

Abstract
Murine leukemia virus (MLV)-based retroviral vectors are the most frequently used gene delivery vehicles. However, the current vectors are still not fully optimized for gene expression and viral titer, and many genetic and biochemical features of MLV-based vectors are poorly understood. We have previously reported that the retroviral vector MFG, where the gene of interest is expressed as a spliced mRNA, is superior in the level of gene expression with respect to other vectors compared in the study. As one approach to developing improved retroviral vectors, we have systematically performed mutational analysis of the MFG retroviral vector. We demonstrated that the entire gag coding sequence, together with the immediate upstream region, could be deleted without significantly affecting viral packaging or gene expression. To our knowledge, this region is included in all currently available retroviral vectors. In addition, almost the entire U3 region could be replaced with the heterologous human cytomegalovirus immediately-early promoter without deleterious effects. We could also insert internal ribosome entry sites (IRES) and multicloning sites into MFG without adverse effects. Based on these observations, we have constructed a series of new, improved retroviral constructs. These vectors produced viral titers comparable to MFG, expressed high levels of gene expression, and stably transferred genes to the target cells. Our vectors are more convenient to use because of the presence of multicloning sites and IRESs, and they are also more versatile because they can be readily converted to various applications. Our results have general implications regarding the design and development of improved retroviral vectors for gene therapy.
Murine leukemia virus (MLV)-based retroviral vectors are the most widely used gene delivery vehicles in gene therapy clinical trials, being employed in almost 70% of approved protocols (3, 27). However, despite its frequent use for gene transfer, many of the biochemical and genetic properties of MLV, such as cis and trans factors important for gene expression, viral assembly, and packaging, are not completely understood. Indeed, there are many problems with the retroviral vectors currently in clinical use, such as MFG (6, 8, 13, 20, 32) and LN-based vectors (1, 4, 7, 29, 33, 34). For example, all retroviral vectors contain sequences that are also present in the packaging lines. Recombination between the packaging genome and the vector can result in the generation of replication-competent retrovirus (RCR). Second, most retroviral vectors use either the LTR from MLV or a related LTR such as that from myeloid proliferation-stimulating virus, murine sarcoma virus, or a heterologous internal promoter. Although the LTR works efficiently in certain cell types, its activity can be down-regulated and its presence can affect expression from internal promoters (8, 15). Third, the viral titers achieved with the vectors in the current packaging lines, although improving, are still not sufficiently high enough for many therapeutic applications. Furthermore, MLV-based vectors, when packaged in murine packaging lines, are susceptible to complement-mediated inactivation in vivo, which limits their utility for in vivo applications (11, 39). Finally, it has been difficult to produce a virus at a reasonable titer for targeting a specific cell type or tissue by direct, in vivo delivery (21, 22). For retroviral vectors to be clinically viable-forms of gene delivery, some or all of these current limitations have to be addressed.
We have previously compared the relative levels of gene expression from several different types of retroviral vectors currently used in gene therapy clinical trials (10). Our results suggested that the MFG retroviral vector was superior in conferring gene expression after transduction of a variety of target cells, consistent with the previous results of others (23, 32, 37). However, MFG still contains many features that should be modified to improve gene expression and titers as well as safety. Therefore, we subjected the MFG retroviral vector to a systematic analysis of certain parameters. We demonstrated that the entire gag coding region could be removed without any effect on the packaging efficiency and that almost the entire U3 region could be replaced with heterologous promoter elements without affecting the viral life cycle. Furthermore, internal ribosome entry sites and multiple cloning sites could be introduced into MFG, making it easier to use. When a vector containing all these modifications was constructed, the resulting construct worked as well as or, in some case, better than the starting vector MFG. Our results have general implications regarding the development of more sophisticated and improved retroviral vectors.
MATERIALS AND METHODS
Cells.
NIH 3T3 (CRL1658) and U937 (CRL1593) cells were obtained from the American Type Culture Collection (Rockville, Md.), while CEM-SS (no. 776) and H9 (no. 87) cells were from the NIH AIDS Research and Reference Reagent Program (Rockville, Md.). CRIP (12) and BING cells were provided by Warren Pear (Massachusetts Institute of Technology, Cambridge, Mass.). The latter is the amphotropic cell line derived from 293T cells (14), similar to the ecotropic BOSC23 packaging cell line (35). NIH 3T3 and CRIP cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% calf serum. BING was grown in DMEM supplemented with 10% fetal bovine serum. CEM-SS, H9, and U937 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum. Each medium used in this study was supplemented with 120 μg of penicillin G per ml (Sigma P-3032; 1,690 U/mg) and 200 μg of streptomycin sulfate per ml (Sigma S-9137; 750 U/mg).
Plasmids.
Many plasmids used in this study were constructed by PCR with proofreading Pfu DNA polymerase (Stratagene, La Jolla, Calif.). The nucleotide sequences of final constructs were determined to confirm that no mutations were introduced by this amplification step. To determine the minimum length of packaging signal sequence, deletions were introduced as follows. Ten oligonucleotide primers based on MFG-LacZ (5) were used for amplification of two types of fragments, groups I and II. Group I fragments were obtained by PCR with the primer HindIIIr and one of the four primers L228, L377, L523, and L739. The HindIII linker was attached to HindIIIr, while the XhoI linker was attached to these L series primers. Group II fragments were generated by PCR with the primer ClaIl and one of the four primers R371, R527, R743, and R1016. The ClaI and XhoI linkers were attached to the respective primers. The nucleotide sequence of primers used in this experiment is as follows (restriction sites are underlined): HindIIIr: GCATTAAAGCTTTGCTCT HindIII L228: GCCTCGAGATAAGTTGCTGGCCAG XhoI L377: GCCTCGAGTCCCTGGGACGTCTCC XhoI L523: GCCTCGAGCAAAAATTCAGACGGA XhoI L739: GCCTCGAGCAGAAGGTAACCCAA XhoI R371: GCCTCGAGGGACTTCGGGGGCCGT XhoI R527: GCCTCGAGGTTTGGGACCGAAGCC XhoI R743: GCCTCGAGAATGGCCAACCTTTAA XhoI R1016: GCCTCGAGCCCTCACTCCTTCTCT XhoI ClaIl: ACGCTCATCGATAATTTC ClaI
The eight fragments from group I and II were amplified and cloned into plasmid pCR II (Invitrogen, San Diego, Calif.), resulting in a series of four pCR II-I and four pCR II-II constructs. The XhoI-XhoI fragments were isolated from the series of pCR II-II plasmids and then inserted into the XhoI site of the series of pCR II-I, generating a series of pMΔ gag constructs. The HindIII-ClaI fragment was isolated from pMΔ gag and used to replace the HindIII-ClaI fragment (including the 5′ LTR) of MFG-LacZ, resulting in a series of pMΔLacZ constructs, now containing deletions between the splice donor (SD) and the splice acceptor (SA). Altogether, nine deletion mutants were constructed (see Fig. 2A).
FIG. 2.
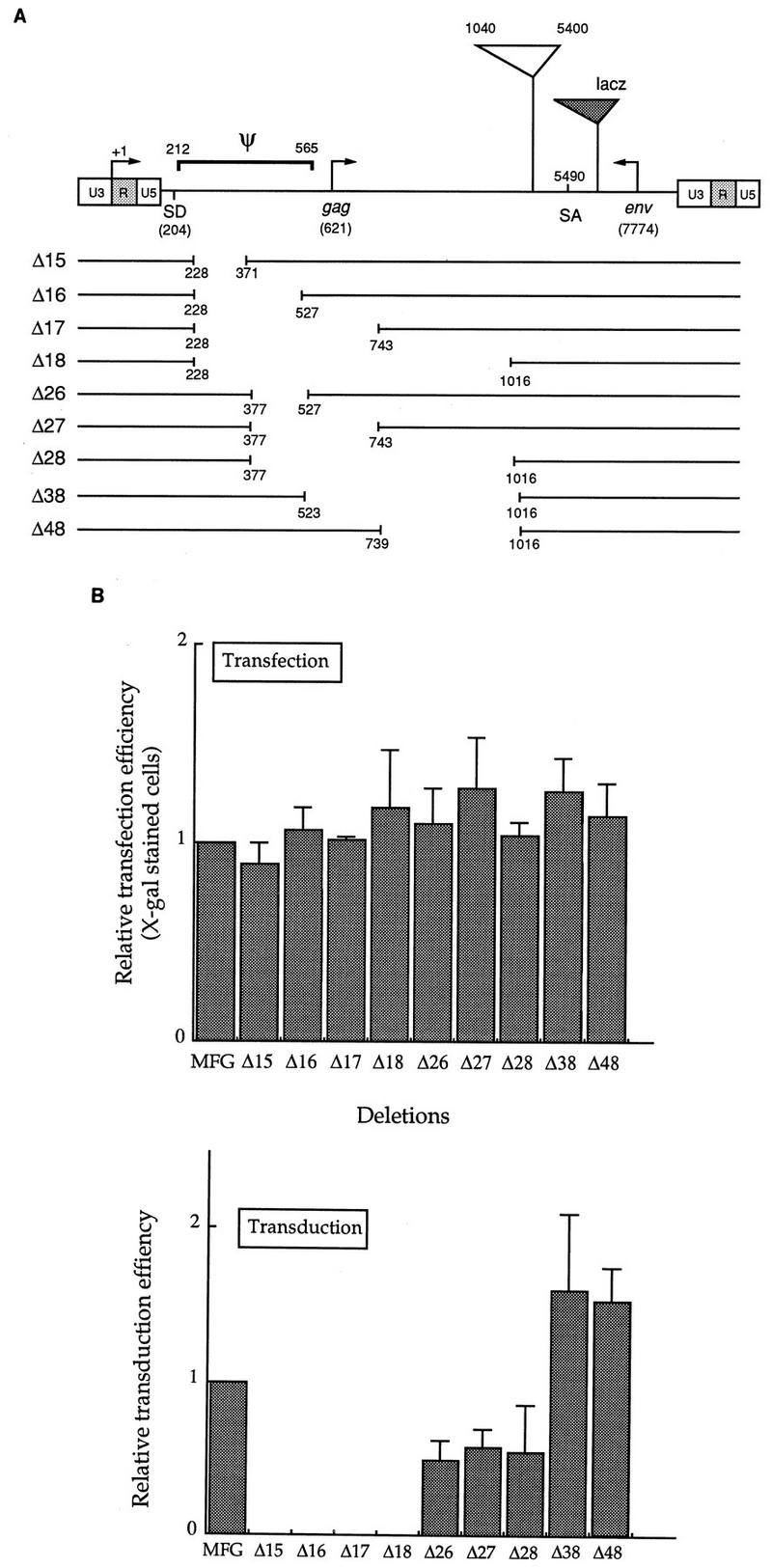
Localization of the packaging signal sequence. (A) Summary of deletions. Nine deletions were constructed as described in Materials and Methods. Ψ indicates the packaging sequence previously defined by Mann et al. (25), which includes the gag coding region as well as the entire sequence between SD and the start codon for gag. The numbering system is based on that of Shinnick et al. (38). The region between positions 1040 and 5400 includes gag and pol coding sequences and is missing from MFG. lacZ was used as a reporter gene in this study, and its relative position is shown as a dotted triangle. Note that the vector is not drawn to scale. (B) Effects of deletion on gene expression and viral titers. Deletion constructs, together with the parental vector MFG-lacZ, were transfected to the packaging line BING or CRIP. (In this figure, only the result for BING is shown.) After 3 days, culture supernatants were filtered through 0.45-μm-pore-size filters, while cells were stained with X-Gal to measure transfection efficiency. Duplicate dishes were also prepared for some constructs and subjected to the ONPG assay for β-galactosidase activity. Cell-free viral supernatants were used to transduce NIH 3T3 cells, and after 3 days the cells were stained with X-Gal to determine the viral titer. The transfection and transduction efficiency of MFG were set to 1, and those of others were normalized to it. More than five transfections and transductions were performed at separate times. In one independent experiment, four to six transfections and transductions were carried for each mutant.
To remove the residual env sequences, PCR was performed with MFG-NEO (10), using primers M3L-52 and M3L-31 (the restriction linkers attached to each primer are underlined): M3L-52: AAAGGATCCATTTAGTCT BamHI M3L-31: GAATTCATGTGAAAGGCGGCCGCTGA EcoRI
The amplified product covered the polypurine tract and the entire 3′ LTR. The amplified fragment was then cloned back into pCR II, resulting in CR-M3L. The BamHI-EcoRI fragment from CR-M3L replaced the same BamHI-EcoRI fragment of MFG-NEO, generating MΔE-NEO. The NcoI-BamHI neo gene was replaced with the NcoI-BamHI chloramphenicol acetyltransferase (CAT) sequence from MFG-CAT (10), resulting in MΔE-CAT.
The chimeric promoters containing the human cytomegalovirus (HCMV) immediate-early (IE) promoter elements in the MLV LTR were constructed as follows. First, four HCMV IE promoter elements were amplified by PCR with six primers (see Fig. 4) and cloned into the plasmid pCR II. To the 5′ and 3′ ends of each primers, restriction sites that are naturally present in the U3 of MLV were added as indicated. The nucleotide sequences of these primers are as follows: C5NH: GCTAGCGGGACTTTCCATTGACGT NheI C3KP: GGGTACCCGGGCGACTCAGTCAATCGGAGGAGGA KpnI CCI-5: CGATCGCCGCGTTACATAAC PvuII CCI-3: TCTAGAGGAAACTCCCGTAAG XbaI CCII-5: TCTAGAGGTTTGACTCACGG XbaI CCII-3: GAGCTCCCTACCGCCCATTT SacI
FIG. 4.

Construction of chimeric U3. (A) Four HCMV MIEP fragments were used to substitute the U3 sequence. They are CR, CCI, CCII, and CP. (B) Schematic diagram of chimeric LTRs. Four restriction sites (NheI, PvuII, XbaI, and SacI) are naturally present in U3, and their coordinates are shown in parentheses. These sites were used to clone the four HCMV MIEP fragments. The relative positions of the CAAT and TATA boxes of U3 are indicated. The numbers shown above the LTR are the lengths of U3 (unshaded) or HCMV MIEP (shaded) that replaced a part of U3, while those in parentheses are the coordinates of MLV based on the numbering of Shinnick et al. (38). Note that the promoter is not drawn to scale.
Second, the cloning vector SP65 (Promega, Madison, Wis.) was changed to RPX68 by removing the region between HindIII and PvuII, leaving HindIII intact, and filling in with XbaI and SacI sites, for the convenience of further manipulation. Third, plasmid RPX68-M5L, the RPX68 containing the entire 5′ LTR of MLV, was constructed by amplifying the same region from pMLV (38). The nucleotide sequences of primers used in amplifying the 5′ LTR of MLV are as follows: HHIR: AAGCTTATGTGAAAGACCCCTCCTG HindIII 5LBG: AGATCTGGCGCCTAGAGAAGG BglII
RPX68-M5L was subjected to four different restriction digestions. Each restriction site used in the digestions is unique, and they all cut the sites inside the 5′ LTR. Four HCMV IE promoter fragments were then isolated from the pCR II constructs containing these fragments (pCRII-CCI, pCRII-CCII, pCRII-CR, and pCRII-CP) and used to substitute the PvuII-XbaI, XbaI-SacI, PvuII-SacI, and NheI-KpnI fragments of the LTR, generating four plasmids (RPX68-hybrid 5′ LTR). The HindIII-BglII fragments were isolated from these plasmids and then used to replace the HindIII-BglII fragment of MFG-CAT containing the 5′ LTR, resulting in four M5L-chimeric CAT plasmids, M5LMCP1-CAT, M5LMCP2-CAT, M5LMCP3-CAT, and M5LCP-CAT. The chimeric promoters constructed this way are summarized in Fig. 4B.
To insert the HCMV IE promoter fragments into the 3′ LTR, RPX68-M3L was constructed by amplifying the 3′ LTR from pMLV and cloning it into RPX68. The oligonucleotide primers used in this amplification are M3L-51 and M3L-31, with the latter used to construct MΔE-CAT. The nucleotide sequence of M3L-51 is as follows: M3L-51: AAAGGATCCGATTAGTCCAATTTG BamHI
As with RPX68-M5L, RPX68-M3L was subjected to four different restriction digestions and the retroviral LTR fragments were replaced with four HCMV IE promoter fragments to generate RPX68-hybrid 3′ LTR in the same manner as for RPX68-hybrid 5′ LTR. The four BamHI-EcoRI fragments from RPX68-hybrid 3′ LTR were then used to substitute the BamHI-EcoRI fragment containing the 3′ LTR of MFG-CAT, resulting in four M3L-chimeric CAT plasmids, M3LMCP1-CAT, M3LMCP2-CAT, M3LMCP3-CAT, and M3LCP-CAT (see Fig. 5).
FIG. 5.
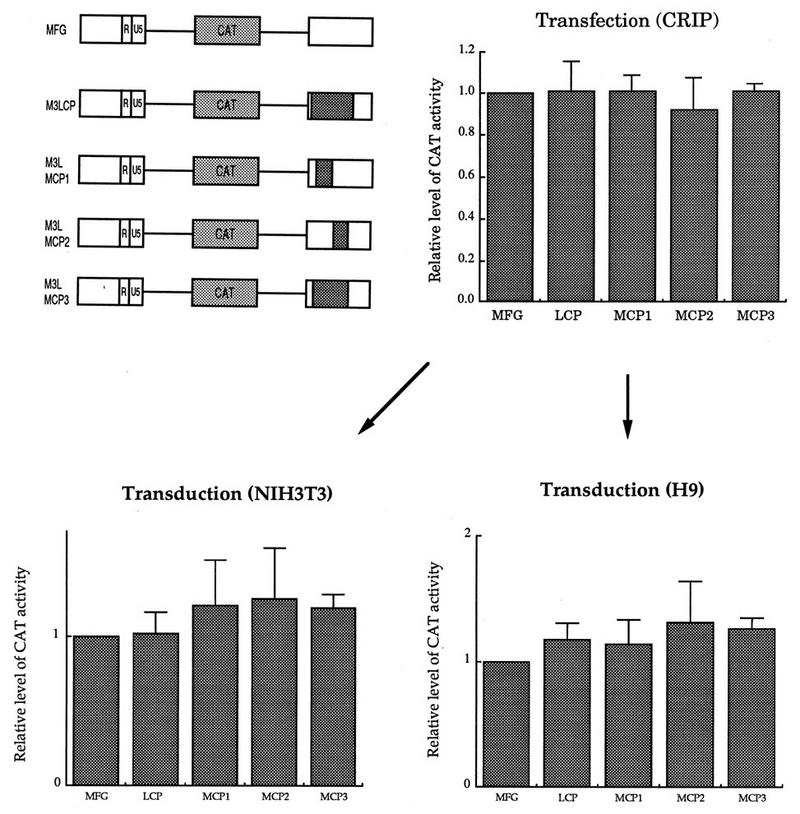
Effect of the chimeric 3′ LTR on gene expression. The five constructs, including the parental MFG-CAT, were transfected to the packaging line CRIP, and cell-free viral supernatant was used to transduce NIH 3T3 cells and the human T-lymphoid line H9. Both transfected and transduced cells were subjected to CAT analysis. Expression of MFG-CAT was set to 1, and the others were normalized to it.
To construct retroviral vectors containing hybrid promoters, CAT, IRES, and NEO, three plasmids (pMLV, M3LMCP1-CAT, and M3LMCP3-CAT) were amplified with the M3L-52 and M3L-31 primers used for the construction of MΔE-CAT. The amplified fragments containing BamHI and EcoRI linkers at each end were used to replace the BamHI-EcoRI fragment of MFG-CAT including the 3′ LTR, resulting in MΔE-CAT, MΔEMCP1-CAT, and MΔEMCP3-CAT. The HindIII-BamHI fragments of the last three plasmids containing the 5′ LTR were replaced with the HindIII-BamHI fragment amplified from MCC-CAT. The nucleotide sequences of the primers are as follows: SALDGAG: AAGCTTGTCGACATGAGATCTTATATGGGG HindIII SalI CATSTOP: GGATCCTTACGCCCCGCCCTGCCA BamHI
The small HindIII-SalI fragments of the three intermediate plasmids (ΔGE-CAT, ΔGEMCP1-CAT, and ΔGEMCP3-CAT) were replaced by the HindIII-XhoI fragments amplified from the three plasmids MLV, M5LMCP1, and M5LCP, resulting in the four plasmids SFG-CAT, SCP1-CAT, KCP1-CAT, and KCP3-CAT. The primers used in this step were HindIIIR and L523. Finally, the BamHI-BamHI cassette containing the encephalomyocarditis virus (EMCV) IRES/NEO (see below) was inserted into the BamHI sites of the four plasmids, generating retroviral constructs containing hybrid promoters at both the 5′ and 3′ ends. The CAT gene was linked with NEO through the EMCV IRES. For the structures of the final four constructs, see Fig. 6.
FIG. 6.
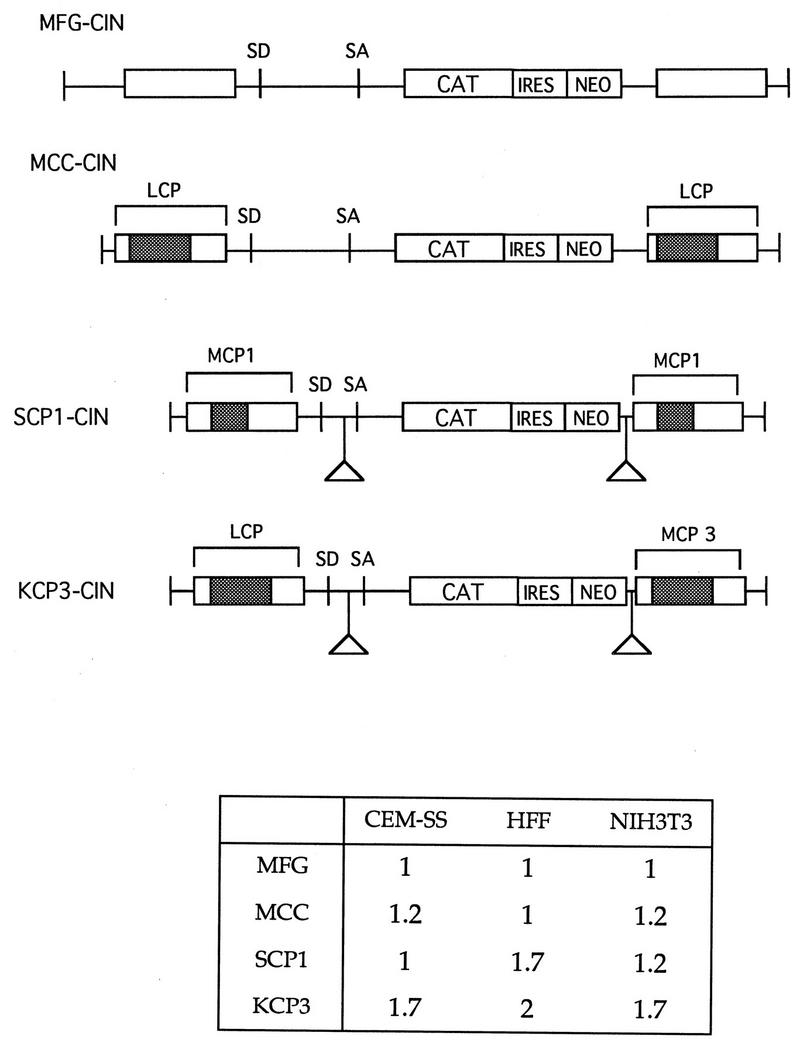
Comparison of retroviral vectors containing IRES and neo. Retroviral vectors were constructed to contain chimeric U3 at both the 5′ and 3′ LTRs, deletions in gag and env coding sequences, and the selectable marker NEO gene linked to CAT through IRES. Again, for each cell line, expression of MFG-CAT was set to 1 and those of the other constructs were normalized to it. Because the assay conditions were different for the different cell lines, direct comparison between cell lines based on the above numbers should be avoided. Transductions were performed at least three to five times for each line at separate times. Here the result from one representative experiment is shown.
The BamHI-BamHI EMCV IRES/NEO cassette was constructed with pCITE, containing EMCV IRES (Novagene) and pSVTK-neo (Stratagene). First, the XbaI site of pCITE was converted to BamHI. Second, the BstXI-BamHI neo fragment was prepared by PCR from pSVTKneo. Third, the BstXI-BamHI neo fragment was inserted into the BstXI-BamHI site of pCITE-XB, whose EcoRI fragment was subsequently converted to BamHI, resulting in pCBIN.
SCP1-mGM/CSF (see Fig. 7) was constructed by replacing the NcoI-BamHI CAT sequence in the SCP1-CIN with the NcoI-BamHI mGM/CSF from pCRII-GM/CSF (10). The two plasmids KCP3-WNIN and KCP3-WXIN (see Fig. 8), which were used to test the requirement for NcoI in MFG, were constructed as follows. First, KCP3-WNIN was constructed by replacing the NcoI-BamHI CAT sequence with the NcoI-BamHI erythropoietin (EPO) fragment from pCRII-EPO (10). Second, to construct a retroviral vector lacking a NcoI site, the NcoI site of EPO was filled in by the Klenow fragment, and this filled NcoI-BamHI EPO gene was then inserted into the filled XbaI-BamHI site of KCP3-WNIN, resulting in KCP3-WXIN.
FIG. 7.

Comparison of expression levels of mGM-CSF between MFG and SCP1. The experimental conditions were identical to the others, except that one of the target cells was the human skin fibroblast cell line and the level of mGM-CSF, instead of CAT, was measured. The two retroviral vectors expressing mGM-CSF were transfected to CRIP cells. NIH 3T3 and human foreskin fibroblasts were transduced and then selected in the presence of G418. The same number of drug-resistant cells were plated on 6-cm plates, grown for another 3 days, and subjected to enzyme-linked immunosorbent assay. Expression of MFG–mGM-CSF was set to 1.
FIG. 8.
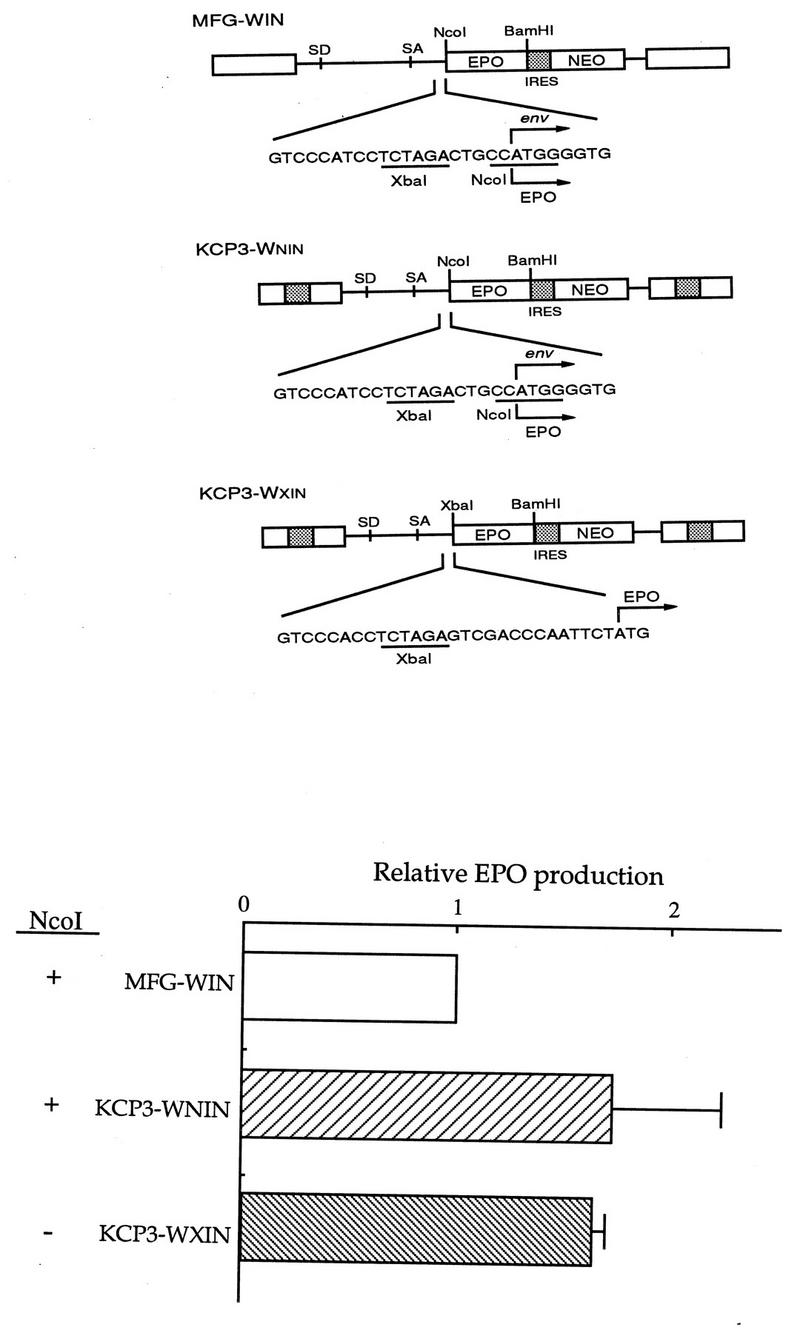
Effect of removal of NcoI in MFG. (A) Retroviral vector construction. MFG-WIN and KCP3-WNIN contain the NcoI site, while KCP3-WXIN does not. The nucleotide sequence around the NcoI site is shown. This sequence is identical in MFG-WIN and KCP3-WNIN. The human EPO cDNA sequence was used as a reporter gene. (B) Effect of gene expression. The three constructs were transfected to CRIP cells, cell-free viral supernatant was harvested to transduce NIH 3T3 cells, and the cells were selected in the presence of G418. The same number of drug-resistant cells were plated on 6-cm plates, grown for another 3 days, and subjected to enzyme-linked immunosorbent assay. Only the results from transduction assays are shown. Expression of MFG-WIN was set to 1, and those of the others were normalized to it.
To construct the series of retroviral vectors incorporating all the features found in this study, the CAT sequence was removed from KCP3-CAT and the XbaI site was converted to BamHI (with pCRII-M5LCP), resulting in HCP3. The primers used in this step are HHIR and XB5L3 (the former primer was used for construction of RPX68-M5L). The nucleotide sequence of XB5L3 is as follows: XB5L3: GGATCCTCTAGAGGATGGTC BamHI XbaI
The BamHI-BglII fragment containing the foot-and-mouth disease virus (FMDV) IRES (16) was inserted into the BamHI site of HCP3, generating COI. Subsequently, the BamHI-SalI fragment containing the EMCV IRES was inserted into the BamHI-SalI site of COI, resulting in CTI. The FMDV IRES was removed from CTI by restriction digestion with HpaI and StuI, followed by filling in and ligation, generating COE.
To construct similarly improved retroviral vectors but under the control of the original U3 of the MLV LTR, the XbaI-BamHI fragment containing the CAT sequence was first removed from SFG-CAT to generate HFG by amplifying the region between the 5′ end of U3 and the naturally occurring XbaI site, just upstream from the start codon of env, with primers HHIR and XB5L3 (used for the construction of HCP3), fusing this HindIII-XbaI fragment to the large HindIII-XbaI fragment of SFG-CAT. The BamHI-BglII fragment containing the FMDV IRES was then isolated from the pCRII-FMDV IRES and inserted into the BamHI site of HFG, resulting in MOI. Subsequently, the BamHI-SalI fragment containing the EMCV IRES was inserted into the BamHI-SalI site of MOI, producing MTI. MOE was constructed by cutting MTI with HpaI and XhoI and filling in these sites, so that MOE contains only the EMCV IRES.
Transfection.
BING and CRIP cells were transfected by a calcium phosphate-DNA coprecipitation method as previously described in detail (10, 28, 35). A total of 10 μg of DNA in 500 μl of CaCl2 · H2O (124 mM CaCl2) was mixed with 500 μl of 2× HBS (280 mM NaCl, 10 mM KCl, 1.5 mM Na2HPO4 · 2H2O, 12 mM dextrose, 50 mM HEPES) with constant bubbling, and within 1 to 2 min this solution was added to the cells with 25 μg of chloroquine per ml. The transfection efficiency was measured in most experiments by 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) staining with the same culture plates or duplicate dishes, if necessary.
Transduction.
Supernatants from the transfected packaging cells were collected, usually 48 h after transfection, filtered through a 0.45-μm-pore-size filter, and used for transduction of target cells. For transduction of CEM-SS, H9, and U937 cells, 5 × 106 cells were harvested, resuspended with 5 ml of viral supernatant in the presence of 8 μg of Polybrene per ml, and incubated in a 37°C incubator (5% CO2) with occasional stirring for 5 h. Fresh medium was then added to maintain the cell density at 5 × 105 to 6 × 105/ml, and the cells were grown for another 36 to 44 h. NIH 3T3 cells were also transduced with 3 ml of viral supernatant in the presence of 8 μg of Polybrene per ml for 5 h followed by the addition of fresh medium. The following day, the cells were refed with fresh medium containing G418 as required. When needed, the viral titer was determined as described by Byun et al. (9, 10).
Enzyme and cytokine assays.
CAT assays were performed by standard procedures as previously described by Byun et al. (10). Two days after transfection or transduction, the cells were harvested, washed once with phosphate-buffered saline, and resuspended in 0.25 M Tris-HCl (pH 7.5). Total proteins were prepared by four or five freeze-thaw cycles followed by incubation at 65°C for 7 min. Equivalent amounts of protein were assayed for CAT activity. The percent conversion of [14C]chloramphenicol to its acetylated forms was determined by quantitating the intensity of each spot with a phosphorimager (FUJIX BAS 1000).
The levels of human EPO and murine granulocyte-macrophage colony-stimulating factor (GM-CSF) production were determined by enzyme-linked immunosorbent assay with commercially available kits from R & D Systems Inc. (Minneapolis, Minn.), i.e., DEP00 for hEPO and MGM00 for mGM-CSF.
The activity of β-galactosidase expressed in cells containing the lacZ gene was measured by the o-nitrophenyl-β-d-galactopyranoside (ONPG) assay. The cells with an introduced lacZ gene were stained with X-Gal as described previously (24).
PCR of genomic DNA.
To test whether the retroviral sequences were preserved in transduced target cells, total DNA was prepared by lysing transduced and selected NIH 3T3 cell lines with TES (10 mM Tris.HCl [pH 7.8], 1 mM EDTA, 0.7% sodium dodecyl sulfate) and then treating them with 400 μg of proteinase K per ml at 50°C for 1 h and subjecting them to phenol-chloroform extraction and ethanol precipitation. PCR was performed with 5 μg of total genomic DNA and oligonucleotide primers specific to various region of the retroviral vector (see Fig. 10). The nucleotide sequences of the primers are as follows: MLba: TCGCGAGTTCGAAGAGAACCATCAGATG L228: GCCTCGAGATAAGTTGCTGGCCAG C5NH: GCTAGCGGGACTTTCCATTGACGT EPC5: CCATGGGGCTGCAGAAT EPC3: GGATCCTCATTTTTGGACTGG
FIG. 10.

Test for preservation of retroviral sequences in transduced cells. Total cellular DNAs were prepared from G418-resistant NIH 3T3 cells transduced with MFG-EPO, MOIN-EPO, and COIN-EPO. The three pairs of oligonucleotide primers were used to amplify the regions indicated in the figure. SD, splice donor; SA, splice acceptor.
The samples were amplified through 30 cycles of denaturation at 94°C for 1 min, primer annealing at 55°C for 1 min, and primer extension at 72°C for 1 min 30 s. The amplified DNA fragments were analysed by agarose gel electrophoresis.
Helper virus assay.
The BAG mobilization assay was carried out as described by Pear et al. (35). Supernatant (3 ml) from producer lines was used to infect BAG cells (36), and the cells were passaged 1:10 every 3 or 4 days. When passages 3 of the infected BAG cells had reached approximately 50% confluence, the medium was changed, and 24 h later the supernatant was filtered through a 0.45-μm-pore-size membrane. A 3-ml portion of the filtrate was used to infect NIH 3T3 cells, and 48 h later the cells were divided into two portions; one was stained for β-galactosidase, and the other underwent G418 selection. To determine the titer of the virus used to infect BAG cells, 1 ml of the viral supernatant from the virus-producer cells was used in parallel to infect NIH 3T3 cells; this was followed by G418 selection.
The amphotropic retroviral env gene was also amplified by PCR from recombinant viral and transduced cellular genomes. Virus-producing cells were seeded at 5 × 106 per 100-mm-diameter dish, and virus-containing medium was harvested 48 h later. Recombinant viruses were harvested by ultracentrifugation at 35,000 × g for 2 h in an SW50.1 rotor after 0.45-μm-pore-size syringe filtration. The viral pellet was resuspended in 200 μl of TES, 100 μg of proteinase K was added, and the samples were incubated for 30 min at 37°C. After a phenol-chloroform extraction, 5 U of RNase-free DNase (Promega) was added to the samples, which were incubated at 37°C for 30 min. After one more phenol-chloroform extraction, RNAs were precipitated with ethanol and the pellets were resuspended with 50 μl of diethylpyrocarbonate-treated water. Viral cDNAs were synthesized from the viral RNAs with avian myeloblastosis virus reverse transcriptase (Promega). Reverse transcription was initiated from the MLV 3′ LTR-specific oligomer MLhe (TCGCGAGCGGCCGCTTGCCAAACCTACG) and incubated with deoxynucleoside triphosphates and RNase inhibitor at 42°C for 1 h. Synthesized cDNAs were used as a template for PCR. The env gene of recombinant viral and transduced cellular genome were amplified with MLV-E5 and MLV-E3 primers, whose oligonucleotide sequences are AAGCTTATGGCGCGTTCAACGCTCTCA and AAGCTTCTATGGCTCGTACTCTATAGG, respectively.
RESULTS
Defining the packaging sequence in MFG.
We used MFG as a starting vector for systematic deletion analysis and modification. We and other have previously demonstrated that MFG functions as well as or better than other current retroviral vectors with respect to levels of gene expression and virus titer (10, 23, 32). MFG contains gag sequences up to the NarI site at position 1040 followed by a splice acceptor fragment from the NdeI site (position 5402) to the XbaI site (position 5766) in MLV (Fig. 1). An adapter oligonucleotide was used to insert an NcoI site at the natural ATG of the env gene at position 5777 followed by the sequences from the ClaI site at position 7675, converted to a BamHI site, to the end of MLV. The gene inserted at the NcoI site is expressed from a spliced mRNA, resembling the normal spliced env mRNA following MLV infection.
FIG. 1.

Schematic representation of the retroviral vector MFG. In MFG, the gene of interest (dotted box) is cloned into the NcoI site, containing the start codon in it, and expressed as a spliced mRNA. MFG contains the 420- and 99-bp coding sequences for gag and env, respectively. U3 of Moloney MLV is 448 bp long. ATG, start codon of gag.
Initially we were interested in determining if the gag and env regions of the vector were required (Fig. 1). The former is thought to contain the sequence necessary for viral packaging, whereas the latter does not seem to be needed for any retroviral vector functions. These sequences will enhance the frequency of recombination between the packaging genome and the vector, increasing the possibility of producing RCR. Furthermore, deletion of unnecessary sequences will allow the insertion of larger DNA fragments into the vector.
To determine the minimum length of nucleotide sequence needed for packaging, a series of deletions between the SD and SA were generated, as summarized in Fig. 2A, and their effects on packaging and transduction efficiencies were tested with the lacZ gene as a reporter. The MFG deletion constructs were transfected to the NIH 3T3-based packaging line CRIP or the 293-based amphotropic packaging line BING, the resulting viral supernatants were used to transduce NIH 3T3 cells, and X-Gal-stained cells were counted to estimate the packaging efficiency. Transfection efficiency was determined by measuring both lacZ activity and the number of X-Gal-stained cells in the transfected packaging line. All mutant constructs gave comparable numbers of blue cells with virtually identical intensity as well as similar levels of lacZ activity (Fig. 2B), demonstrating that the deletions did not affect gene expression.
The relative titers of each of the deletion constructs are shown in Fig. 2B. The deletion constructs can be divided into three classes, depending on their effects on packaging efficiency. First, four mutant constructs containing the deletion from positions 228 to 371 (Δ15, Δ16, Δ17, and Δ18) completely lost the packaging function. Second, the sequence from 377 to 527 appeared to be necessary but not essential for the optimal packaging efficiency, since the titers of these mutant constructs were consistently lower than those of the control. Third, there are two mutant constructs that reproducibly showed a maximally twofold increase in packaging efficiency. The mutant construct Δ38, which always gave the highest titer, contains a 500-bp deletion removing the entire gag coding sequence present in MFG.
Deletion of the residual env sequence.
MFG also has approximately 140 bp between the stop codon of the foreign gene and the 5′ end of U3 (Fig. 3). This region contains the 99-bp env coding sequence, which can be used as a template for recombination with the same sequence in the packaging line. We deleted 113 bp, including the entire residual env coding sequence, but left the polypurine tract intact (MΔE, Fig. 3). To allow comparison of the levels of gene expression, the bacterial CAT gene was used as a reporter(MFG-CAT, MΔE-CAT). The level of CAT activity was measured after either transfection of packaging lines or transduction of various cell types including the human monocytic U937 and T lymphoid CEM lines, to determine the effects on gene expression (Fig. 3). MΔE-CAT always produced levels of CAT activity similar to those produced by MFG-CAT in both transfected packaging lines and transduced target cells, suggesting that deletion of residual env sequences did not significantly affect gene expression, as observed by others (17, 29).
FIG. 3.

Effect of deletion of the residual env coding sequence. (A) In MΔE-CAT, 113 bp, including the entire env coding sequence (99 bp), was deleted but the polypurine tract remained intact. In this experiment, the bacterial CAT sequence was used as a reporter gene. The vector is not drawn to scale. (B) Effect on gene expression. MFG-CAT and MΔE-CAT constructs were transfected to the packaging line CRIP, cell-free viral supernatants were used to transduce the human promonocytic line U937 and T-lymphoid line CEM-SS, and all the cells were subjected to the CAT assay. Other experimental conditions are as described in the legend to Fig. 2. The expression of MFG was set to 1.
Deletion of the LTR U3 sequence.
To test whether the nucleotide sequence present in the LTR is essential for viral function other than as a promoter and also whether the LTR could be substituted with a heterologous promoter sequence, we constructed four hybrid LTRs in which retroviral sequences were deleted and replaced with heterologous promoter fragments of similar lengths (Fig. 4). As a model system, we isolated the four fragments from the HCMV major IE promoter (MIEP), which contains sequences interacting with various cellular transcription factors such as NF-κB, ATF, and AP1 (Fig. 4A). Various lengths of U3 were deleted, and four fragments from MIEP were then added to the respective sites (Fig. 4B). MCP1 contains the 264-bp HCMV IE promoter fragments in the region between -330 (PvuII) and -152 (XbaI) of U3. In MCP2, 117 bp of U3 (XbaI-SacI) was replaced with the 144-bp MIEP. In MCP3, the U3 region from -330 (PvuII) to -36 (SacII) was substituted with the 490-bp HCMV promoter. In MCP2 and MCP3, the retroviral TATA box but not the CAAT sequence is intact. LCP has the 422-bp HCMV promoter, which contains full promoter activity. In this construct, the entire U3 except for 30 bp at the 5′ end was deleted from the hybrid LTR, resulting in an LTR where gene expression is essentially under the control of the HCMV IE promoter. The original 3′ LTR in MFG-CAT was then replaced with these hybrid LTRs, as shown in Fig. 5.
The four CAT constructs containing hybrid promoters in the 3′ LTR, together with the parental vector MFG-CAT, were transfected to CRIP cells, and cell-free supernatants were used to transduce various human cell lines. The level of CAT activity was measured after either transfection of the packaging line or transduction of various cell lines. Because all constructs have the MLV LTR at the 5′ end in transfected cells, the levels of CAT activity in transfected CRIP cells were always comparable (Fig. 5). The level of CAT activity was also quite similar following transduction of NIH 3T3 and H9 cells. This result suggested that almost all the U3 sequence could be deleted from the LTR without any deleterious effects on retroviral functions.
Expression of the two genes by a single transcriptional unit.
The original version of MFG does not contain the selectable marker. However, expression of more than one gene would make a retroviral vector more versatile in its application to various in vitro experiments or gene therapy trials. We and others have shown that IRES elements can be inserted into MFG, allowing for the expression of multiple genes from a single polycistronic mRNA (31, 41). In the following study, we constructed a series of retroviral vectors expressing CAT and NEO linked by the FMDV IRES, as well as harboring modifications within gag, env, 5′ LTR, and 3′ LTR, and compared them with MFG, which also contains the two genes using the same IRES (Fig. 6).
Retroviral constructs were transfected to CRIP cells, cell-free viral supernatants were used to transduce various target cells, and the levels of CAT activity in the transduced cells were determined. One representative result is summarized in Fig. 6. The new constructs generally produced levels of CAT activity comparable to those of the parental construct, suggesting that the two genes could be efficiently expressed in the modified vectors.
The above experiments were performed with the CAT sequence as a reporter gene. To demonstrate that our observation was not restricted to a specific reporter gene, we also inserted the mGM-CSF gene into the SCP1 retroviral vector (Fig. 7). The transduced cells were selected with G418, and the levels of mGM-CSF were compared to those for the parental vector. The newly constructed retroviral vector generally gave slightly higher levels of mGM-CSF in all cell lines tested, confirming the above result based on CAT activity.
Role of NcoI in gene expression.
As described above, it has been speculated that the use of the NcoI site at the env ATG in MFG is necessary to achieve high levels of protein production. To test whether the initiation codon of a foreign gene has to coincide with the ATG in the NcoI cloning site, the NcoI site was deleted and the EPO reporter gene was inserted downstream, resulting in KCP3-WXIN (Fig. 8). Transduced NIH 3T3 cells were selected with G418, and EPO levels between the parental vector and the new construct lacking the NcoI site were compared. The level of EPO produced from the construct lacking the NcoI site was always comparable to that from the parental vector, indicating that NcoI has marginal, if any, effects on gene expression in the cell lines tested.
Improved retroviral vectors.
Based on above results, we constructed a series of retroviral vectors which accommodated the above observations; i.e., retroviral vectors in which the gag sequence unnecessary for packaging was deleted; the U3 sequence not essential for retroviral functions were replaced with heterologous promoter elements; the IRES was used to express more than one gene; and the NcoI expression site was replaced with multicloning sites. Two examples of such a vector are shown in Fig. 9. To demonstrate that these new vectors function as expected, the EPO and neo genes were inserted and compared with the parental MFG-based construct for levels of gene expression and viral titer. Transduced cells were selected with G418, and the levels of EPO in the culture supernatants were compared. The improved vector always gave higher levels of EPO.
FIG. 9.
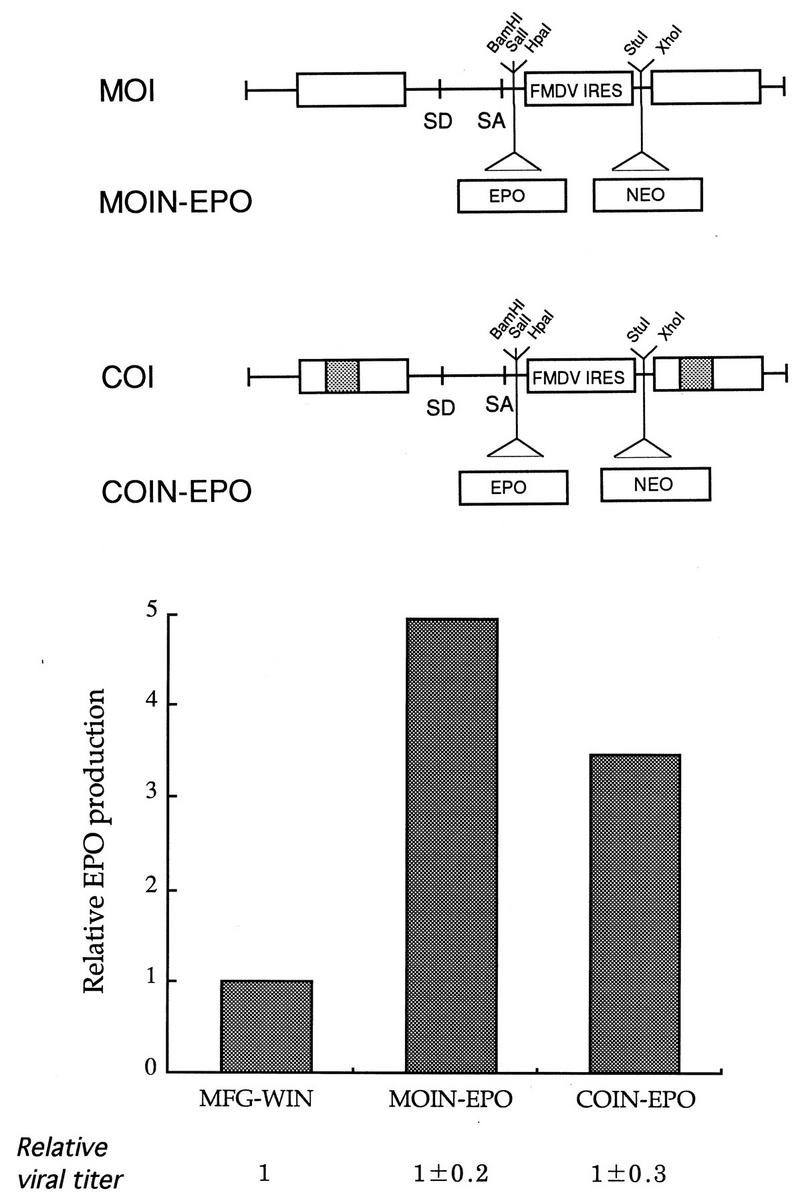
Construction of improved retroviral vectors. Based on the results shown from Fig. 2 to 8, improved vectors were constructed and tested for their performance with EPO. In this example, the two constructs COI and MOI are shown. The former has a chimeric U3, identical to that of LCP at the 5′ LTR and MCP3 at the 3′ LTR, while the latter contains the original U3 from MLV. Both vectors have deletions around the gag region (like Δ38 in Fig. 2), no env coding sequence (Fig. 3), the convenient restriction site for the gene of interest, and IRES and NEO as selectable markers. The NEO sequence was added to the XhoI site of MOI and COI, resulting in MOIN and COIN, respectively. The EPO cDNA sequence was subsequently cloned into the BamHI site of MOIN and COIN, generating MOIN-EPO and COIN-EPO, respectively. The three vectors, including the parental construct MFG-WIN, were transfected into CRIP or BING cells, and cell-free viral supernatants were harvested to transduce NIH 3T3 cells. Viral titer were determined 3 days posttransduction as described by Byun et al. (9). Cells were selected in the presence of G418 to be close to the actual situation. Drug-resistant populations were obtained, and identical numbers of cells were plated on 6-cm culture plates. After 3 days, the levels were determined. To compare viral titers between vectors, G418-resistant cultures were obtained after transfection of PA317 cells with the above vectors and grown to similar densities on 10-cm culture plates. Viral titers were determined by the conventional and new methods (9, 10).
To confirm that these newly constructed vectors lacking the entire gag coding sequence could indeed produce viral titers comparable to MFG, we transfected the amphotropic packaging line PA317 with MFG-, MOI-, and COI-based retroviral vectors expressing EPO. G418-resistant PA317 populations were generated and compared for viral titers at similar cell concentrations. As indicated in Fig. 9, the viral titers were always comparable for the three vectors, confirming the previous finding that the deletion of the gag coding sequence has no significant effect on viral packaging.
We have demonstrated, using PCR, that the nucleotide sequence in the retroviral vectors was preserved in the transduced target cells. Total DNAs were prepared from transduced, G418-selected cells followed by PCR with the oligonucleotide primer as shown in Fig. 10. If the retroviral vectors stably transfer the retroviral sequences to the target cells, these primers would amplify 380, 582, and 622 bp of the 5′ region of the viral genome, EPO, and the 3′ LTR from MFG-WIN or MOIN-EPO, respectively, and 655, 582, and 617 bp from COIN-EPO. DNA fragments of the expected lengths were present in all cells, suggesting that the new vectors can stably transfer the foreign gene to target cells.
Tests for replication-competent virus.
The producer lines containing MFG-, MOI-, and COI-based retroviral vectors were shown to be free of RCR by a BAG mobilization assay and reverse transcription-PCR of the retroviral env gene. The PA317-based producer lines were obtained by transfection with these vectors expressing EPO followed by G418 selection. Antibiotic-resistant populations were passaged at least 10 times prior to the RCR assays. First, BAG cells were incubated with cell-free culture supernatants (105 to 106/ml) from the three producer lines. These cells contain an integrated β-galactosidase provirus that can be rescued by any packaging functions including gag, pol, and env. Cells were split every 3 to 4 days at 1:10, and 3-ml portions of the supernatants were used to infect NIH 3T3 cells after passage 3 of infected BAG cells; this was followed by X-Gal staining or G418 selection. No X-Gal-stained or G418-resistant cells were found from any producer lines tested in this experiment, suggesting that at a given sensitivity of the assay, no RCR was produced from the newly constructed vectors. This result was also confirmed by reverse transcription-PCR of culture supernatants from the producer lines with the oligonucleotide primers that can amplify the retroviral env gene (data not shown).
DISCUSSION
As an approach to developing the improved retroviral vectors, we used MFG as a starting vector, since we and others have demonstrated previously that it consistently gave higher titers and gene expression. In this study, we focused on constructing retroviral vectors which are safer, more versatile, and more convenient to use than the parental vector. We have demonstrated that all coding sequences for gag and env present in the vector could be deleted without any significant effects on packaging efficiency and gene expression, thus minimizing the frequency of RCR generation by homologous recombination in the packaging cell line. Indeed, it should now be possible to design the retroviral vectors and the expression plasmids for gag-pol and env used in the packaging line in such a way that no viral sequence is overlapped between them. Because almost all U3 sequence could be replaced with other promoter sequences, the newly made vectors would have a very short retroviral sequence. For example, our new vector COI contained only 1,230 bp of retroviral sequence, consisting of U3 (30 bp for 5′ LTR and 156 bp for 3′ LTR), R (68 bp), U5 (78 bp), the packaging signal (378 bp), and the region downstream from SA (410 bp).
Our mutational analysis of the packaging signal produced the three types of phenotype (no packaging, decreased packaging, and increased packaging) and defined at least three regions involved in packaging. The first group of mutants, all of which contained a deletion in region A (positions 228 to 371), showed absolutely no packaging function, which is consistent with previously reported results (2, 25, 26). The second phenotype is characterized by a twofold decrease in packaging, localizing the regions that are not essential but necessary for the maximum packaging function. This group of mutants contain a deletion from positions 377 to 527 (region B). The third group of deletion mutants, Δ38 and Δ48, reproducibly showed maximally twofold-higher packaging efficiency than did the parental type, suggesting the possible presence of the sequence interfering with the packaging function, probably at positions 739 to 1016 (region C). In Δ38, which contains almost a 500-bp deletion, the entire gag coding sequence was removed but showed no decrease in packaging efficiency. However, when the deletion was extended to position 377 (region B) as in Δ28, the packaging efficiency was decreased substantially.
In summary, there seems to be a complex array of sequences that are involved in viral packaging: region A is essential for viral packaging, regions B is which is necessary but not essential for optimal packaging, and region C probably interferes with packaging. When both regions B and C were deleted, the B phenotype was shown. These results suggest that the entire N-terminal gag sequence was not necessary for efficient viral packaging in the context of the MFG vector. This is somewhat unexpected because this region has previously been thought to contain an extended packaging signal. According to a literature survey, the presence of nucleotide sequence in the gag coding region which may be involved in the packaging was first reported by Armentano et al. (4) and Bender et al. (6) with the same N2 vector system. In their works, the packaging efficiency of the retroviral vector, which contains the gag coding region, was at least 40 times higher than that of the vector lacking this region. This region has also been shown to efficiently package nonretroviral transcriptional units (1). However, similar work carried out by Guild et al. (18) indicated that the presence of the gag coding region has only marginal effects on packaging. One possible explanation is that the SA site plays a role in levels of packagable RNA. The gag coding region included in the N2 vector system contains the so-called cryptic packaging sequence, while the SA present upstream from the env coding region was present in our vectors. It is also important to note that our assay for packaging requires gene expression following transduction. It is possible that certain deletions could be affecting, either positively or negatively, other processes independent of packaging, such as reverse transcription or RNA stability. Whatever the actual mechanism for packaging, our retroviral vector lacking the gag coding region produced viral titers and levels of gene expression similar to those produced by the vector containing this sequence even when various reporter genes including CAT, EPO, and GM-CSF were used.
We have also demonstrated that almost the entire U3 could be replaced without any effects on retroviral functions. The U3 region of the Moloney MLV LTR is almost 450 bp long, similar to the size of many viral and cellular promoters. Therefore, it would be possible to design a retroviral vector containing only the heterologous promoter. It has been demonstrated that the enhancer within the U3 region could be replaced with other viral and cellular enhancers or hypersensitive regions (19, 30, 40). Riviere et al. (37) reported that both the enhancer and promoter of MLV could be replaced with U3 from myeloid proliferation-stimulating virus or Friend MLV, but the nucleotide sequences of these U3s are very similar. Our results suggest that almost the entire U3 could be replaced with the full-length heterologous promoter containing completely different nucleotide sequences without affecting any essential viral functions. Although the TATA box of the 3′ MLV LTR may be thought to be important for polyadenylation, clearly it can be deleted and replaced with heterologous sequences. Whether sequences within the HCMV MIEP are acting as a polyadenylation site needs to be determined.
We have also attempted to constructed a retroviral vector more convenient to use and more versatile than MFG by adding both IRES elements and multicloning sites. Although MFG has been used clinically in the absence of selection because of the high titers, it would be desirable in actual human applications to select and enrich transduced cells for therapeutic effects. Similar to previous results, we have found that IRES elements could be used effectively in our modified MFG vectors. All the vectors constructed in this study reproducibly gave comparable levels of gene expression in transiently transduced cells but substantially higher levels in selected population. Furthermore, we also found that fusion of the ATG of the gene of interest to the env ATG in MFG at the NcoI site is not necessary for high levels of gene expression, making it possible to use multicloning sites.
Based on our observations, we constructed a series of retroviral vectors containing some or all of the identified modifications. Two vectors (COI and MOI) performed as expected, producing viral titers comparable to those of MFG and driving high-level gene expression. Our vectors do not contain coding sequences for gag and env and can be manipulated to have virtually no U3 sequences. To our knowledge, the N-terminal gag coding sequence is present in all currently available retroviral vectors. Consequently, our vectors are safer because they should give rise to RCR by homologous recombination in the producer line at a much lower frequency than other existing vectors do. Our observation that almost all of the U3 sequences could be replaced with the heterologous promoter demonstrates that one can construct a retroviral vector containing full-size heterologous promoters, allowing therapeutic genes to be regulated by their natural promoters. Taken together, our results should allow the design and construction of safer, more sophisticated, and more versatile retroviral vectors.
ACKNOWLEDGMENTS
This work was supported in part by research grants from the Korean Ministry of Science and Technology (S.K.) and the Korean Science and Engineering Foundation (S.K.) and by Public Health Service grants CA59371 and DK44935 from the National Cancer Institute.
We thank S.T. Kim (Chung Book University) for providing FMDV-IRES.
REFERENCES
- 1.Adam M A, Miller A D. Identification of a signal murine retrovirus that is sufficient for packaging of nonretroviral RNA into virions. J Virol. 1988;62:3802–3806. doi: 10.1128/jvi.62.10.3802-3806.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alford R L, Honda S, Lawrence C B, Belmont J T. RNA secondary structure analysis of the packaging signal for Moloney murine leukemia virus. Virology. 1991;183:611–619. doi: 10.1016/0042-6822(91)90990-s. [DOI] [PubMed] [Google Scholar]
- 3.Ali M, Lemoine N R, Ring C J A. The use of DNA viruses as vectors for gene therapy. Gene Ther. 1994;1:367–384. [PubMed] [Google Scholar]
- 4.Armentano D, Yu S-F, Kantoff P W, von Ruden T, Anderson W F, Gilboa E. Effect of internal viral sequences on the utility of retroviral vectors. J Virol. 1987;61:1647–1650. doi: 10.1128/jvi.61.5.1647-1650.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bandara G, Muller G M, Galea-Lauri J, Georgescu H I, Suchanek M K, Hung G L, Glorioso J C, Robbins P D, Evans C H. Intraarticular expression of biologically active interleukin 1-receptor-antagonist protein by ex vivo gene transfer. Proc Natl Acad Sci USA. 1993;90:10764–10768. doi: 10.1073/pnas.90.22.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bender M A, Palmer T D, Gelinas R E, Miller A D. Evidence that the packaging signal of Moloney murine leukemia virus extends into the gag region. J Virol. 1987;61:1639–1646. doi: 10.1128/jvi.61.5.1639-1646.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boggs S S, Patrene K D, Muller G M, Evans C H, Doughty L A, Robbins P D. Prolonged systemic expression of huma IL-1 receptor antagonist hIL-1ra in mice reconstituted with hematopoietic cells transduced with retrovirus carrying the hIL-1ra cDNA. Gene Ther. 1995;2:632–638. [PubMed] [Google Scholar]
- 8.Bowtell D D L, Copy S, Johnson G R, Gonda T J. Comparison of expression in hematopoietic cells by retroviral vectors carrying two genes. J Virol. 1988;62:2464–2473. doi: 10.1128/jvi.62.7.2464-2473.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byun J, Kim J-M, Kim S-H, Yim J, Robbins P D, Kim S. A simple and rapid method for determination of recombinant retrovirus titer by G418 selection. Gene Ther. 1996;3:1018–1020. [PubMed] [Google Scholar]
- 10.Byun J, Kim S H, Kim J M, Yu S S, Robbins P D, Yim J, Kim S. Analysis of the relative level of gene expression from different retroviral vectors used for gene therapy. Gene Ther. 1996;3:780–788. [PubMed] [Google Scholar]
- 11.Cosset F-L, Takeuchi Y, Battini J-L, Weiss R A, Collins M K L. High-titer packaging cells producing recombinant retroviruses resistant to human serum. J Virol. 1995;69:7430–7436. doi: 10.1128/jvi.69.12.7430-7436.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dannos O, Mulligan R C. Safe and efficient generation of recombinant retroviruses with amphotropic and ecotropic host range. Proc Natl Acad Sci USA. 1988;85:6460–6464. doi: 10.1073/pnas.85.17.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dranoff G, Jaffe E M, Lasenby A, Golumbeck P, Levitesky H, Brose K, Jackson V, Hamada H, Pardol D M, Mulligan R C. Vaccination with irradiated tumor cells engineered to secrete murine GM-CSF stimulates potent, specific and long lasting antitumor immunity. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DuBridge R B, Tang P, Hsia H C, Phaik-Mooi L, Miller J H, Calos M P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol Cell Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Emerman M, Temin H M. Genes with promoters in rerovirus vectors can be independently suppressed by an epigenetic mechanism. Cell. 1984;39:449–467. [PubMed] [Google Scholar]
- 16.Escarmis C, Toja M, Medina M, Domingo E. Modifications of the 5′ untranslated region of foot-and-mouth disease virus after prolonged persistence in cell culture. Virus Res. 1992;26:113–125. doi: 10.1016/0168-1702(92)90151-x. [DOI] [PubMed] [Google Scholar]
- 17.Faustinella F, Kwon H H, Serrano F, Belmont J W, Caskey C T, Aguilar-Cordova E. A new family of murine retroviral vectors with extended multiple cloning sites for gene insertion. Hum Gene Ther. 1994;5:307–312. doi: 10.1089/hum.1994.5.3-307. [DOI] [PubMed] [Google Scholar]
- 18.Guild B C, Finer M H, Housman D E, Mulligan R C. Development of retrovirus vectors useful for expressing genes in cultured murine embryonal cells and hematopoietic cells in vivo. J Virol. 1988;62:3795–3801. doi: 10.1128/jvi.62.10.3795-3801.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilberg F, Stocking C, Ostertag W, Grez M. Functional analysis of a retroviral host-range mutant: altered long terminal repeat sequences allow expression in embryonal carcinoma cells. Proc Natl Acad Sci USA. 1987;84:5232–5236. doi: 10.1073/pnas.84.15.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaffe E M. High efficiency gene transfer into primary human tumor explants without cell selection. Cancer Res. 1993;53:2221–2226. [PubMed] [Google Scholar]
- 21.Kabat D. Targeting retroviral vectors to specific cells. Science. 1995;269:417. doi: 10.1126/science.7618110. [DOI] [PubMed] [Google Scholar]
- 22.Kasahara N, Dozy A M, Kan Y W. Tissue-specific targeting of retroviral vectors through ligand-receptor interactions. Science. 1994;266:1373–1376. doi: 10.1126/science.7973726. [DOI] [PubMed] [Google Scholar]
- 23.Krall W J, Skelton D C, Yu X-J, Riviere I, Lehn P, Mulligan R C, Kohn D B. Increased levels of spliced RNA account for augmented expression from the MFG retroviral vector in hematopoietic cells. Gene Ther. 1996;3:37–48. [PubMed] [Google Scholar]
- 24.Lee S-G, Kim S, Robbins P D, Kim B-G. Optimization of environmental factors for the production and handling of retroviruses. Appl Microbiol Biotechnol. 1996;45:477–483. doi: 10.1007/BF00578459. [DOI] [PubMed] [Google Scholar]
- 25.Mann R, Baltimore D. Varying the position of a retrovirus packaging sequence results in the encapsidation of both unspliced and spliced RNAs. J Virol. 1985;54:401–407. doi: 10.1128/jvi.54.2.401-407.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mann R, Mulligans R C, Baltimore D. Construction of a retrovirus packaging mutant and its use to produce helper-free defective retrovirus. Cell. 1983;33:153–159. doi: 10.1016/0092-8674(83)90344-6. [DOI] [PubMed] [Google Scholar]
- 27.Marshall E. Gene therapy’s growing pains. Science. 1995;269:1050–1055. doi: 10.1126/science.7652552. [DOI] [PubMed] [Google Scholar]
- 28.Miller A D, Buttimore C. Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol Cell Biol. 1986;6:2895–2902. doi: 10.1128/mcb.6.8.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller A D, Roseman G J. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 30.Moore K A, Scarpa M, Kooyer S, Utter A, Caskey C T, Belmont J W. Evaluation of lymphoid-specific enhancer addition or substitution in a basic retrovirus vector. Hum Gene Ther. 1991;2:307–315. doi: 10.1089/hum.1991.2.4-307. [DOI] [PubMed] [Google Scholar]
- 31.Morgan R A. Retroviral vectors containing putative internal ribosome entry sites: development of a polycistronic gene transfer system and application to human gene therapy. Nucleic Acids Res. 1992;20:1293–1299. doi: 10.1093/nar/20.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohashi T, Boggs S, Robbins P D, Bahnson A, Patrene K, Wei F, Wei J, Li L, Lucht L, Fei Y, Clark S, Kimak M, He H, Mowery-Ruchton P, Barranger J. Efficient transfer and sustained high expression of the human glucocerebrosidase gene in mice and their functional macrophages following transplantation of bone marrow transduced by a retroviral vector. Proc Natl Acad Sci USA. 1992;89:11332–11336. doi: 10.1073/pnas.89.23.11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osborne W R A, Miller A D. Design of vectors for efficient expression of human purine nucleotide phosphorylase in skin fibroblasts from enzyme-deficient humans. Proc Natl Acad Sci USA. 1988;85:6851–6855. doi: 10.1073/pnas.85.18.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmer T D, Thomson A R, Miller A D. Production of human factor IX in animals by genetically modified skin fibroblast: potential therapy for hemophilia B. Blood. 1989;73:438–445. [PubMed] [Google Scholar]
- 35.Pear W S, Nolan G P, Scott M L, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Price J, Turner D, Cepko C. Lineage analysis in the vertebrate nervous system by retrovirus-mediated gene transfer. Proc Natl Acad Sci USA. 1987;84:156–160. doi: 10.1073/pnas.84.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riviere I, Brose K, Mulligan R C. Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc Natl Acad Sci USA. 1995;92:6733–6737. doi: 10.1073/pnas.92.15.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shinnick T M, Lerner R A, Sutcliffe J G. Nucleotide sequence of Moloney murine leukaemia virus. Nature. 1981;293:543–548. doi: 10.1038/293543a0. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi Y, Cosset F-L C, Lachmann P J, Okada H, Weiss R A, Collins M K. Type C retrovirus inactivation by human complement is determined by both the viral genome and the producer cell. J Virol. 1994;68:8001–8007. doi: 10.1128/jvi.68.12.8001-8007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valerio D, Einerhand M P, Wamsley P M, Bakx T A, Li C L, Verma I M. Retrovirus-mediated gene transfer into embryonal carcinoma and hemopoietic stem cells: expression from a hybrid long terminal repeat. Gene. 1989;14:419–427. doi: 10.1016/0378-1119(89)90516-7. [DOI] [PubMed] [Google Scholar]
- 41.Zitvogel L, Tahara H, Cai Q, Storkus W J, Muller G, Wolf S F, Gately M, Robbins P D, Lotze M T. Construction and characterization of retroviral vectors expressing biologically active human interleukin-12. Hum Gene Ther. 1994;5:1493–1506. doi: 10.1089/hum.1994.5.12-1493. [DOI] [PubMed] [Google Scholar]