cell cycle, or differentiate are based on internal and external environmental stimuli. Progression through the cell cycle is dependent on the phosphorylation of key regulatory proteins by cyclin-dependent kinases (CDKs), which in turn are regulated in a complex fashion by association with cyclins, by phosphorylation and dephosphorylation, and by CDK-inhibitory proteins, CKIs (reviewed in reference 60). Progression through the cell cycle is monitored and DNA synthesis or mitosis is delayed if the integrity of the cell is compromised (5).
Mammalian cells commit to cell division during mid-G1, termed the restriction point (66), following phosphorylation of pRb, the product of the retinoblastoma tumor suppressor gene (13, 66). At least two cyclin-CDK complexes phosphorylate Rb, cyclin D-CDK4 or -CDK6 and cyclin E-CDK2 (22, 34, 60). While Rb appears to be an exclusive target of cyclin D-associated kinases, cyclin E probably targets additional factors necessary for cell cycle progression (49, 50). Ectopic expression of cyclins D1 and E have been shown to accelerate the G1/S transition (49, 50, 54), and they appear to control two different rate-limiting events (55). Rb becomes inactivated by phosphorylation and releases the transcription factor E2F. Free E2F transactivates numerous S-phase gene promoters prior to the movement of cells into S phase (14, 41). Among the targets of E2F transactivation are cyclins D, E, and A (48).
A number of human papillomaviruses (HPV) infect epithelial cells in the genital tract and are classified based on their oncogenic potential: the low-risk viruses (e.g., types 6 and 11) are most often associated with benign genital warts, whereas the high-risk viruses (e.g., types 16 and 18) are frequently found in cervical carcinomas (74). The in vivo biology of HPVs is recapitulated in their potential to immortalize primary human cells in vitro (37). The E6 and E7 genes are invariably retained and expressed in tumors, while all the other viral genes are dispensable, and E6 and E7 together efficiently immortalize cells. In response to growth arrest signals such as DNA damage or transforming growth factor β (TGF-β), cells expressing HPV-16 but not HPV-6 oncoproteins continue to proliferate (15). Replication of papillomaviruses requires a cis-acting origin and a virally encoded origin binding protein with helicase activity, while other factors required for the biosynthesis of DNA are cellular products (reviewed in references 44 and 63). Thus, HPVs have evolved functions to ensure that infected cells enter S phase.
The E7 proteins of several HPVs bind to the retinoblastoma tumor suppressor, p105Rb (18), and other Rb family members such as p107 and p130 (10) through a well-defined motif, LxCxE. HPV-16 E7 binds Rb with higher affinity both in vitro (46) and in vivo (26) than HPV-6 E7 does. The E7-Rb interaction results in the release of E2F. Accordingly, the expression of E7 in NIH 3T3 cells leads to a rapid induction of cyclin E (71), which was shown to be dependent on the E2F site in the cyclin E promoter (3). In addition, HPV-16 E7 expression results in a reduction in the levels of Rb protein (16), although the combined expression of E6 and E7 results in high levels of Rb (16). E7 has been shown to induce cells to enter S phase (65), including suprabasal epithelial cells, which are normally quiescent (2, 6, 15, 29). HPV-16 E6 binds to p53 (67) in concert with a cellular ubiquitin-conjugating enzyme, E6AP, and targets p53 for degradation (58). HPV-6 E6 binds to p53 much more weakly (9, 24, 67) and does not target p53 for degradation in vitro, and the intracellular level of p53 in cells expressing 6E6 is unaffected (24).
To further analyze how papillomavirus oncoproteins disrupt cell cycle control, we introduced HPV-6 and HPV-16 E6 and E7 genes into primary human cells and examined various aspects of the cell cycle machinery. HPV-16 E7 expression had a profound effect on levels of several cyclins, CDKs, and CKIs. Interestingly, cyclin E regulation was disrupted both transcriptionally by the increase in E2F activity and posttranscriptionally by additional mechanisms resulting in altered cell cycle distribution.
MATERIALS AND METHODS
Cell culture and recombinant retroviral plasmids.
Primary human epithelial and fibroblast cells were prepared from human foreskin samples and infected with amphotropic retroviruses as previously described (30). The retroviral vector LXSN (45) contained the HPV-6 or HPV-16 E6 or E7 oncogenes singularly or in combination and a gene which confers neomycin resistance (30). Retroviral constructs expressing mutated HPV-16 E7 proteins have also been described (15). The vector containing the cDNA encoding human cyclin E (49) was kindly provided by Jim Roberts (Fred Hutchinson Cancer Research Center). Infected cells were selected with G418 (1 mg/ml) for 7 to 10 days. Epithelial cells were maintained in keratinocyte serum-free medium (GIBCO). Fibroblasts were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS; HyClone) and antibiotics.
Western blots and kinase assays.
Cell monolayers were washed with phosphate-buffered saline (PBS) and scraped from culture plates. Total-cell lysates were prepared by lysing packed cells in lysis buffer (50 mM Tris-HCl [pH 7.5], 250 mM NaCl, 5 mM EDTA, 0.1% sodium dodecyl sulfate [SDS], 1% Nonidet P-40, 1% deoxycholic acid, 20% glycerol) containing 50 mM NaF, 0.5 mM sodium orthovanadate, 80 mM β-glycerophosphate, 10 μg of aprotinin per ml, 10 μg of pepstatin per ml, 25 μg of leupeptin per ml, 0.5 mg of Pefablock per ml, and 1 mM dithiothreitol. The lysates were sonicated in a cup horn sonicator (Branson Sonifier 450), and the supernatants were diluted in sample buffer (0.25 M Tris-HCl [pH 7.5], 8% [wt/vol] SDS, 40% [vol/vol] glycerol, 20% [vol/vol] β-mercaptoethanol, 0.05% [wt/vol] bromophenol blue). The proteins were resolved on Tris-glycine SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes (Dupont). Cyclin A (1:2,500) and CDC2 (1:3,000) polyclonal rabbit antibodies were kindly provided by Jim Roberts. Affinity-purified goat anti-rabbit immunoglobulin Gs IgGs (peroxidase conjugated) were used as secondary antibodies (1:20,000; Boehringer Mannheim). Monoclonal antibodies to cyclins B1 (1:1000), D1 (1:200), E (1:5,000), and A (1:600) (Pharmingen) and to CDK2 (1:250) and CDK4 (1:250) (Transduction Laboratories) were used. Anti-mouse horseradish peroxidase conjugate was used as a source of secondary antibodies (Jackson Immunoresearch Laboratories). Membranes were developed by chemiluminescence (Renaissance; NEN).
Histone H1 kinase assays were performed as previously described (38, 39) with polyclonal antibodies against cyclins A and E, CDK2, and CDC2 provided by Jim Roberts. Briefly, extracts (100 μg) were subjected to immunoprecipitation with cyclin E antibodies (1 μl) on ice for 20 min. Immunocomplexes were collected on protein A-Sepharose (Pharmacia Biotech) and washed three times with H1 wash buffer and once with H1 kinase buffer. The H1 kinase reaction mixtures were incubated at 37°C for 30 min in kinase buffer in the presence of histone H1 (40 μg; Sigma) and [γ-32P]ATP (10 μCi per reaction) (Dupont/NEN). The reaction products were resolved on SDS–12% polyacrylamide gels and analyzed by autoradiography.
RNase protection assays.
Total-cell RNA was extracted by lysing cells directly on culture plates with 1.5 ml of guanidinium solution (4M guanidinium isothiocyanate, 20 mM sodium acetate [pH 5.2], 0.1 mM dithiothreitol, 0.5% N-lauroylsarcosine [Sarkosyl]). The lysates were scraped from the culture dishes and sheared, and the entire cell lysate was layered onto a cushion of 5.7 M cesium chloride (CsCl). The samples were fractionated at 150,000 × g for 16 to 24 h at 18°C. RNA was resuspended in TES buffer (10 mM Tris [pH 7.5], 5 mM EDTA [pH 7.5], 1% SDS] and precipitated twice on a mixture of dry ice and ethanol. Riboprobe templates were prepared by cloning cyclin E (390 bp), A (342 bp), and D1 (320 bp) coding fragments into the PBS(+) plasmid expression vector. The RNA loading control riboprobe was synthesized from pGem-4z containing a 220-bp PstI fragment of 36B4 (a gift of Robert Dickson and Sharyl Nass, Lombardi Cancer Center, Georgetown University) (42, 47). The plasmids were linearized with EcoRI or NcoI. RNA antisense strands were synthesized with RNA polymerase T3 or T7 (Boehringer Mannheim) and [α-32P]UTP (800 Ci/mmol; Dupont/NEN). The riboprobes were treated with DNase I and purified through Sephadex G-50 columns (Pharmacia Biotech).
RNase protection assays were conducted by following the protocol of the Ribonuclease Protection Assay kit (RPA II; Ambion). Protected RNA fragments were separated on 5% acrylamide–8 M urea gels. The gels were quantitated with a PhosphorImager (Molecular Dynamics).
Cell synchronization and sorting and cell cycle analysis.
Primary human foreskin fibroblasts (HFF) were synchronized either by a combination of density arrest and serum starvation (D/S) or by just serum starvation (S). Either the cells were grown to density for 2 days and incubated in 0.5% FBS for 3 days (D/S) or subconfluent cell populations were incubated in 0.2% FBS for 3 days (S). The cell cycle phases were measured by flow cytometry (FACScan; Becton Dickinson) and analyzed with Cell Quest software (Becton Dickinson) and MultiCycle (PHOENIX Flow Systems, San Diego, Calif.).
Human foreskin keratinocytes (HFK) were trypsinized from culture plates and resuspended in 5 ml of PBS-ABC (1× phosphate-buffered saline, 1 mM CaCl2, 1 mM MgCl2). Nuclear staining was performed by staining the cells with 5 μg of Hoechst 33342 per 106 cells per ml. To eliminate nonviable cells from the sorted population, the cells were counterstained with propidium iodide (PI) at a final concentration of 5 μg/ml and gated out. HFKs were sorted into G1, S, and G2/M fractions on the FACS Vantage (Becton Dickinson). The cell cycle fractions (6 × 104 cells) were sorted into sterile Eppendorf tubes. Flow cytometric data were collected and analyzed with Cell Quest and MultiCycle. The cell pellets were lysed in lysis buffer, sonicated, and centrifuged to remove cellular debris. Sample buffer was added before the entire cellular lysates were loaded onto SDS-polyacrylamide gel electrophoresis (PAGE) gels.
RESULTS
HPV-16 E7 alters the levels and activities of cyclins, CDKs, and CKIs.
LXSN-based amphotropic retroviruses expressing the E6 and E7 genes of HPV-6 and HPV-16 were used to infect HFK and HFF cultures derived from neonatal foreskin. HFKs were chosen because they represent the natural targets of HPV infection; HFFs were chosen because cell cycle proteins have been frequently studied in this cell type because it is easier to grow in culture and easier to synchronize. The cultures were selected for neomycin resistance, and expression of the viral proteins was documented by radioimmunoprecipitation with specific antisera (23). As demonstrated previously (16, 29, 30), each of the papillomavirus proteins was expressed abundantly; E7 was expressed at a reduced level from E6/E7 retrovirus compared to the E7 retrovirus, as shown previously (16, 21, 29), because of transcriptional repression of the retroviral long terminal repeat LTR by E6 (21).
To begin to examine the effects of the HPV oncogenes on the cell cycle, expression of a panel of G1, S and G2/M regulatory control proteins was examined. Figure 1 shows individually probed Western blots for several cyclins and CDKs in asynchronously growing HFKs (Fig. 1A) and HFFs (Fig. 1B) and in HFFs arrested in G0/G1 by growth to density and serum starvation (HFF-D/S) (Fig. 1C). No changes in any of the cell cycle proteins were detected as a result of the low-risk oncogenes. The only noticeable change in cells expressing HPV-16 E6 was a reduction in the level of cyclin D1. In cells expressing HPV-16 E7, several cyclins and CDKs were more abundant; the most striking and consistent change was the increased level of cyclin E. In asynchronously growing populations of HFKs or HFFs, cyclin E levels were elevated approximately threefold. A more dramatic elevation in the level of cyclin E protein, ranging from 5- to 20-fold, was observed in quiescent cells (Fig. 1C) (see below). Elevated levels of cyclin A were not consistently detected in asynchronously growing cells expressing E7 but were detected in synchronous populations released from quiescence (see below).
FIG. 1.

Cyclin and CDK protein levels in asynchronous and G0/G1-arrested cells. Total-cell lysates were prepared from asynchronous HFKs and HFFs (A and B) or G0/G1-arrested fibroblasts (C) expressing the designated viral gene. Proteins (20 μg/lane) were fractionated on individual SDS-PAGE gels (12% polyacrylamide) and transferred to PVDF membranes. The membranes were probed with antibodies, as described in Materials and Methods, and were visualized by enhanced chemiluminescence. (D) The cells were fixed, processed for PI immunofluorescence, and monitored for their position in the cycle by FACScan analysis.
Among asynchronously growing HFFs, the distribution of cells in G1, S, and G2/M was not significantly affected by the HPV oncogenes whereas constitutive overexpression of cyclin E from a retroviral promoter reduced the number of cells in G1 as previously reported (49). HFKs responded more dramatically to increased cyclin E levels, with fewer cells in the G1 phase in the E7-expressing population and an even greater reduction in the cyclin E-overexpressing population. Growth arrest by either depletion of serum (S) or density arrest followed by serum depletion (D/S) generally resulted in an almost complete accumulation of LXSN cells in G0/G1, with only 1 to 2% of the cells being found in the S phase (Fig. 1D); however, the E7-expressing cells were never as tightly arrested, with an S-phase population of 5 and 10% in the D/S- and S-arrested cells, respectively.
At least two families of CDK inhibitors, the CIP/KIP family and the INK4 family, mediate the G1/S transition by controlling the kinase activity of the cyclin D- and cyclin E-associated CDKs (60). We had previously observed that p53 protein levels were elevated three- to fivefold in HPV-16 E7-expressing cells and that p53 protein was nearly undetectable in E6-expressing cells, although the RNA levels were unchanged (16, 17). p21cip1 is transcriptionally activated by p53 (19, 20); thus, the elevated levels of p53 in E7-expressing cells may result in increased p21 production. Figure 2A shows that the levels of p21cip1 parallel the levels of p53 in the HPV oncogene-expressing cells; the levels of p21 protein reflected the level of p21 mRNA (data not shown). p16ink4 specifically regulates cyclin D-associated kinases, which in turn mediate phosphorylation of Rb and release of E2F (reviewed in references 34, 36, and 60). High levels of p16 protein were observed in 16E7-expressing cells (Fig. 2B), consistent with the hypothesis that the requirement for cyclin D-associated kinase activity is obviated by the ability of E7 to release E2F from Rb. p27kip1 associates with G1 cyclin complexes and mediates G1 arrest in response to antimitogenic signals such as TGF-β or contact inhibition (40, 53). Neither p27 protein (Fig. 2C) nor RNA levels (data not shown) varied significantly in E6- or E7-expressing cells, indicating that transcription of p27 is not regulated by p53 or E2F and that HPV oncogenes did not affect the levels of p27. These observations indicate that cells expressing E7 continue to cycle in the presence of high levels of at least two CKIs, p21 and p16.
FIG. 2.

CKI expression and CDK activity in proliferating cells. (A to C) Whole-cell lysates were prepared from retrovirally transduced asynchronous HFKs. Lysates (20 μg) were resolved on SDS-PAGE gels (12% polyacrylamide) and probed with monoclonal antibodies to p53 and p21 (A), p16 (B), and p27 (C). (D) Extracts were prepared from asynchronous HFKs expressing the indicated HPV oncogenes, immunoprecipitated with antibodies to cyclin A, cyclin E, CDK2, or CDC2 with protein A-Sepharose, washed, and tested for H1 histone kinase activity.
Lysates were prepared from asynchronously growing HFKs, immunoprecipitated with antibodies to various cyclins or CDKs, and assayed for the ability to phosphorylate histone H1 (Fig. 2D). Cells expressing the low-risk viral oncogenes had kinase activity comparable to levels in uninfected or vector-infected cells. In contrast, the cyclin A-, cyclin E-, CDK2-, and CDC2-associated kinase activity was elevated in the HPV-16 E6- or E7-expressing cells. In all cases, kinase activity was highest in the E6-expressing cells, either alone or with E7, suggesting that the reduction of p21 had a profound effect on CDK activity in cycling cells. E7-expressing cells also showed a significant enhancement of CDK activity compared to vector-infected HFKs, despite the presence of elevated levels of p21. Elevated levels of cyclin E probably contributed to the increased cyclin E-associated kinase activity; however, other mechanisms may be involved as well.
Transcriptional and posttranscriptional regulation of cyclin E.
The promoter for the cyclin E gene contains E2F-responsive sites (14, 27), and transcription of cyclin E is elevated in cells with inactive Rb protein (32, 71); therefore, we examined levels of cyclin E RNA in HPV-16 E7-expressing cells. Although Northern blotting had detected elevated levels of cyclin E RNA in established rodent cell lines (32, 71), that approach was not sensitive enough in HFK cells, although cyclin D RNA was detectable in the HFKs and cyclin E RNA was detectable in NIH 3T3 cells by Northern blot analysis (data not shown). Therefore, RNase protection assays were carried out with RNA from asynchronously growing cells expressing the vector or the HPV-16 oncogenes (Fig. 3). Each hybridization contained total RNA; a single cyclin probe for cyclin E, D1, or A; and an internal loading control probe, 36B4. Fivefold more RNA was required to detect the cyclin E RNA than the cyclin D RNA, and 2.5-fold more was required to detect the cyclin E RNA than the cyclin A RNA. In the asynchronously growing HFKs expressing E7, less than a twofold elevation in cyclin E RNA levels was seen. These results suggested that transcriptional activation by E2F at least partially contributed to the increased level of cyclin E in E7-expressing cells, but posttranscriptional mechanisms may also contribute.
FIG. 3.

mRNA expression of cyclins E, D1, and A in primary human keratinocytes. Total-cell RNA was extracted from asynchronous populations of HFKs expressing the high-risk HPV oncogenes. RNA (25, 10, and 5 μg) was used for hybridization with [α-32P]UTP riboprobes for cyclins E, A and D1, respectively. Protected fragments and the internal loading control 36B4 were run on the same gel and exposed for the same length of time.
Because cyclin E levels were more dramatically elevated in growth-arrested cells, we sought to investigate the cell cycle-regulated expression of cyclin E. Two complementary approaches were used. First, HFFs were growth arrested, either by growth to confluence followed by removal of serum (Fig. 4) or by serum depletion of sparse cultures (Fig. 5), and then they were stimulated and examined periodically for 36 h. Arrest of primary human fibroblasts, particularly those expressing E7, was difficult, requiring prolonged periods of growth at confluence (2 days) and without serum (3 days). Consequently, a substantial fraction of the population did not reenter the cycle. Because reentry into the cell cycle from a G0/G1 arrest and/or mitogenic stimulation can complicate interpretation of the cell cycle profile, as a second approach asynchronously growing HFKs were separated into G1, S, and G2/M fractions by FACS sorting and cell cycle proteins were analyzed (Fig. 6).
FIG. 4.
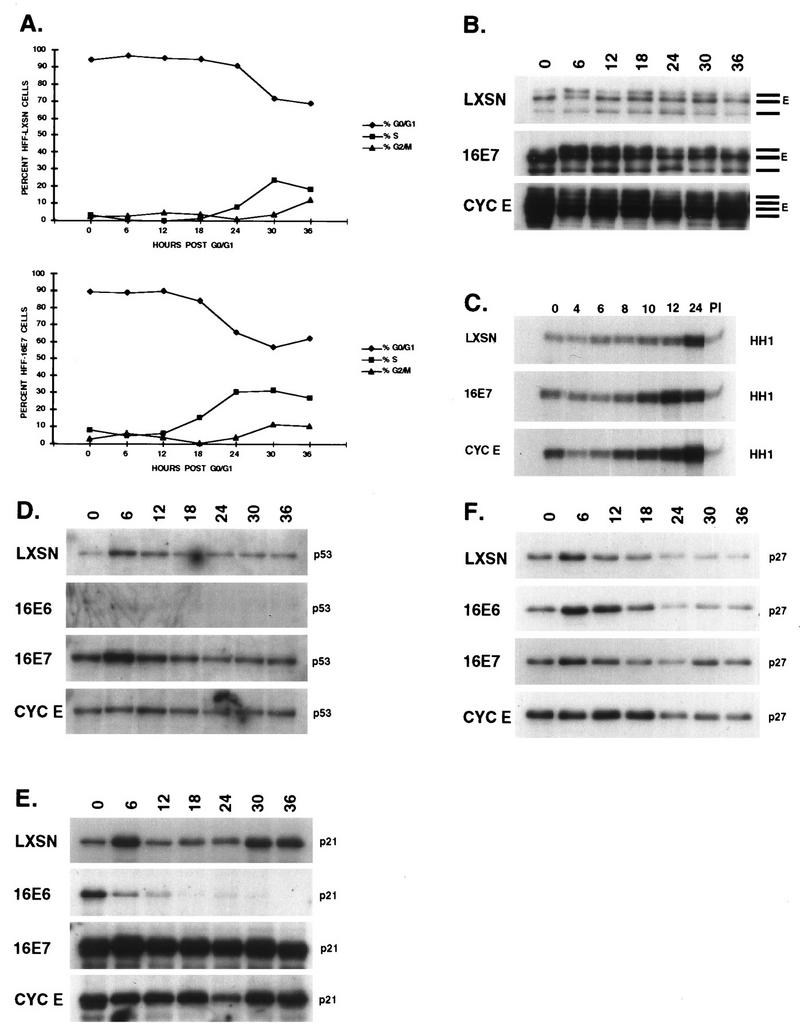
Cell cycle progression following density and serum starvation arrest. HPV-16 E6, E7, cyclin E, and LXSN vector were retrovirally transduced into HFFs, and the cells were G0/G1 arrested by a combined growth to density and serum deprivation. The cells were restimulated to enter the cycle by being plated subconfluently in 10% FBS-supplemented media and harvested at 0, 6, 12, 18, 24, 30, and 36 h after release. (A) Cells were processed for PI immunofluorescence and monitored for cell cycle position by FACScan. (B to F) Cyclin E protein levels (B) and associated kinase activity (C) was determined, and protein lysates were probed for p53 (D), p21 (E), and p27 (F).
FIG. 5.

Cell cycle arrest following serum starvation. HPV-16 E7- and vector-expressing HFFs were starved of serum for 48 h, restimulated with 10% FBS, and monitored at intervals for 30 h after release. (A to D) The cells were harvested and prepared for FACS analysis (A), protein lysates were immunoblotted for cyclin E (B) and cyclin A (C), and RNA samples were analyzed by RNase protection for cyclins E and A; 36B4 was used as a control (D). (E) RNA levels were quantitated by PhosphorImager analysis.
FIG. 6.
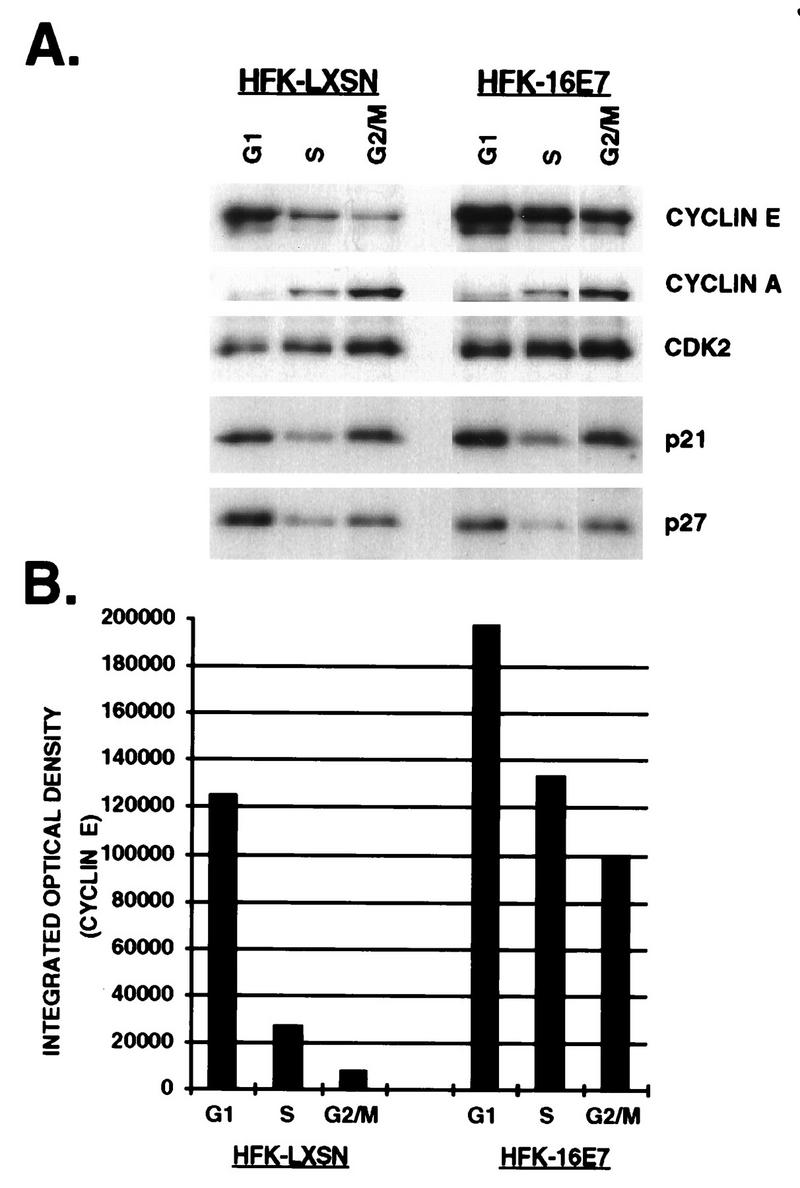
Cyclin and CKI levels in different cell cycle compartments. Asynchronous HFKs expressing E7 or vector were harvested at 106 cells/ml, stained with the DNA binding dye Hoechst 33342 (500 μg/ml), and counterstained with PI (100 μg/ml). G1, S, and G2/M cell populations were isolated at 6 × 104 cells per phase. Protein lysates were probed with either anti-cyclin E, A, CDK2, p21, or p27 (A), and the amount of cyclin E was quantitated by PhosphorImager analysis (B).
After replating and addition of serum, the LXSN-expressing D/S HFFs began to enter S phase after 18 h; by 30 h the S-phase population was maximal and some cells had begun to enter G2 (Fig. 4A). The progression was accelerated in E7-expressing D/S cells; they began to enter S by 12 h, were maximally in S by 24 h with cells entering G2, and were asynchronous by 30 h. When the cells were arrested by serum deprivation only, the same relative profiles were observed except that entry into S phase occurred 4 h earlier and G0/G1 arrest of the E7 cells was less complete (Fig. 5A).
In the LXSN-expressing cells, the levels of cyclin E were low in the D/S-arrested cells and peaked by 12 to 18 h just prior to entry into S phase (Fig. 4B). In the serum-starved cells, an increase in the level of cyclin E protein was detected at 16 h, with a decrease to G1 levels by 20 h (Fig. 5B). The E7-expressing cells arrested with high levels of cyclin E, which were elevated from 6 to 12 h after release; the level of cyclin E protein decreased by 16 to 18 h but was elevated in comparison to that of LXSN. Cells that constitutively overexpressed cyclin E had high levels of cyclin E protein that did not oscillate with the cell cycle. It is interesting that multiple bands of cyclin E were observed in all of the arrested cells (Fig. 4B and 5B) whereas the asynchronously growing cells had fewer bands (Fig. 1 and 6). Alternative forms of cyclin E were previously noted in cyclin E-overexpressing cells (28, 50) and are shown here to be related not only to high levels of cyclin E protein but also to growth arrest conditions. The slowest-migrating species of cyclin E predominated in asynchronously growing cells, whereas a faster-migrating species was more abundant in G0/G1-arrested cells.
Transcription of cyclin RNA was examined after release from serum starvation (Fig. 5D). In the LXSN-expressing cells, cyclin E mRNA levels rose following the addition of serum, were highest as the cells entered S, and dropped in late S phase. The cyclin E mRNA level was threefold higher in the serum-deprived E7-expressing cells, although this may simply represent the greater proportion of cells in the S phase. A similar, though less pronounced, oscillation of the cyclin E mRNA level was observed as the E7-expressing cells entered and then passed through the S phase. Cyclin E RNA and protein levels oscillated less than twofold throughout the cell cycle; however, the difference in cell cycle expression was obscured by the large percentage of arrested cells expressing cyclin E which never reentered the cycle.
A clearer pattern emerged from the cyclin E-associated kinase activity (Fig. 4C). The rise in cyclin E-associated kinase activity occurred a few hours before cells detectably entered the S phase despite high levels of cyclin E protein in the E7- and cyclin E-expressing cells during G1. Taken together, these data showed that quiescent E7-expressing HFFs had at least 10-fold more cyclin E protein than normal growth-arrested HFFs, which resulted only partially from increased transcription of cyclin E. Cyclin E transcription and protein levels increased prior to entry into S phase, coincident with an increase in cyclin E-associated kinase activity.
Serum-starved LXSN HFFs arrested with undetectable levels of cyclin A, which remained low throughout G1, rose continuously throughout S, and had the highest level at 24 h, when cells were in G2 (Fig. 5C). Cyclin A RNA levels paralleled the levels of protein (Fig. 5D). The serum-starved E7-expressing cells had more abundant cyclin A protein, probably reflecting cells in S phase due to incomplete arrest. Levels of cyclin A protein and RNA began to rise about 4 h earlier in the E7-expressing cells, consistent with the accelerated entry into S phase. At a time when most cells were in S and G2, the difference in the cyclin A levels was less than twofold between the LXSN- and E7-expressing cells.
Arrested LXSN cells had low levels of p53 that increased in early G1 or upon mitogenic stimulation and then returned to low levels (Fig. 4D). The level of p53 protein was higher in the E7-expressing cells, but the pattern of expression was similar throughout the cell cycle. E6-expressing cells had almost undetectable levels of p53, although a slight mitogenic stimulation was noticeable. The pattern of p21 expression did not entirely parallel the levels of p53, indicating both p53-dependent and p53-independent regulation of p21 expression (Fig. 4E). LXSN-expressing cells were arrested with a low level of p21, which increased with the addition of serum or entry into G1, as seen for p53; the levels remained low in late G1 and S and then increased in G2/M without a parallel increase in the p53 protein level. p21 levels were greatly elevated in the E7-expressing cells. A shorter exposure of the blot also showed a decline in p21 levels during late G1 and S, with higher levels by G2/M (data not shown). Interestingly, E6-expressing cells arrested with high levels of p21 in the absence of detectable p53; no burst in early G1 or upon mitogenic stimulation was seen, nor was there a detectable increase in G2/M. p27 protein levels were equivalent in all of the arrested cells, increased slightly as the cells were restimulated to enter G1, and declined as the cells entered S (Fig. 4F). By 30 h, when the E7-expressing cells were in late G2/M, the levels of p27 increased.
Figure 6 shows the distribution of cell cycle proteins among an equivalent number of flow-sorted G1, S, and G2/M cells expressing vector or E7. As anticipated, vector-expressing cells had the highest level of cyclin E in the G1 phase of the cell cycle, which decreased in S and continued to decline in G2/M. Cyclin E levels were also highest in the G1 population of E7-expressing cells, but the amount of protein was elevated less than twofold compared to that in vector-expressing cells. Although the levels of cyclin E declined in S and G2/M, the abundance of protein was 5.5- and 20-fold greater, respectively, than the S and G2/M fractions of LXSN-expressing cells. These data suggest that the cell cycle regulation of cyclin E was disrupted in E7-expressing cells. In comparison, cyclin A levels were lowest in G1, increased in S, and were highest in G2/M, as expected. Both the pattern of expression and the protein levels were similar in LXSN- and E7-expressing cells, indicating that the disregulation of cyclin turnover was not perturbed globally in E7-expressing cells but was specific for cyclin E. CDK2 protein levels were more or less constant throughout the cell cycle, and the levels were similar in both populations (data not shown). The pattern of expression of p21 and p27 had a similar distribution throughout the cell cycle. Protein levels were highest in G1, decreased in S, and were elevated in G2/M. The G2/M-associated increase was less prominent for p27 than for p21. The distribution of these proteins was the same in the E7-expressing cells, although the amount of p21 was elevated two- to threefold in all three cell cycle compartments in the E7-expressing cells compared with the LXSN-expressing cells, indicating that the cell cycle regulation of p21 and p27 was not disrupted in the E7-expressing cells.
Elevation of the cyclin E level requires the CR1 and CR2 domains of E7.
To begin to understand the mechanism by which E7 expression resulted in deregulation of cyclin E levels and cell cycle distribution, we examined the regions of E7 responsible for increasing the levels of cyclin E protein. The amino terminus of E7 contains two regions of homology to other DNA viral oncoproteins. The CR2 domain contains the well-recognized LxCxE motif (residues 22 to 26), which mediates binding to Rb and other related “pocket” proteins. Residues in the CR1 domain have been shown to be required for cellular transformation (1, 7) and for bypass of growth arrest signals (15); however, the mechanisms responsible for these activities are unclear. Unlike E1A, the CR1 region of E7 apparently is not involved in the release of E2F from Rb (33). Although complementation studies with E1A and E7 mutants provided some evidence for similar functions encoded by the N termini, no E7-p300 complexes were detected, nor did the mutants complement in reciprocal crosses (11). Two mutations in CR1, Δ6–10 and H2P, and three mutations in CR2, Δ21–24, C24G, and E26G, were tested for their ability to increase the levels of cyclin E. Retroviral transduction of the mutated E7 genes resulted in comparable levels of all the E7 proteins (15). Figure 7 demonstrates that only wild-type E7 resulted in elevated levels of cyclin E, which indicated a requirement for both the CR1 and CR2 domains of E7. Interestingly, elevation of p53 protein levels also required both CR1 and CR2. These results further underscore the notion that mechanisms in addition to E2F-mediated transcriptional activation are involved in the elevation of cyclin E levels in E7-expressing cells.
FIG. 7.

The CR1 and CR2 domains of E7 are required for increased cyclin E levels. Total-cell lysates were prepared from asynchronous HFK expressing the designated mutated E7 gene product. Proteins (20 μg/lane) were fractionated on SDS-PAGE gels (12% polyacrylamide), transferred to PVDF membranes, probed with the designated antibodies, and visualized by enhanced chemiluminescence.
DISCUSSION
HPV-6 infection typically causes benign proliferative lesions with detectable viral particles, and in at least some cases, basal cell hyperplasia is present. Thus, HPV-6 is clearly able to replicate in epithelial cells and probably induces cellular replication. However, in this study we did not detect any alteration in cell cycle proteins as a consequence of HPV-6 E6 or E7 gene expression, consistent with earlier observations in which HPV-6 E7 failed to stimulate the proliferation of epithelial cells grown in organotypic culture (15, 30) or to bypass growth arrest signals (15, 16). An early study examining the mitogenic activity of various E7 proteins in Rat 3Y1 cells indicated that HPV-6 E7 stimulated DNA replication approximately threefold less well than did HPV-16 E7 (65), and they and others (46) found that in vitro binding of HPV-6 E7 to Rb was 10- to 20-fold weaker than the HPV-16 E7-Rb interaction. The in vivo association of HPV-6 E7 with Rb was also much weaker than that of HPV-16 E7 (26). Thus, it is likely that weak stimulation of replication by HPV-6 in vivo has a profound impact on cellular replication over a prolonged period; however, in our assays we were not able to detect less than a twofold difference.
HPV-16 E6 did not substantially affect the levels of cyclins or CDKs. As shown previously (24, 67), HPV-16 E6 dramatically reduced the level of p53 (Fig. 2A). In asynchronously growing keratinocytes or fibroblasts, E6 expression resulted in greatly reduced levels of p21, suggesting that in these proliferating cells, p21 expression is dependent predominantly on p53 transactivation. Expression of other CKIs including p27 and p16 was unchanged. Additionally, there was a global increase in CDK activity in HPV-16 E6- and E6/E7-expressing cells (Fig. 2D). CDK complexes in proliferating cells have been reported to contain p21, and the p21-associated CDK complexes can be active (68, 73). Our data would suggest that the elimination of p21 increased CDK activity, suggesting either that p21-containing complexes are less active than complexes that do not contain p21 or that some p21-containing CDK complexes in proliferating cells are inactive. Xiong et al. (69) examined the composition, but not the activity, of CDK complexes in proliferating HFFs expressing HPV-16 E6 and found that although p21 mRNA was undetectable, p21 protein was apparently present in cyclin D-CDK4 and cyclin A-CDK2 complexes but not in cyclin B-CDC2 complexes (cyclin E-CDK2 complexes were not examined); none of the CDK complexes contained proliferating-cell nuclear antigen. It is also notable that G0-arrested E6-expressing cells had high levels of p21 in the absence of p53 (Fig. 4E and D), indicating that the induction of p21 in response to depletion of serum and/or confluence was not p53 dependent. The expression of p21 in E6-expressing cells, although always greatly reduced, was somewhat variable (for example, the HFKs in Fig. 2A), which could reflect some degree of growth arrest resulting in p53-independent expression of p21.
HPV-16 E7 had a profound effect on several aspects of the cell cycle machinery. (i) A number of cyclins and CDKs had elevated levels. (ii) Cyclin E expression was upregulated both transcriptionally and posttranscriptionally and persisted at high levels in S and G2/M; transit through G1 was shortened. (iii) We had observed previously that Rb levels were reduced and p53 levels were increased (16, 17); both changes were due to protein half-life rather than changes in transcription (reference 15 and data not shown). (iv) Despite the elevation of the levels of at least two CKIs, p21 and p16, CDK activity was elevated.
Other groups (57, 71) have also reported that cyclin E levels were elevated in E7-expressing cells, but conclusions about the mechanism by which the steady-state levels of cyclin E protein were increased were contradictory. In NIH 3T3 cells expressing adenovirus E1A (62) or HPV-16 E7 (71), E2F binding sites in the promoter of cyclin E were necessary for induction, and mutants of E7 that failed to bind Rb were not able to stimulate cyclin E expression (71). Rb−/− murine fibroblasts were also shown to have increased levels of cyclin E (32), confirming the prediction that disruption of the Rb-E2F pathway will lead to increased transcription of E2F-responsive genes. Our finding that both CR1 and CR2 were required for increased cyclin E may point to differences in the regulation of cyclin E between primary human cells and established murine cell lines. Another discrepancy is that the cyclin A and CDC2 promoters have also been shown to contain E2F-responsive elements (14, 59), yet overexpression of cyclin A or CDC2 was not reproducibly detected in asynchronous populations expressing E7. Although E7 cells released from quiescence had elevated cyclin A RNA and protein levels (Fig. 5), the apparent overexpression could be explained by the increased number of E7 cells in S phase. Interestingly, although the promoter of cyclin D also contains an E2F-responsive site, neither this report nor previous studies with E7-, E1A-, or Rb-deficient murine fibroblasts (32, 62, 71) found any evidence of overexpression of cyclin D1. Furthermore, E7 did not elevate the level of cyclin E in the U2OS cell line, which contains functional Rb protein, arguing against transcriptional activation of the cyclin E gene (57).
Our results indicate that cyclin E regulation was disrupted in multiple ways. The increase in cyclin E transcription due to E2F activation was not sufficient to account for the increased levels of protein. In asynchronous E7-expressing cells, the cyclin E RNA level increased less than twofold in comparison with vector whereas the cyclin E protein level increased three- to fivefold (Fig. 1 and 3). In synchronous populations, the RNA level increased approximately 3-fold in G1 whereas the protein level increased 5- to 20-fold (Fig. 5). In contrast, the increase in the level of cyclin A protein was proportional to the increase in the RNA level. Thus, in normal human epithelial and fibroblast cells, the cyclin E level is elevated both transcriptionally and posttranscriptionally. The cyclin E protein level has been shown to peak at the G1/S boundary and to decline as the S phase progresses (39) by degradation via the ubiquitin-proteasome system (8). Consistent with that, we observed that cyclin E levels were highest in cells in the G1 phase; however, while the levels dropped sharply in S and G2/M in vector-expressing cells, cyclin E protein levels declined by less than twofold in other cell cycle compartments in the E7-expressing cells (Fig. 6). This indicated that the mechanisms restricting cyclin E to a specific period in the cycle were perturbed by HPV-16 E7. These effects were specific to cyclin E since the patterns of cyclin A, p21, and p27 expression were unaltered.
The levels of cyclin E protein peaked several hours earlier following release from arrest in E7-expressing cells than in vector-expressing cells (Fig. 4B and 5B). Ohtsubo et al. (49) noted that cells engineered to overexpress cyclin E by retroviral transduction entered S phase several hours early following restimulation and that even though the cyclin E protein level was constitutively elevated throughout the cell cycle, kinase activity did not increase until late G1. HPV-16 E7 cells had cyclin E protein levels that were intermediate between those in vector and cyclin E overexpressers; accordingly, the timing of cyclin E kinase activity and entry into S phase was intermediate (Fig. 4). These data provide further support for the notion that cyclin E kinase activity is rate-limiting for entry into S phase, even in cells where the Rb pathway has been inactivated. Alternatively, it is possible that E7-expressing cells arrest in response to serum withdrawal or density at a point in G1 that is closer to the restriction point, or the G1/S boundary, than the arrest point for vector-containing cells.
Entry into S phase is dependent on the release of E2F from Rb; however, disruption of the E2F-Rb interaction by E7 did not block the majority of E7-expressing cells from becoming quiescent, indicating that some growth arrest pathways are still intact in the E7-expressing cells. To achieve >95% G1 arrest of E7-expressing cells, prolonged growth arrest regimens were required, with the result that a major fraction of both vector- and E7-expressing cells failed to reenter the cycle (Fig. 4A and 5A). We do not know whether there are differences between cells that remain arrested and those that reenter the cycle, but they are not related to E7, since a substantial proportion of the vector population remained arrested too. Under less severe growth arrest conditions, E7-expressing cells were not as tightly arrested as vector-expressing cells. Similarly, we do not know why only some E7-expressing cells escape from growth arrest induced by serum withdrawal or density. The levels of E7 protein may vary, but only slightly, since all cells after selection in G418 harbor at least one copy of E7, and the copy number in retrovirally transduced cells is low, one to a few. When E7-expressing cells are treated with a DNA-damaging agent or TGF-β, they enter S phase as well as do untreated cells (15, 16), arguing that every cell expresses a sufficient amount of E7 to bypass those growth arrest signals.
We had previously noted that the expression of E7 reduced the levels of Rb protein and that DNA damage of E7 expressing cells further reduced the level of protein (16). Jones and Munger (35) found that reduction of Rb levels was impaired when E7 proteins mutated in either CR1 or CR2 were used and that the reduction of Rb protein levels correlated with the ability to bypass DNA damage-induced growth arrest. The mechanism responsible for increased Rb turnover is unclear, or it may be cell type specific. Ubiquitin-mediated proteolysis was implicated in breast epithelia expressing HPV-16 E7 (4) but was not found to mediate degradation in HPV-16 E7-expressing RKO cell lines (35). Importantly, when HPV-16 E6 was coexpressed with HPV-16 E7, Rb levels did not decline (16), and the reduction in Rb levels may be an indirect consequence of E7, depending on elevated levels of p53 or other parameters. It is intriguing that the CR1- and CR2-mutated E7 proteins also failed to elevate the levels of p53 (unpublished data).
Studies with rodent cell lines or transgenic mice have indicated that the release of E2F by the viral oncogenes SV40T, E1A, or HPV-16 E7 in cells expressing wild-type p53 led to apoptosis, particularly when the cells were simultaneously confronted with growth arrest signals (12, 51, 64). Similarly, E7- but not E6-immortalized human uroepithelial cells underwent apoptosis in response to irradiation (56). However, the E7-expressing HFFs did not detectably undergo apoptosis in response to withdrawal of serum, suggesting that human fibroblasts may be particularly refractory to apoptosis (31).
HPV-16 E7 expression elevated the levels of p53 protein three- to fivefold in a wide variety of cell types by a posttranscriptional mechanism (16, 17, 24, 35) with a concomitant increase in p21 protein levels (Fig. 2A and 4), due to increased transcription (data not shown). No increase in p27 protein (Fig. 2 and 6) or mRNA (data not shown) levels was observed. Despite elevated levels of p21, the cell cycle expression of p21 and p27 was not altered. Soos et al. (61) found that the amount of p27 in the MANCA cell line was constant during the cell cycle, whereas our data indicated that in primary human cells the levels of both p21 and p27 were lowest during S phase, consistent with a role for these CKIs in controlling entry into the S and M phases of the cell cycle. p16 was also elevated in E7-expressing cells, as observed in the absence of functional Rb (52, 70). In cells in which cyclin D-CDK4 phosphorylation of Rb is required for S phase entry, Rb represses p16 mRNA transcription in a feedback loop to maintain CDK activity (43).
It is intriguing that despite high levels of p21 inhibitor, CDK activity was globally elevated in E7-expressing HFKs (Fig. 2D). Although the activity was not quite as high as in E6-expressing cells that had greatly reduced levels of p21, the CDK activity was substantially higher than in vector-expressing cells or in cells expressing the low-risk HPV oncoproteins. When the established colorectal tumor cell line RKO was engineered to express HPV-16 E7, no increase in cyclin E-associated kinase activity was observed (35). It is possible that the establishment of a cell line or the development of a tumor perturbs cyclin E regulation and hence that expression of E7 would have no additional effects. The mechanism by which E7 bypasses p21 inhibition is complex and incompletely understood. E7 proteins that failed to bind Rb were unable to bypass p21-mediated growth arrest, suggesting a mechanism in which the E2F-mediated increase in the cyclin E level could result in a shift in the number of cyclin E-CDK2 complexes needed to titrate p21 (15). However, E7 proteins that were able to bind Rb but were mutated in other domains also failed to bypass p21-mediated growth arrest. Recently the C-terminal half of E7 has been shown to bind to both p27 (72) and p21 (25) and to restore CDK activity to p27- and p21-inhibited complexes, providing evidence for new pathways by which E7 can bypass growth arrest signals. Understanding the mechanisms used by HPV-16 E7 to perturb cell cycle regulation may have broad importance for our understanding of the disruption of these processes in many human neoplasias.
ACKNOWLEDGMENTS
We thank Jim Roberts and members of his laboratory for many reagents and for helpful suggestions, Tim Knight and Andrew Berger for assistance with fluorescence-activated cell sorter and image analysis, and Scott Foster and Jens Oliver Funk for discussions and critical reading of the manuscript.
This work was supported by grant CA64795 from the National Cancer Institute.
REFERENCES
- 1.Banks L M, Edmonds C, Vousden K H. Ability of the HPV16 E7 protein to bind RB and induce DNA synthesis is not sufficient for efficient transforming activity in NIH3T3 cells. Oncogene. 1990;5:1383–1389. [PubMed] [Google Scholar]
- 2.Blanton R A, Coltrera M D, Gown A M, Halbert C L, McDougall J K. Expression of the HPV16 E7 gene generates proliferation in stratified squamous cell cultures which is independent of endogenous p53 levels. Cell Growth Differ. 1992;3:791–802. [PubMed] [Google Scholar]
- 3.Botz J, Zerfass-Thome K, Spitkovsky D, Delius H, Vogt B, Eilers M, Hatzigeorgiou A, Jansen-Durr P. Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol Cell Biol. 1996;16:3401–3409. doi: 10.1128/mcb.16.7.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyer S N, Wazer D E, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4624. [PubMed] [Google Scholar]
- 5.Carr A M. Checkpoints take the next step. Science. 1996;271:314–315. doi: 10.1126/science.271.5247.314. [DOI] [PubMed] [Google Scholar]
- 6.Cheng S, Schmidt-Grimminger D C, Murant T, Broker T R, Chow L T. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995;9:2335–2349. doi: 10.1101/gad.9.19.2335. [DOI] [PubMed] [Google Scholar]
- 7.Chesters P M, Vousden K H, Edmonds C, McCance D J. Analysis of human papillomavirus type 16 open reading frame E7 immortalizing function in rat embryo fibroblast cells. J Gen Virol. 1990;71:449–453. doi: 10.1099/0022-1317-71-2-449. [DOI] [PubMed] [Google Scholar]
- 8.Clurman B E, Sheaff R J, Thress K, Groudine M, Roberts J M. Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 1996;10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- 9.Crook T, Tidy J A, Vousden K H. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell. 1991;67:547–556. doi: 10.1016/0092-8674(91)90529-8. [DOI] [PubMed] [Google Scholar]
- 10.Davies R, Hicks R, Crook T, Morris J, Vousden K. Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J Virol. 1993;67:2521–2528. doi: 10.1128/jvi.67.5.2521-2528.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davies R C, Vousden K H. Functional analysis of human papillomavirus type 16 E7 by complementation with adenovirus E1A mutants. J Gen Virol. 1992;73:2135–2139. doi: 10.1099/0022-1317-73-8-2135. [DOI] [PubMed] [Google Scholar]
- 12.Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 1993;7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- 13.DeCaprio J A, Ludlow J W, Lynch D, Furukawa Y, Griffin J D, Piwnica-Worms H, Huang C M, Livingston D M. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell. 1989;58:1085–1095. doi: 10.1016/0092-8674(89)90507-2. [DOI] [PubMed] [Google Scholar]
- 14.DeGregori J, Kowalik T, Nevins J R. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demers G W, Espling E, Harry J B, Etscheid B G, Galloway D A. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol. 1996;70:6862–6869. doi: 10.1128/jvi.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demers G W, Foster S A, Halbert C L, Galloway D A. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc Natl Acad Sci USA. 1994;91:4382–4386. doi: 10.1073/pnas.91.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demers G W, Halbert C L, Galloway D A. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology. 1994;198:169–174. doi: 10.1006/viro.1994.1019. [DOI] [PubMed] [Google Scholar]
- 18.Dyson N, Howley P M, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 19.El-Deiry W S, Harper J W, O’Connor P M, Velculescu V E, Canman C E, Jackman J, Pietenpol J A, Burrell M, Hill D E, Wang Y, Wiman K G, Mercer W E, Kastan M B, Kohn K W, Elledge S J, Kinzler K W, Vogelstein B. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- 20.El-Deiry W S, Tokino T, Velculescu V E, Levy D B, Parsons R, Trent J M, Lin D, Mercer E, Kinzler K W, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 21.Etscheid B G, Foster S A, Galloway D A. The E6 protein of human papillomavirus type 16 functions as a transcriptional repressor in a mechanism independent of the tumor suppressor protein, p53. Virology. 1994;205:583–585. doi: 10.1006/viro.1994.1684. [DOI] [PubMed] [Google Scholar]
- 22.Ewen M E. The cell cycle and the retinoblastoma protein family. Cancer Metastasis Rev. 1994;13:45–66. doi: 10.1007/BF00690418. [DOI] [PubMed] [Google Scholar]
- 23.Firzlaff J M, Hsia C N L, Halbert C L, Jenison S A, Galloway D A. Polyclonal antibodies to human papillomavirus type 6b and type 16 bacterially derived fusion proteins. Cancer Cells. 1987;5:105–113. [Google Scholar]
- 24.Foster S A, Demers G W, Etscheid B G, Galloway D A. The ability of human papillomavirus E6 proteins to target p53 for degradation in vivo correlates with their ability to abrogate actinomycin D-induced growth arrest. J Virol. 1994;68:5698–5705. doi: 10.1128/jvi.68.9.5698-5705.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Funk J O, Waga S, Harry J B, Espling E, Stillman B, Galloway D A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997;11:2090–2100. doi: 10.1101/gad.11.16.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gage J R, Meyers C, Wettstein F O. The E7 proteins of the nononcogenic human papillomavirus type 6b (HPV-6b) and of the oncogenic HPV-16 differ in retinoblastoma protein binding and other properties. J Virol. 1990;64:723–730. doi: 10.1128/jvi.64.2.723-730.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geng Y, Eaton E N, Picon M, Roberts J M, Lundberg A S, Gifford A, Sardet C, Weinberg R A. Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene. 1996;12:1173–1180. [PubMed] [Google Scholar]
- 28.Gray-Bablin J, Zalvide J, Fox M P, Knickerbocker C J, DeCaprio J A, Keyomarsi K. Cyclin E, a redundant cyclin in breast cancer. Proc Natl Acad Sci USA. 1996;93:15215–15220. doi: 10.1073/pnas.93.26.15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halbert C L, Demers G W, Galloway D A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halbert C L, Demers G W, Galloway D A. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J Virol. 1992;66:2125–2134. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawkins D S, Demers G W, Galloway D A. Inactivation of p53 enhances sensitivity to multiple chemotherapeutic agents. Cancer Res. 1996;56:892–898. [PubMed] [Google Scholar]
- 32.Herrera R E, Sah V P, Williams B O, Makela T P, Weinberg R A, Jacks T. Altered cell cycle kinetics, gene expression, and G1 restriction point regulation in Rb-deficient fibroblasts. Mol Cell Biol. 1996;16:2402–2407. doi: 10.1128/mcb.16.5.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang P S, Patrick D R, Edwards G, Goodhart P J, Huber H E, Miles L, Garsky V M, Oliff A, Heimbrook D C. Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Mol Cell Biol. 1993;13:953–960. doi: 10.1128/mcb.13.2.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunter T, Pines J. Cyclins and cancer II: cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 35.Jones D L, Munger K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol. 1997;71:2905–2912. doi: 10.1128/jvi.71.4.2905-2912.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamb A. Cell-cycle regulators and cancer. Trends Genet. 1995;11:136–140. doi: 10.1016/s0168-9525(00)89027-7. [DOI] [PubMed] [Google Scholar]
- 37.Kaur P, McDougall J K. HPV-18 immortalization of human keratinocytes. Virology. 1989;173:302–310. doi: 10.1016/0042-6822(89)90247-x. [DOI] [PubMed] [Google Scholar]
- 38.Koff A, Cross F, Fisher A, Schumacher J, Leguellec K, Philippe M, Roberts J M. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell. 1991;66:1217–1228. doi: 10.1016/0092-8674(91)90044-y. [DOI] [PubMed] [Google Scholar]
- 39.Koff A, Giordano A, Desai D, Yamashita K, Harper J W, Elledge S J, Nishimoto T, Morgan D O, Franza B R, Roberts J M. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257:1689–1694. doi: 10.1126/science.1388288. [DOI] [PubMed] [Google Scholar]
- 40.Koff A, Ohtsuki M, Kornelia P, Roberts J M, Massague J. Negative regulation of G1 in mammalian cells: inhibition of cyclin E-dependent kinase by TGF-beta. Science. 1993;260:536–539. doi: 10.1126/science.8475385. [DOI] [PubMed] [Google Scholar]
- 41.Kowalik T F, DeGregori J, Schwarz J K, Nevins J R. E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J Virol. 1995;69:2491–2500. doi: 10.1128/jvi.69.4.2491-2500.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laborda J. 36B4 cDNA used as an estradiol-independent mRNA control is the cDNA for human acidic ribosomal phosphoprotein PO. Nucleic Acids Res. 1991;19:3998. doi: 10.1093/nar/19.14.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Nichols M A, Shay J W, Xiong Y. Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product. Cancer Res. 1994;54:6078–6082. [PubMed] [Google Scholar]
- 44.Matlashewski G. The cell biology of human papillomavirus transformed cells. Anticancer Res. 1989;9:1447–1456. [PubMed] [Google Scholar]
- 45.Miller A D, Rosman G J. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 46.Munger K, Werness B A, Dyson N, Phelps W C, Harlow E, Howley P M. Complex formation of the human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nass S J, Dickson R B. Detection of cyclin messenger RNAs by nonradioactive ribonuclease protection assay: a comparison of four detection methods. BioTechniques. 1977;19:772–776. [PubMed] [Google Scholar]
- 48.Ohtani K, DeGregori J, Nevins J R. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci USA. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohtsubo M, Roberts J M. Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science. 1993;259:1908–1912. doi: 10.1126/science.8384376. [DOI] [PubMed] [Google Scholar]
- 50.Ohtsubo M, Theodoras A M, Schumacher J, Roberts J M, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. 1995;15:2612–2624. doi: 10.1128/mcb.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan H, Griep A E. Temporally distinct patterns of p53-dependent and p53-independent apoptosis during mouse lens development. Genes Dev. 1995;9:2157–2169. doi: 10.1101/gad.9.17.2157. [DOI] [PubMed] [Google Scholar]
- 52.Parry D, Bates S, Mann D J, Peters G. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. EMBO J. 1995;14:503–511. doi: 10.1002/j.1460-2075.1995.tb07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polyak K, Kato J-Y, Solomon M J, Sherr C J, Massague J, Roberts J M, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-b and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 54.Quelle D E, Ashmun R A, Shurtleff S A, Kato J-Y, Bar-Sagi D, Roussell M F, Sherr C J. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 1993;7:1559–1571. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- 55.Resnitzky D, Reed S I. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol Cell Biol. 1995;15:3463–3469. doi: 10.1128/mcb.15.7.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reznikoff C A, Belair C, Savelieva E, Zhai Y, Pfeifer K, Yeager T, Thompson K J, De Vries S, Bindley C, Newton M A, Sekhon G, Waldman F. Long-term genome stability and minimal genotypic and phenotypic alterations in HPV16 E7-, but not E6-, immortalized human uroepithelial cells. Genes Dev. 1994;8:2227–2240. doi: 10.1101/gad.8.18.2227. [DOI] [PubMed] [Google Scholar]
- 57.Ruesch M N, Laimins L A. Initiation of DNA synthesis by human papillomavirus E7 oncoproteins is resistant to p21-mediated inhibition of cyclin E-cdk2 activity. J Virol. 1997;71:5570–5578. doi: 10.1128/jvi.71.7.5570-5578.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheffner M, Huibregtse J M, Vierstra R D, Howley P M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 59.Schulze A, Zerfass K, Spitkovsky D, Middendorp S, Berges J, Helin K, Jansen-Durr P, Henglein B. Cell cycle regulation of the cyclin A gene promoter is mediated by a variant E2F site. Proc Natl Acad Sci USA. 1995;92:11264–11268. doi: 10.1073/pnas.92.24.11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherr C J. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 61.Soos T J, Kiyokawa H, Yan J S, Rubin M S, Giordano A, DeBlasio A, Bottega S, Wong B, Mendelsohn J, Koff A. Formation of p27-CDK complexes during the human mitotic cell cycle. Cell Growth Differ. 1996;7:135–146. [PubMed] [Google Scholar]
- 62.Spitkovsky D, Jansen-Durr P, Karsenti E, Hoffman I. S-phase induction by adenovirus E1A requires activation of cdc25a tyrosine phosphatase. Oncogene. 1996;12:2549–2554. [PubMed] [Google Scholar]
- 63.Tommasino M, Crawford L. Human papillomavirus E6 and E7: proteins which deregulate the cell cycle. Bioessays. 1995;17:509–518. doi: 10.1002/bies.950170607. [DOI] [PubMed] [Google Scholar]
- 64.Van Dyke T A. Analysis of viral-host protein interactions and tumorigenesis in transgenic mice. Semin Cancer Biol. 1994;5:47–60. [PubMed] [Google Scholar]
- 65.Watanabe S, Sato H, Komiyama N, Kanda T, Yoshiike K. The E7 functions of human papillomaviruses in rat 3Y1 cells. Virology. 1992;187:107–114. doi: 10.1016/0042-6822(92)90299-5. [DOI] [PubMed] [Google Scholar]
- 66.Weinberg R A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 67.Werness B A, Levine A J, Howley P M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 68.Xiong Y, Hannon G J, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 69.Xiong Y, Kuppuswamy D, Li Y, Livanos E M, Hixon M, White A, Beach D, Tlsty T D. Alteration of cell cycle complexes in human papillomavirus E6- and E7-expressing fibroblasts precedes neoplastic transformation. J Virol. 1996;70:999–1008. doi: 10.1128/jvi.70.2.999-1008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yeager T, Stadler W, Belair C, Puthenveettil J, Olopade O, Reznikoff C A. Increased p16 levels correlate with pRb alterations in human urothelial cells. Cancer Res. 1995;55:493–497. [PubMed] [Google Scholar]
- 71.Zerfass K, Schulze A, Spitkovsky D, Friedman V, Henglein B, Jansen-Durr P. Sequential activation of cyclin E and cyclin A gene expression by human papillomavirus type 16 E7 through sequences necessary for transformation. J Virol. 1995;69:6389–6399. doi: 10.1128/jvi.69.10.6389-6399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz J W, Jansen-Durr P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene. 1996;13:2323–2330. [PubMed] [Google Scholar]
- 73.Zhang H, Hannon G J, Beach D. p21-containing cyclin kinases exist in both active and inactive states. Genes Dev. 1995;8:1750–1758. doi: 10.1101/gad.8.15.1750. [DOI] [PubMed] [Google Scholar]
- 74.zur Hausen H. Human papillomaviruses in the pathogenesis of anogenital cancer. Virology. 1991;184:9–13. doi: 10.1016/0042-6822(91)90816-t. [DOI] [PubMed] [Google Scholar]