

Abstract
The interferon (IFN)-induced promyelocytic leukemia (PML) protein is specifically associated with nuclear bodies (NBs) whose functions are yet unknown. Two of the NB-associated proteins, PML and Sp100, are induced by IFN. Here we show that overexpression of PML and not Sp100 induces resistance to infections by vesicular stomatitis virus (VSV) (a rhabdovirus) and influenza A virus (an orthomyxovirus) but not by encephalomyocarditis virus (a picornavirus). Inhibition of viral multiplication was dependent on both the level of PML expression and the multiplicity of infection and reached 100-fold. PML was shown to interfere with VSV mRNA and protein synthesis. Compared to the IFN mediator MxA protein, PML had less powerful antiviral activity. While nuclear body localization of PML did not seem to be required for the antiviral effect, deletion of the PML coiled-coil domain completely abolished it. Taken together, these results suggest that PML can contribute to the antiviral state induced in IFN-treated cells.
Interferons (IFNs) are a family of secreted proteins with antiviral, antiproliferative, and immunomodulatory activities. The molecular basis of the IFN response, in particular the antiviral and antiproliferative effects, is not yet fully understood. More than 100 genes are known to be IFN induced, but the physiological roles of the majority of their products are not yet recognized. Only a few IFN-induced proteins, namely, the p68 protein kinase, the 2′,5′-oligoadenylate (2′5′A) synthetase, and certain Mx family proteins, have been shown to display intrinsic antiviral activities (reviewed in references 37, 39 and 41). While IFN-treated cells are resistant to a large variety of virus infections, these three known effectors confer protection only against some RNA viruses, implying the existence of complementary pathways.
The PML (promyelocytic leukemia) gene has been identified through its fusion to the RARα gene in the t(15;17) translocation found in patients with acute promyelocytic leukemia (reviewed in reference 44). The PML protein shares a C3HC4 (RING finger) zinc binding motif (14) with a large group of polypeptides which perform heterogeneous functions ranging from transactivation of viral genes to DNA repair or peroxisome assembly (reviewed in references 4 and 15). PML belongs to a subfamily of nine proteins defined by the additional presence of one or two other cysteine-rich motifs, the B boxes, as well as a very long coiled-coil region (35), which is implicated in PML homodimerization (21, 32).
PML has a speckled nuclear expression pattern which is the consequence of the localization of the protein to nuclear bodies (NBs) (10, 12, 23, 45). PML colocalizes on these structures with an autoantigen of primary biliary cirrhosis, Sp100 (43). The functions of NBs are unknown, but they seem not to be sites of replication, transcription, or splicing (42). Analysis of the 5′ genomic sequences of PML revealed both a functional IFN-α/β-stimulated response element, ISRE, and an IFN-γ activation site, GAS (40), demonstrating that PML is a primary target gene of IFNs. That the two NB-associated proteins PML (8, 24, 40) and Sp100 (19) are IFN induced suggests a role for this nuclear structure in the IFN response. An important point is to find which, if any, of the biological effects of IFN could be mediated by PML. Recently, we and others have shown that overexpression of PML suppresses the growth of some cell lines (22). At present, the molecular basis of the antiproliferative effect of PML is not understood. These findings could allow PML to be included in the pathways responsible for IFN-induced cell growth suppression.
Here we demonstrate that in the absence of IFN, constitutive overexpression of PML but not of Sp100 confers resistance to infection by vesicular stomatitis virus (VSV) and influenza A virus but not by encephalomyocarditis virus (EMCV), identifying a novel pathway in the mechanism of IFN antiviral action.
MATERIALS AND METHODS
Cell cultures.
Human glioblastoma astrocytoma U373 MG, Chinese hamster ovary (CHO) cells, mouse GP+E−86 cells (26), and L929 cells were grown at 37°C in Dulbecco’s modified Eagle’s medium. The human histocytic lymphoma cell line U937 was grown in RPMI 1640. All media were supplemented with 10% fetal calf serum. CHO cells, GP+E−86 cells (transfected with the empty or PML encoding vector), and CHO cells overexpressing Sp100 were kept in medium supplemented with 0.5 mg of hygromycin (GIBCO) per ml. U373 MG control cells (transfected with the empty vector) or the same cells overexpressing PML were kept in medium supplemented with 0.5 mg of G418 per ml. Swiss 3T3 mouse cells transfected by the empty vector or vector expressing MxA (31) were a kind gift from J. Pavlovic and were grown in the same medium supplemented with 0.5 mg of G418 per ml.
Construction of expression vectors and cell lines.
The PML cDNA was inserted in different vectors: the BglII-BamHI fragment (positions 48 to 2084 [11]) was ligated into the retroviral vector M3P-SVhygro (17), whereas the complete cDNA on an EcoRI fragment was inserted in the pSG5 vector or the bicistronic pCIN vector (neomycin resistance) (36). The C terminal PML mutant, PML Stop 504, was constructed by inserting an oligonucleotide with an in-frame stop codon at the SacI site of the PML cDNA (in the pSG5 construct). The coiled-coil PML mutant PMLΔ(216–333) was created by total digestion of the pSG5 PML vector with BssHII and religation. The RING finger PML mutant Q59C60/EL was described previously (21). This mutation results in a change of the amino acids glutamine and cysteine at positions 59 and 60 into aspartic acid and leucine, respectively. The cytoplasmic PML mutant, Stop 381, results from insertion of three stop codons in the unique Sse8387 I site (nucleotide 1228 of the PML insert) of the pSG5 PML vector. For the Sp100 construct, the region containing bp 32 to 1548 was amplified by PCR from the original construct (43) with the nucleotides -5′ oligo (5′gaagatctgccgccATGGCAGGTGGGGGCGGC3′) and -3′ oligo (5′GAGGGTCAGGTAAAGAAGATTAGagatcttc3′) and inserted in the BglII site of the pSG5 vector. This was done to remove an in-frame upstream stop codon, to optimize the ATG, and finally to create flanking BglII sites (underlined) for easy cloning. The amplified Sp100 used was completely verified by sequence analysis.
Stable transfections of CHO, GP+E−86, and U373 MG cells.
Stable CHO or GP+E−86 clones were obtained by lipofection (Gibco/BRL) with pSG5 constructs cotransfected with DSPhygro or M3P-SVhygro-derived constructs and subsequent hygromycin selection (final concentration, 0.5 mg/ml). Stable PML-expressing U373 MG clones were obtained via transfection with the pCIN-PML construct and subsequent neomycin selection (final concentration, 0.5 mg/ml). Control cells were generated in the same way with the empty vectors. Resistant colonies were examined for PML or Sp100 expression by indirect immunofluorescence, and positive pools were subjected to a round of subcloning by limiting dilution. As a consequence of the antiproliferative effect of PML, some of these clones tend to lose their expression. Therefore, expression of the clones was verified every six passages by immunofluorescence and Western blot analysis. The apparent molecular weight of the PML mutant proteins was in agreement with the molecular weight of the mutations made.
Interferons and anti-interferon antibodies.
Human IFN-β was from Triton Biosciences (Alameda, Calif.), and anti-human IFN-α (G-030-501-553), IFN-β (G-028-501-568), and IFN-γ (G-034-501-565) antibodies were from the National Institutes, of Health. U373 MG PML cells were grown for two passages in the presence of 104 neutralizing units of anti-IFN-α/β/γ antibodies per ml.
Virus stocks and virus yield assay.
Stocks of the WSN strain of influenza A virus (4 × 108 PFU/ml), VSV (6 × 108 PFU/ml), or EMCV (8 × 108 PFU/ml) were prepared from supernatants of virus-infected CHO cells. Cells were seeded in 24-well plates for 5 h at 37°C and then infected with virus at a multiplicity of infection (MOI) ranging from 0.1 to 3. At the times indicated in the legends, cultures were frozen and thawed three times and centrifuged to remove cell debris. The supernatants were serially diluted, and the virus titers were measured by a plaque assay on L929 or CHO cells and expressed as plaque-forming units per milliliter of supernatant.
Determination of IFN titers.
Culture media from mouse GP+E−86 control and GP+E−86 PML cells infected or not infected with VSV or influenza virus at a MOI of 0.1 for 16 h were subjected to titer determination on L929 cells, and those from U373 MG control and U373 MG PML cells were subjected to titer determination on HeLa cells. To inactivate the virus present, the culture supernatants from infected cells were brought to pH2 for 24 h and neutralized before the titer determination. All the cells were challenged with VSV. IFN titers, determined as the amounts of IFN required to produce 50% inhibition of the cytopathic effect, were expressed in relation to the human IFN-α reference (G-023-902-527) or mouse IFN reference (Ga 02 901 511) of the National Institutes of Health.
Immunofluorescence and Western blot analysis.
Rabbit and mouse anti-human PML and rabbit anti-human Sp100 antibodies were produced as described previously (10, 34). Anti-VSV antibodies were a kind gift from D. Blondel. The cells were fixed in 4% paraformaldehyde for 15 min at 4°C and then 100% methanol for 5 min at 4°C. Immunofluorescence was performed with anti-PML, anti-Sp100, or anti-VSV antibodies diluted 1/500 to 1/1,000 and revealed with fluorescein isothiocyanate (FITC)-conjugated secondary antibodies. For Western blot analysis, the cells were scraped in phosphate-buffered saline, centrifuged, and lysed in 0.25 ml of 125 mM Tris (pH 7)–1% sodium dodecyl sulfate–10% glycerol–0.75 mM phenylmethylsulfonyl fluoride. Each sample was sonicated to reduce the viscosity. Western blot analysis was performed by standard procedures (20). A 40-μg portion of total-cell extracts was analyzed with rabbit polyclonal anti-PML or anti-VSV antibodies, each diluted 1/2,000 and revealed by enhanced chemiluminescence (Amersham).
Northern blot analysis.
Total RNA was extracted with a Bioprobe Systems RNA extraction kit standard. After the RNA was blotted on nitrocellulose membranes (Scheicher & Schuell), the Northern blot analysis was performed by random priming (Boehringer Mannhein) radiolabelled VSV N, NS, M, G, or glyceraldehyde-3-phosphate dehydrogenase probes. VSV N, NS, M, and G probes were generated as previously described from plasmids (a gift from D. Blondel) pGN2 (13), pGNS1 (13), pKOM1 (5), and pSVG (18), respectively.
RESULTS
IFN-β inhibits VSV and influenza virus replication in human monocytic cell line U937 in the absence of the MxA protein.
The IFN-induced human MxA protein inhibits the multiplication of VSV and influenza virus by an unknown mechanism (31, 41). The level of this known anti-VSV and anti-influenza virus mediator is not increased in the monocytic cell line U937 cells after IFN-β treatment (38). To assess the capacity of human IFN-β to induce PML expression and to inhibit VSV and influenza virus replication in these cells, U937 cells were treated with 1,000 U of IFN-β per ml for 48 h and infected at an MOI of 0.1 with VSV or influenza virus. Double immunofluorescence was performed with anti-PML and anti-VSV antibodies. The results presented in Fig. 1 show that IFN-β increases PML levels and inhibits VSV antigen expression. The virus yields in IFN-β-treated U937 cells compared to control infected cells were 750 times lower (6 × 104 and 4.5 × 107 PFU/ml, respectively) for VSV and 125 times lower (8 × 104 and 107 PFU/ml, respectively) for influenza virus. The absence of MxA induction by IFN in U937 cells (38) suggests that human IFNs protect cells from VSV and influenza virus infections by a pathway which is independent of MxA protein expression.
FIG. 1.

IFN-β induces PML protein synthesis and inhibits VSV antigen expression in the human monocytic cell line U937. U937 cells were treated with 1,000 U of human IFN-β per ml. After 48 h at 37°C, control (C) and IFN-treated cells were infected with VSV at a MOI of 0.1. Double immunofluorescence were performed 24 h postinfection with mouse anti-PML antibodies visualized with Texas red and rabbit anti-VSV antibodies followed by FITC labelling.
Overexpression of PML confers resistance to infections by VSV and influenza virus.
Since the PML gene product was shown to have growth-suppressing properties (22, 25, 30) and thus could be a candidate for antiproliferative IFN actions, it may also mediate other IFN-induced biological effects. To test a possible antiviral effect of PML, cell lines stably expressing PML were constructed in hamster CHO and mouse GP+E−86 cells. PML expression was verified by immunofluorescence and Western blot analysis (see Fig. 5B and 8B; data not shown). CHO control cells (transfected with the empty vector) and those overexpressing PML (CHO PML3) were infected with a picornavirus (EMCV), an orthomyxovirus (influenza A virus), or a rhabdovirus (VSV) at an MOI of 0.1. At 16 h later, the cells were stained with carbol methylene blue or used for the determination of virus yields. Compared to the effect in CHO control cells, which underwent nearly 100% cell death, the overexpression of PML in CHO PML3 conferred resistance to lysis by VSV and influenza virus but not EMCV (Fig. 2). No difference in virus yield was found between parental CHO cells and CHO cells transfected with the empty vector (data not shown). Inhibition of the cytopathic effect was accompanied by a decrease in virus multiplication, as shown by the viral titers (Fig. 2). The highest inhibitions obtained by CHO PML3 with VSV and influenza A virus were 125- and 100-fold, respectively, while no effect on EMCV multiplication was observed. Similarly, in GP+E−86 PML cells, 100- and 80-fold decreases in VSV and influenza virus growth, respectively, compared to those in infected cells harboring the empty vector were observed (data not shown).
FIG. 5.

(A) PML levels parallel inhibition of virus multiplication. CHO control cells (transfected with the empty vector) and CHO PML 1, 2, and 3 clones were infected with VSV or influenza A virus at an MOI of 0.1. After 16 h, viral titers were determined as described in Materials and Methods. (B) PML levels parallel the inhibition of VSV antigen expression. CHO control cells (transfected with the empty vector) and CHO PML 1, 2, and 3 clones were infected for 13 h with VSV at an MOI of 0.1. Western blot analysis of the extracts of these cells was done as described in Materials and Methods. (Top) Revealed with rabbit anti-PML antibodies; (middle) revealed with anti-VSV antibodies (VSV antigens are indicated at the right); (bottom) Coomassie brilliant blue (CBB)-stained proteins.
FIG. 8.

(A) Description of PML mutants. The structures of PML (including the C3HC4 zinc finger motif, the two B boxes and the coiled-coil) and of four PML mutants, the C-terminal PML mutant (PML Stop 504), the coiled-coil PML mutant [PMLΔ(216–333)], the RING finger PML mutant (Q59C60/EL), and the cytoplasmic PML mutant (Stop 381), are shown. aa, amino acids; NLS, nuclear localization signal. (B) Western blot analysis of PML in stably transfected CHO and GP+E−86 cells as well as the four PML mutants; also shown is the overexpression of Sp100 in CHO cells. (C) Analysis of PML domains involved in the inhibition of VSV antigens. CHO control (transfected with the empty vector), CHO PML wt (CHO PML3), and the four mutants of PML were infected with VSV for 13 h at an MOI of 0.1. Swiss 3T3 neo and 3T3 MxA were infected with VSV under the same conditions and used as control. Western blot analysis of the extracts of these cells was done as described in Materials and Methods. (Top) Revealed with anti-VSV antibodies (VSV antigens are indicated at the right); (bottom) Coomassie brilliant blue (CBB)-stained proteins. (D) Description of subcellular distributions in stably transfected CHO cells and the antiviral potentials of wild-type and mutant forms of human PML protein. Cells were infected with VSV at an MOI of 0.1. After 16 h, viral titers were determined as described in Materials and Methods.
FIG. 2.
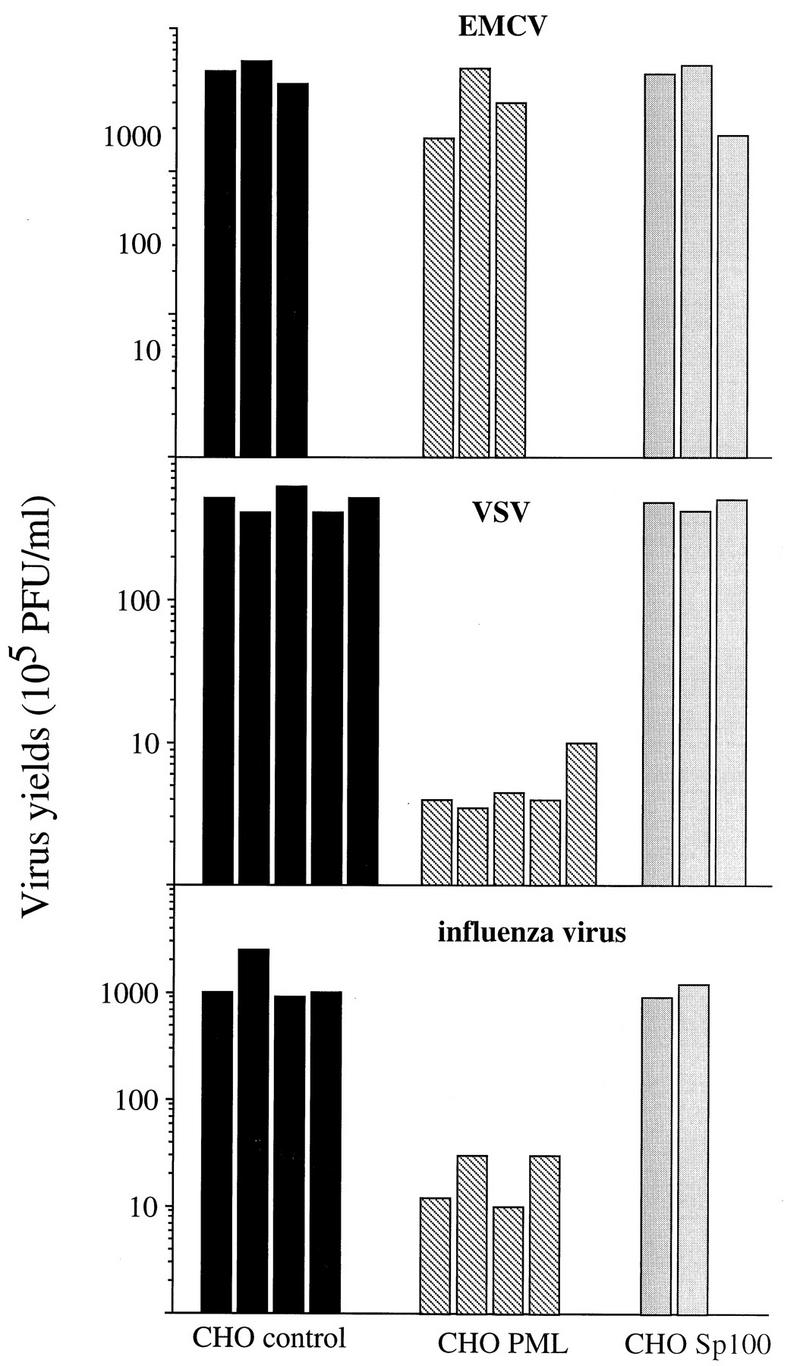
Overexpression of PML, and not of Sp100, confers resistance against infections by VSV and influenza virus. CHO control (transfected with the empty vector), CHO PML3, or CHO Sp100 cells were infected with EMCV, VSV, or influenza A virus at an MOI of 0.1. After 16 h, viral titers were determined as described in Materials and Methods.
Overexpression of another NB-associated protein, Sp100, does not confer antiviral resistance.
Since Sp100 colocalizes with PML onto NBs (23) and is also IFN induced (19), our demonstration that PML has antiviral properties raised the question whether Sp100 could display similar properties. We generated stable CHO clones which overexpress Sp100 (see Fig. 8B). The capacity of CHO Sp100 to inhibit virus growth was tested and compared to that of CHO PML. After 16 h of infection with VSV, influenza virus, or EMCV at an MOI of 0.1, no protective effect against either of these viruses was found in CHO Sp100, as revealed by carbol methylene blue staining (data not shown) or by determination of VSV, influenza virus, or EMCV titers (Fig. 2). Hence, Sp100 does not inhibit their multiplication, making a direct role of this protein in the IFN-induced antiviral state against these three viruses unlikely. Taken together, these results establish that overexpression of PML specifically inhibits the multiplication of VSV and influenza A virus.
Inhibition of virus replication in IFN-treated U373 MG cells and U373 MG PML.
To compare levels of PML expression in transfected cells to those induced in IFN-treated cells, we have overexpressed PML in human cells. This is because no hamster IFN is available and because our anti-PML antibodies recognize only human PML on Western blots. Three PML expression vectors (pSG5, M3P-SVhygro, or pCIN neo [see Materials and Methods]) were tranfected in three different human cell lines (U937, HeLa, and U373 MG). In nearly all cases, we were unable to isolate clones that expressed PML uniformly, confirming that overexpression of this protein interferes with cell proliferation (22), particularly in human cell lines. However, after several assays, we succeeded in isolating clones of human U373 MG with the pCIN PML construct.
PML levels in these transfected U373 MG cells were compared to those induced by IFN in control cells. U373 MG cells were treated for 48 h with 1,000 U of human IFN-β per ml. Equal amounts of total protein extracts from control, IFN-treated cells and U373 MG PML were analyzed by Western blotting. Figure 3A shows that no detectable band of PML was found in untreated U373 MG cells whereas, as previously described (8), different forms of PML, arising from alternative splicing of a single gene, were induced upon IFN treatment. Molecular imaging analysis (system GS525; Bio-Rad) revealed that comparable levels of PML were found in IFN-treated and transfected cells.
FIG. 3.
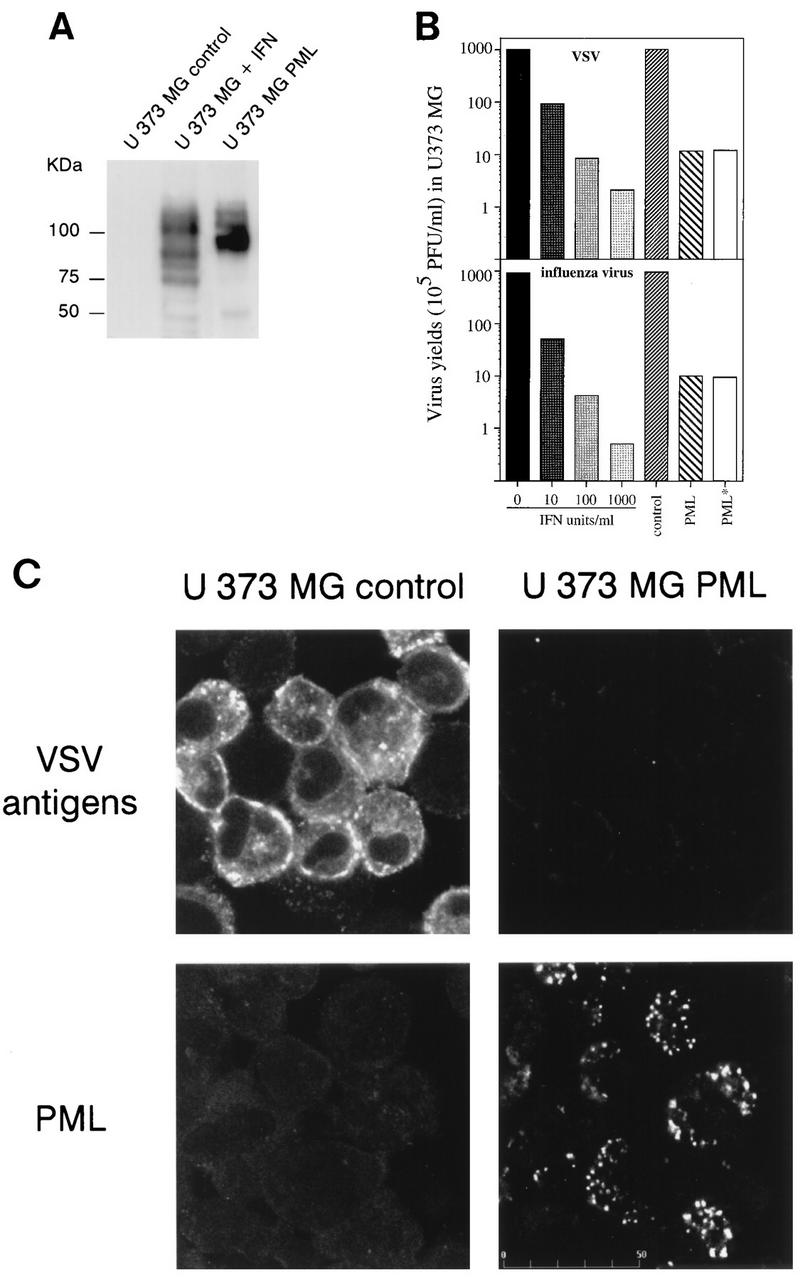
(A) PML level in IFN-treated U373 MG cells and U373 MG PML. U373 MG cells were treated for 48 h with 1,000 U of IFN-β per ml. Samples (50 μg) of extracts of control IFN-treated cells and U373 MG PML were analyzed by Western blotting and revealed with rabbit anti-PML antibodies. Note that all bands visualized by anti-PML antibodies are likely to be isoforms derived from alternative splicing of unique gene. Molecular size markers are indicated on the left. (B) Inhibition of virus replication in IFN-treated U373 MG cells and U373 MG PML. One series of U373 MG cells was treated for 48 h with 10, 100 or 1,000 U of IFN-β per ml. The second series of cells, U373 MG control (transfected with the empty vector), U373 MG PML, and U373 MG PML* (+ anti-IFN-α/β/γ antibodies [see Materials and Methods]) was seeded at 37°C for 5 h. The two series were then infected with VSV or influenza A virus at an MOI of 0.1. After 16 h, viral titers were determined as described in Materials and Methods. (C) Expression of PML in U373 MG cells inhibits the expression of VSV antigens (Top) Expression of VSV antigens in infected U373 MG control cells (transfected with the empty vector) and U373 MG PML. Immunofluorescence with rabbit anti-VSV antibodies was performed 13 h after infection with VSV at an MOI of 0.1 and revealed by FITC labelling. (Bottom) Expression of PML in U373 MG and U373 MG PML cells revealed by immunofluorescence with mouse anti-PML antibodies visualized with Texas red.
Then we tested whether U373 MG cells overexpressing PML, like CHO PML and GP+E−86 PML, display resistance to VSV and influenza virus. U373 MG PML cells grow much more slowly than the parental cell line transfected with the empty vector (data not shown). To avoid interference of growth rates by viral replication, all the experiments were done within 24 h. U373 MG and U373 MG PML cells (2 × 105 cells in each case) were seeded for 5 h in Dulbecco’s modified Eagle’s medium containing 10% serum and G418 and then were infected at an MOI of 0.1 with VSV or influenza virus for 16 h. At that time point, there were no observable differences in growth in uninfected control and U373 PML cells (data not shown). However, a clear difference in VSV and influenza virus replication was found in infected U373 MG and U373 MG PML cells. Overexpression of PML leads to a 90-fold decrease in VSV or influenza virus yield compared to control cells (Fig. 3B). VSV antigen expression was monitored in these cells by immunofluorescence (Fig. 3C). The PML-induced antiviral state was associated with a lower VSV protein expression.
To compare the degree of inhibition of VSV and influenza virus replication in U373 MG PML to that obtained in IFN-treated cells, U373 MG cells were treated for 48 h with 10, 100, or 1,000 U of IFN-β per ml. Then, control cells, IFN-treated cells, and U373 MG PML were infected with VSV or influenza virus at an MOI of 0.1 for 16 h. As shown in Fig. 3B, inhibition of VSV or influenza virus replication in U373 MG PML was comparable to that obtained in control cells treated with concentrations of IFN between 10 and 100 U/ml.
Resistance in the PML-expressing cells was not due to the presence of low IFN levels for the following reasons. (i) When culture media from mouse GP+E−86 control and GP+E−86 PML cells infected or not infected with VSV or influenza virus at an MOI of 0.1 for 16 h were subjected to titer determination on L929 cells and those from U373 MG control and U373 MG PML were subjected to titer determination on HeLa cells, their IFN titers were below the detection limit (less than 2 U/ml); therefore, PML-expressing cells before and after viral infection did not produce sufficient IFN to be protective. (ii) A mixture of mouse or human anti-IFN-α/β/γ antibodies was unable to reverse the resistance of GP+E−86 PML or U373 MG PML cells to VSV or influenza virus infections (Fig. 3B, PML* and data not shown). (iii) 2′5′A synthetase activity was unaffected by PML overexpression in CHO, GP+E−86, and U373 MG (Fig. 4), whereas it was induced in U373 MG control cells by the addition of 10 U of IFN-β per ml. These experiments demonstrate that overexpression of PML did not lead to the induction and secretion of IFN and that PML alone could contribute to IFN-induced inhibition of viral replication.
FIG. 4.

2′5′ A synthetase activity in IFN-treated cells and cells overexpressing PML. 2′5′ A synthetase activity was determined in cells extracts from CHO control cells (lane 1). CHO PML3 (lane 2), GP+E−86 control cells (lane 3), GP+E−86 PML (lane 4), U373 MG control cells (lane 5), U373 MG PML (lane 6), and U373 MG cells treated for 48 h with 10 U of IFN-β per ml (lane 7). All control cells are cells transfected with the empty vector. The 2′5′ A synthetase activity was determined by chromatographic analysis of the reaction substrate (ATP) and the products, 2′,5′-oligoadenylates (dimer and trimer), as previously described (7).
Resistance to VSV and influenza virus replication is dependent on both PML expression levels and MOI.
To find whether the viral resistance observed above is dependent on the level of PML expression, we selected three CHO PML clones expressing different PML levels, as shown by Western blot analysis (Fig. 5B). These three clones and CHO control cells were infected with VSV and influenza virus at an MOI of 0.1. Figure 5 shows that the PML levels in the CHO PML clones paralleled the resistance to VSV or influenza virus infections as assessed by the determination of viral titers (Fig. 5A).
The effect of overexpression of PML on the inhibition of VSV antigen expression was also confirmed by Western blot analysis with anti-VSV antibodies (Fig. 5B). The five structural proteins of VSV are the major nucleocapside, N (40-kDa), the matrix protein, M (25 kDa), the glycoprotein, G (69 kDa), and two minor proteins, the phosphoprotein, NS (29 kDa), and the polymerase protein, L (24 kDa) (3), but only the G, N, and M proteins were revealed with our rabbit anti-VSV antibodies. As shown in Fig. 5B, the highest inhibition of the VSV antigen expression was obtained with the CHO PML clone expressing the highest PML level. Thus, at least in the case of VSV, PML appears to interfere with the expression of viral proteins, as has been predicted from immunofluorescence analysis (Fig. 3C).
The clone expressing the highest level of PML (CHO PML3) was infected with VSV and influenza virus at different MOIs for 16 h. As the MOI increased from 0.1 to 1. the PML-expressing clone showed decreased resistance to both viruses (Fig. 6A). These data suggest that clones overexpressing PML are less resistant to VSV and influenza virus infection at high MOI. This situation is similar to the effect of an increasing MOI on the antiviral state induced by IFN. The inhibitory effect of PML on VSV multiplication was also tested by Western blot analysis with anti-VSV antibodies (Fig. 6B), which again showed that the degree of inhibition is higher at an MOI of 0.1 than at an MOI of 1.
FIG. 6.
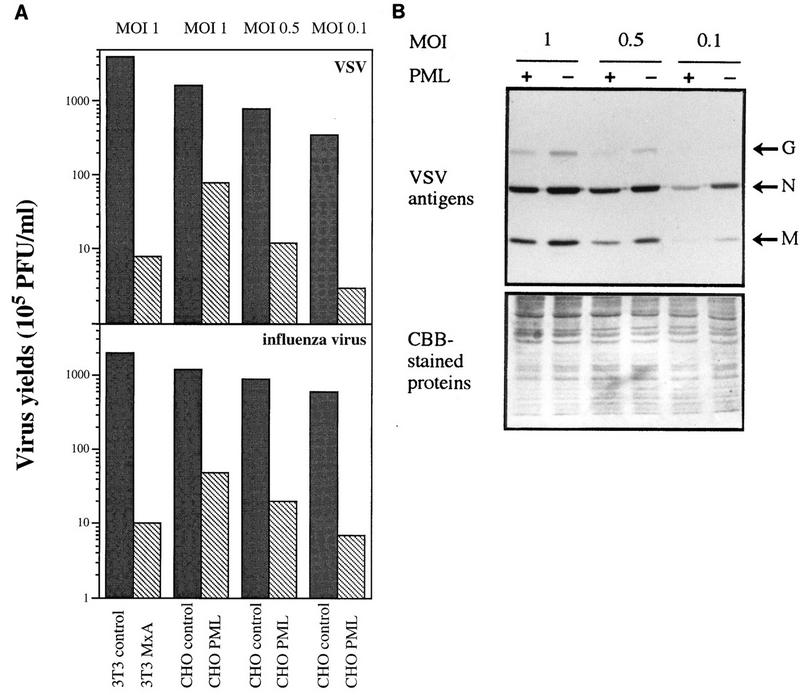
Resistance of PML-expressing clones to VSV and influenza virus is MOI dependent. CHO control cells (transfected with the empty vector) and CHO PML 3 were infected with VSV or influenza A virus at different MOIs as indicated in the figure. Swiss 3T3 control and Swiss 3T3 MxA were infected at an MOI of 1 with VSV or influenza virus. (A) After 16 h, the cells were used for the determination of the viral titers. Antiviral activities are the means of three independent experiments. (B) CHO control cells and CHO PML 3 were infected with VSV for 10 h at different MOIs as indicated in the figure. The results of Western blot analysis are revealed with anti-VSV antibodies. (Top) revealed with anti-VSV antibodies (VSV antigens are indicated at the right); (bottom) Coomassie brilliant blue (CBB)-stained proteins.
Overexpression of human MxA confers a high resistance to VSV and influenza virus infections (31). To compare the ability of PML and MxA to inhibit viral multiplication, CHO control, CHO PML3, Swiss 3T3 control, and Swiss 3T3 MxA were infected with VSV or influenza virus under the same conditions. The overexpression of MxA protein inhibits the replication of these two viruses to a much higher extent than does PML. At an MOI of 1, PML had a small protective effect whereas MxA strongly protected against these viruses (Fig. 6A). Moreover, at higher MOI, MxA still protected against these viruses, while PML had no effect (data not shown). Again, VSV antigen expression was more strongly inhibited by MxA than by PML expression (see Fig. 8C), demonstrating that MxA is a more potent effector than PML.
Viral RNA in PML-expressing cells.
To test if overexpression of PML interferes with viral mRNA synthesis. CHO control and CHO PML3 were infected with VSV at an MOI of 0.5. After 4 h at 37°C, total RNA was isolated. The RNA preparations were analyzed for the presence of VSV mRNA N, NS, M, and G by Northern blot analysis. Figure 7 shows that PML had an inhibitory effect on viral N mRNA synthesis as well as on NS, M, and G mRNAs. A β imager 1200 analysis (Biospace) revealed that the concentrations of VSV N, NS, M, and G mRNAs were about threefold lower in CHO PML3 and U373 MG PML cells than in control cells (Fig. 7 and data not shown), demonstrating that PML interferes with VSV mRNA expression. Moreover, at an MOI of 0.5, the synthesis of viral RNA (Fig. 7) or proteins (Fig. 6B) was less inhibited than was the VSV yield (Fig. 6A) by PML overexpression.
FIG. 7.

CHO control cells (transfected with the empty vector) and CHO PML3 were infected with VSV at an MOI of 0.5 for 4 h. Total RNA was extracted as described in Materials and Methods. Samples (20 μg of RNA per lane) were analyzed for the presence of VSV N, NS, M, and G. GΔPDH, glyceraldehyde-3-phosphate dehydrogenase.
Requirement of the coiled-coil domain of PML for its antiviral activity.
Homodimerization of PML and/or PML-RARα was shown to occur through a long coiled-coil region (amino acids 229 to 360). To see whether the coiled-coil domain, the RING finger domain, or the C-terminal region of PML was involved in the antiviral state, four PML mutants were constructed: the C-terminal PML mutant (PML Stop 504), the coiled-coil PML mutant (PMLΔ216–333), the RING finger PML mutant (Q59C60/EL), and the C-terminal PML mutant (Stop 381). The structures of these mutants compared to wild-type PML are shown in Fig. 8A. These mutants were stably transfected in CHO cells. Their expression was verified by immunofluorescence (Fig. 3C and data not shown), and their product size was compared to that of the wild-type protein by Western blot analysis (Fig. 8B). For both the C-terminal PML Stop 504 and the PML Stop 381 mutants, constructed by the insertion of stop codons, a minor band corresponding to the size of the wild-type PML is expressed, probably involving a readthrough mechanism (see Discussion).
Deletion of the coiled-coil domain led to an altered subnuclear localization characterized by a fine intranuclear network without speckles, whereas absence of the C-terminal region did not impair the targetting of PML Stop 504 onto the NBs. The RING finger PML mutant (Q59C60/EL) is nuclear diffuse, and PML mutant Stop 381 is mostly cytoplasmic (Fig. 8D). No antiviral state against VSV or influenza A virus was induced by the PMLΔ(216–333) coiled-coil mutant, whereas the PML Stop 504, the RING finger PML mutant (Q59C60/EL), and the cytoplasmic PML mutant Stop 381 clones, which all have the coiled-coil domain, were as efficient as clone CHO PML3 in inhibiting the multiplication of VSV and influenza virus, as assessed by viral titers (Fig. 8D) and Western blot analysis (Fig. 8C). Thus, clearly the coiled-coil region of PML is required for its antiviral property.
DISCUSSION
In this study, we provide evidence that overexpression of PML, but not of Sp100, confers resistance to two RNA viruses, VSV and influenza virus, suggesting that PML participates in the antiviral state induced in IFN-treated cells. PML protein has an inhibitory effect on both VSV mRNA and protein synthesis. At an MOI of 0.5, the VSV yield appears to be more highly inhibited than is the synthesis of viral RNA or proteins, suggesting a possible defect in the production of infectious virus in cells overexpressing PML. As expected, this antiviral effect against VSV and influenza virus is dependent both on PML expression and the MOI of the virus. The degree of inhibition of VSV or influenza virus replication by PML was comparable to that obtained in control cells treated with concentrations of IFN between 10 and 100 U/ml. This may lead to the suspicion that overexpression of PML induces IFN secretion, which could in turn inhibit viral replication. However, our results clearly establish that overexpression of PML did not induce IFN secretion even after viral infection.
How PML migh inhibit VSV and influenza virus replication is unknown. Individual expression of previously identified human IFN-mediators has shown that overexpression of 2′5′ A synthetase (6, 9) or p68 kinase (29) confers resistance to EMCV but not to VSV while overexpression of human MxA inhibits VSV, influenza A virus, and other RNA virus multiplication but not that of picornavirus, togavirus, or herpes simplex virus (16, 31, 38). The two known human Mx proteins (MxA and MxB) (31), like rat (Mx2 or Mx3) (27) and mouse (Mx2) (46) Mx proteins, are cytoplasmic. The Mx1 protein has a speckled nuclear localization in both mouse and rat cells (2, 27, 47). While rat Mx3 and human MxB are devoid of antiviral properties, rat Mx1 and human MxA are active against both VSV and influenza virus (28, 31, 41). Mouse Mx1 confers resistance only to influenza virus (47), and Mx2 (mouse or rat) protects against VSV only (2, 28, 46). Thus, PML closely resembles rat Mx1 in both to its localization and its antiviral properties.
Compared to MxA protein, PML was found to have a less powerful antiviral activity against VSV and influenza virus replication. However, it appears that IFNs protect cells against VSV and influenza virus by at least two different pathways, one of which is independent of MxA protein. In human U937 cells, IFN-β inhibits VSV and influenza virus replication (see above) without inducing MxA protein (38). PML could be involved in one of these pathways. The inhibition, in IFN-treated U937 cells, of the VSV yield was 750 times and that of influenza virus was 125 times greater than in control cells. These results suggest that in addition to PML, other mediators could be implicated in inhibiting VSV replication in this cell line. In this sense, it has been shown that the IFN-induced human 9-27 protein could also participate in the inhibition of VSV but not in that of influenza virus (1). The resistance to VSV or EMCV infections conferred by IFN was similar in embryonic fibroblasts derived from PML knockout mice and from wild-type mice (24). This is not surprising for EMCV, since overexpression of PML does not affect the replication of this virus (see above), or for VSV, since Mx2 (46) appears to play a major role, which may mask PML contribution.
Two of the previously identified antiviral IFN effectors have relatively well-defined modes of action: 2′5′A synthetase and p68 kinase (reviewed in references 37 and 39). The molecular targets of the Mx and PML proteins are unknown, although Mx proteins display GTPase activity, which may be required for their antiviral properties (2, 33, 41). PML mutation analysis revealed that both the RING finger PML (Q59C60/EL) and the cytoplasmic PML Stop 381 mutants lost the normal PML localization but still possessed an intact coiled-coil domain and were effective in inhibiting VSV and influenza virus. Deletion of the PML coiled-coil domain abolished the antiviral properties against VSV and influenza virus and altered the punctuate localization of PML onto NBs. Both C-terminal mutants were created by insertion of stop codons, which could be suppressed by translational readthrough, leading to some wild-type PML synthesis. However, the small amount of wild-type PML synthesized cannot explain the protective effect observed. Influenza virus, whose replication and transcription are nuclear, and VSV, whose replication takes place entirely in the cytoplasm, are both inhibited by nuclear PML and cytoplasmic PML Stop 381 proteins. The PML protein, therefore, could inhibit virus multiplication indirectly by modifying other cellular proteins, which may then modulate viral replication in the relevant cellular compartment.
We have shown here that overexpression of human IFN-induced PML affects VSV and influenza virus replication and interferes with viral mRNA and protein synthesis. Thus, PML can contribute to the establishment of the antiviral state in IFN-treated cells. The significant inhibitory effect of PML makes it a member of the family of IFN-induced proteins mediating antiviral properties.
ACKNOWLEDGMENTS
We acknowledge M. C. Guillemin for anti-Sp100 antibodies and D. Blondel for anti-VSV sera and plasmids containing VSV N, NS, M, and G. We thank J. Pavlovic for 3T3 MxA cells, H. Will for the Sp100 cDNA, and S. Rees (Glaxo/Wellcome, Stevenage, United Kingdom) for the pCIN vector. We also thank C. Chopin for technical assistance. The help of B. Boursin with the artwork is greatly appreciated.
This work was supported by grants from Ligue contre le Cancer, Fondation de France, Fondation St. Louis, and ARC.
M.K.C.-A. and F.Q. contributed equally to this work.
REFERENCES
- 1.Alber D, Staeheli P. Partial inhibition of vesicular stomatitis virus by the interferon-induced human 9-27 protein. J Interferon Cytokine Res. 1996;16:375–380. doi: 10.1089/jir.1996.16.375. [DOI] [PubMed] [Google Scholar]
- 2.Arnheiter H, Meier E. Mx proteins: antiviral proteins by chance or by necessity. New Biol. 1990;2:851–857. [PubMed] [Google Scholar]
- 3.Banerjee A K, Chattopadhyay D. Structure and function of the RNA polymerase of vesicular stomatitis virus. Adv virus Res. 1990;38:99–124. doi: 10.1016/s0065-3527(08)60860-x. [DOI] [PubMed] [Google Scholar]
- 4.Barlow P N, Luisi B, Milner A, Elliott M, Everett R. Structure of the C3HC4 domain by 1H-nuclear magnetic resonance spectroscopy. A new structural class of zinc-finger. J Mol Biol. 1994;237:201–211. doi: 10.1006/jmbi.1994.1222. [DOI] [PubMed] [Google Scholar]
- 5.Blondel D, Harmison G G, Schubert M. Role of matrix protein in cytopathogenesis of vesicular stomatitis virus. J Virol. 1990;64:1716–1725. doi: 10.1128/jvi.64.4.1716-1725.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chebath J, Benech P, Revel M, Vigneron M. Constitutive expression of (2′5′) oligo A synthetase confers resistance to picornavirus infection. Nature. 1987;330:587–588. doi: 10.1038/330587a0. [DOI] [PubMed] [Google Scholar]
- 7.Chelbi-Alix M K, Chousterman S. Ethanol induces 2′5′ oligoadenylate synthetase and antiviral activities through interferon β production. J Biol Chem. 1992;267:1741–1745. [PubMed] [Google Scholar]
- 8.Chelbi-Alix M K, Pelicano L, Quignon F, Koken M H M, Venturini L, Stadler M, Pavlovic J, Degos L, de Thé H. Induction of the PML protein by interferons in normal and APL cells. Leukemia. 1995;9:2027–2033. [PubMed] [Google Scholar]
- 9.Coccia E M, Romeo G, Nissim A, Marziali G. A full-length 2-5A synthetase cDNA transfected in NIH-3T3 cells impairs EMCV but not VSV replication. Virology. 1990;179:228–233. doi: 10.1016/0042-6822(90)90292-y. [DOI] [PubMed] [Google Scholar]
- 10.Daniel M-T, Koken M, Romagné O, Barbey S, Bazarbachi A, Stadler M, Guillemin M, Degos L, Chomienne C, de Thé H. PML protein expression in hematopoietic and acute promyelocytic leukemia cells. Blood. 1993;82:1858–1867. [PubMed] [Google Scholar]
- 11.de Thé H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991;66:675–684. doi: 10.1016/0092-8674(91)90113-d. [DOI] [PubMed] [Google Scholar]
- 12.Dyck J A, Maul G G, Miller W H, Chen J D, Kakizuka A, Evans R M. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell. 1994;76:333–343. doi: 10.1016/0092-8674(94)90340-9. [DOI] [PubMed] [Google Scholar]
- 13.Emerson S U, Schubert M. Location of the binding domains for the RNA polymerase L and the ribonucleocapsid template within different halves of the NS phosphoprotein of vesicular stomatitis virus. Proc Natl Acad Sci USA. 1987;84:5655–5659. doi: 10.1073/pnas.84.16.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freemont P, Hanson I, Trowsdale J. A novel cysteine-rich sequence motif. Cell. 1991;64:483–484. doi: 10.1016/0092-8674(91)90229-r. [DOI] [PubMed] [Google Scholar]
- 15.Freemont P S. The RING finger. A novel protein sequence motif related to the zinc finger. Ann NY Acad Sci. 1993;684:174–192. doi: 10.1111/j.1749-6632.1993.tb32280.x. [DOI] [PubMed] [Google Scholar]
- 16.Frese M, Kochs G, Meierdieter U, Siebler J, Haller O. Human MxA protein inhibits tick-borne Thogoto virus but not Dhori virus. J Virol. 1995;69:3904–3909. doi: 10.1128/jvi.69.6.3904-3909.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gäken J, Farzaneh F, Stocking C, Osterlag W. Construction of a versatile set of retroviral vectors confering hygromycin resistance. BioTechniques. 1992;13:32–33. [PubMed] [Google Scholar]
- 18.Guan J-L, Machamer C E, Rose J K. Glycosylation allows cellsurface transport of an anchored secretory protein. Cell. 1985;42:489–496. doi: 10.1016/0092-8674(85)90106-0. [DOI] [PubMed] [Google Scholar]
- 19.Guldner H, Szostecki C, Grotzinger T, Will H. IFN enhances expression of Sp100, an autoantigen in primary biliary cirrhosis. J Immunol. 1992;149:4067–4073. [PubMed] [Google Scholar]
- 20.Harlow, E., and D. Lane. Antibodies. A laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 21.Kastner P, Perez A, Lutz Y, Rochette-Egly C, Gaub M-P, Durand B, Lanotte M, Berger R, Chambon P. Structure, localization and transcriptional properties of two classes of retinoic acid receptor alpha fusion proteins in acute promyelocytic leukemia (APL): structural similarities with a new family of oncoproteins. EMBO J. 1992;11:629–642. doi: 10.1002/j.1460-2075.1992.tb05095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koken M H M, Linares-Cruz G, Quignon F, Viron A, Chelbi-Alix M K, Sobczak-Thépot J, Juhlin L, Degos L, Calvo F, de Thé H. The PML growth-suppressor has an altered expression in human oncogenesis. Oncogene. 1995;10:1315–1324. [PubMed] [Google Scholar]
- 23.Koken M H M, Puvion-Dutilleul F, Guillemin M C, Viron A, Linares-Cruz G, Stuurman N, de Jong L, Szostecki C, Calvo F, Chomienne C, Degos L, Puvion E, de Thé H. The t(15;17) translocation alters a nuclear body in a RA-reversible fashion. EMBO J. 1994;13:1073–1083. doi: 10.1002/j.1460-2075.1994.tb06356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavau C, Marchio A, Fagioli M, Jansen J, Falini B, Lebon P, Grosveld F, Pandolfi P P, Pelicci P G, Dejean A. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene. 1995;11:871–876. [PubMed] [Google Scholar]
- 25.Liu J-H, Mu Z-M, Chang K-S. PML suppresses oncogenic transformation of NIH/3T3 cells by activated neu. J Exp Med. 1995;181:1965–1973. doi: 10.1084/jem.181.6.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markowitz D, Goff S, Bank A. A safe packaging line for gene transfer: separating viral gene on two different plasmids. J Virol. 1988;62:1120–1124. doi: 10.1128/jvi.62.4.1120-1124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meier E, Fäh J, Grob M S, End R, Staeheli P, Haller O. A family of interferon-induced Mx-related mRNAs encodes cytoplasmic and nuclear proteins in rat cells. J Virol. 1988;62:2386–2393. doi: 10.1128/jvi.62.7.2386-2393.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meier E, Kunz G, Haller O, Arnheiter H. Activity of rat Mx proteins against a rhabdovirus. J Virol. 1990;64:6263–6269. doi: 10.1128/jvi.64.12.6263-6269.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meurs E, Watanabe Y, Kadereit S, Barber G, Katze M, Chong K, Williams B, Hovanessian A. Constitutive expression of human double-stranded RNA-activated p68 kinase in murine cells mediates phosphorylation of eukaryotic initiation factor 2 and partial resistance to encephalomyocarditis virus growth. J Virol. 1992;66:5805–5814. doi: 10.1128/jvi.66.10.5805-5814.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mu Z M, Chin K V, Liu J H, Lozano G, Chang K S. PML, a growth suppressor disrupted in acute promyelocytic leukemia. Mol Cell Biol. 1994;14:6858–6867. doi: 10.1128/mcb.14.10.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pavlovic J, Zurcher T, Haller O, Staeheli P. Resistance to influenza virus and vesicular stomatitis virus conferred by expression of human MxA protein. J Virol. 1990;64:3370–3375. doi: 10.1128/jvi.64.7.3370-3375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez A, Kastner P, Sethi S, Lutz Y, Reibel C, Chambon P. PML/RAR homodimers: distinct binding properties and heteromeric interactions with RXR. EMBO J. 1993;12:3171–3182. doi: 10.1002/j.1460-2075.1993.tb05986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pitossi F, Blank A, Schröder A, Schwarz A, Hüssi P, Schwemme M, Pavlovic J, Staeheli P. A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J Virol. 1993;67:6726–6732. doi: 10.1128/jvi.67.11.6726-6732.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puvion-Dutilleul F, Venturini L, Guillemin M-C, de Thé H, Puvion E. Sequestration of PML and Sp100 proteins in an intranuclear viral structure during herpes simplex virus type 1 infection. Exp Cell Res. 1995;221:448–461. doi: 10.1006/excr.1995.1396. [DOI] [PubMed] [Google Scholar]
- 35.Reddy B, Etkin L, Freemont P. A novel zinc finger coiled-coil domain in a family of nuclear proteins. Trends Biochem Sci. 1992;17:344–345. doi: 10.1016/0968-0004(92)90308-v. [DOI] [PubMed] [Google Scholar]
- 36.Rees S, Coote J, Stables J, Goodson S, Harris S, Lee M G. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. BioTechniques. 1996;20:102–110. doi: 10.2144/96201st05. [DOI] [PubMed] [Google Scholar]
- 37.Samuel C E. Antiviral-regulated cellular proteins and their surprisingly selective antiviral activities. Virology. 1991;183:1–11. doi: 10.1016/0042-6822(91)90112-o. [DOI] [PubMed] [Google Scholar]
- 38.Schnorr J J, Schneider-Schaulies S, Simon-Jödicke A, Pavlovic J, Horisberger M A, Meulen V. MxA-dependent inhibition of measles virus glycoprotein synthesis in a stably transfected human monocytic cell line. J Virol. 1993;67:4760–4768. doi: 10.1128/jvi.67.8.4760-4768.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sen G C, Ransohoff R M. Interferon-induced antiviral actions and their regulation. Adv Virus Res. 1993;42:57–102. doi: 10.1016/s0065-3527(08)60083-4. [DOI] [PubMed] [Google Scholar]
- 40.Stadler M, Chelbi-Alix M K, Koken M H M, Venturini L, Lee C, Saïb A, Quignon F, Pelicano L, Guillemin M-C, Schindler C, de Thé H. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene. 1995;11:2565–2573. [PubMed] [Google Scholar]
- 41.Staeheli P, Pitossi F, Pavlovic J. Mx proteins: GTPases with antiviral activity. Trends Cell Biol. 1993;3:268–272. doi: 10.1016/0962-8924(93)90055-6. [DOI] [PubMed] [Google Scholar]
- 42.Stuurman N, de Graaf A, Floore A, Josso A, Humbel B, de Jong L, van Driel R. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J Cell Sci. 1992;101:773–784. doi: 10.1242/jcs.101.4.773. [DOI] [PubMed] [Google Scholar]
- 43.Szostecki C, Guldner H, Netter H, Will H. Isolation and characterization of cDNA encoding a human nuclear antigen predominantly recognized by autoantibodies of patients with primary biliary cirrhosis. J Immunol. 1990;145:4338–4347. [PubMed] [Google Scholar]
- 44.Warrell R, de Thé H, Wang Z, Degos L. Acute promyelocytic leukemia. N Engl J Med. 1993;329:177–189. doi: 10.1056/NEJM199307153290307. [DOI] [PubMed] [Google Scholar]
- 45.Weis K, Rambaud S, Lavau C, Jansen J, Carvalho T, Carmo-Fonseca M, Lamond A, Dejean A. Retinoic acid regulates aberrant nuclear localization of PML/RARα in acute promyelocytic leukemia cells. Cell. 1994;76:345–356. doi: 10.1016/0092-8674(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 46.Zürcher T, Pavlovic J, Staeheli P. Mouse Mx2 protein inhibits vesicular stomatitis virus but not influenza virus. Virology. 1992;187:796–800. doi: 10.1016/0042-6822(92)90481-4. [DOI] [PubMed] [Google Scholar]
- 47.Zürcher T, Pavlovic J, Staeheli P. Nuclear localization of mouse Mx1 protein is necessary for inhibition of influenza virus. J Virol. 1992;66:5059–5066. doi: 10.1128/jvi.66.8.5059-5066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]