

Abstract
The herpes simplex virus type 1 (HSV-1) UL25 gene contains a 580-amino-acid open reading frame that codes for an essential protein. Previous studies have shown that the UL25 gene product is a virion component (M. A. Ali et al., Virology 216:278–283, 1996) involved in virus penetration and capsid assembly (C. Addison et al., Virology 138:246–259, 1984). In this study, we describe the isolation of a UL25 mutant (KUL25NS) that was constructed by insertion of an in-frame stop codon in the UL25 open reading frame and propagated on a complementing cell line. Although the mutant was capable of synthesis of viral DNA, it did not form plaques or produce infectious virus in noncomplementing cells. Antibodies specific for the UL25 protein were used to demonstrate that KUL25NS-infected Vero cells did not express the UL25 protein. Western immunoblotting showed that the UL25 protein was associated with purified, wild-type HSV A, B, and C capsids. Transmission electron microscopy indicated that the nucleus of Vero cells infected with KUL25NS contained large numbers of both A and B capsids but no C capsids. Analysis of infected cells by sucrose gradient sedimentation analysis confirmed that the ratio of A to B capsids was elevated in KUL25NS-infected Vero cells. Following restriction enzyme digestion, specific terminal fragments were observed in DNA isolated from KUL25NS-infected Vero cells, indicating that the UL25 gene was not required for cleavage of replicated viral DNA. The latter result was confirmed by pulsed-field gel electrophoresis (PFGE), which showed the presence of genome-size viral DNA in KUL25NS-infected Vero cells. DNase I treatment prior to PFGE demonstrated that monomeric HSV DNA was not packaged in the absence of the UL25 protein. Our results indicate that the product of the UL25 gene is required for packaging but not cleavage of replicated viral DNA.
Three types of intracellular capsids are found in herpes simplex virus type 1 (HSV-1)-infected cells: A (empty), B (intermediate), and C (full) (15, 20). The three are distinguishable morphologically in electron micrographs, and they can be separated from each other preparatively by sucrose density gradient ultracentrifugation. They differ in the material present inside the capsid cavity. C capsids contain the virus DNA. A and B capsids lack DNA. The B-capsid cavity is filled primarily with VP22a, the cleaved form of the scaffolding protein, while in A capsids the cavity lacks either DNA or protein. B capsids consist of seven major proteins which are the products of the UL18, UL19, UL26, UL26.5, UL35, and UL38 genes (27, 46) and two minor proteins which are the products of the UL6 and UL12.5 genes (10, 38). Replication of viral DNA results in the formation of head-to-tail concatemers which are cleaved at specific sites to generate monomeric HSV DNA (17, 47, 53, 54). The cleaved DNA is packaged into B capsids to generate DNA-containing C capsids. Empty A capsids are thought to result from abortive attempts at DNA encapsidation (39). In studies with HSV-1 mutants, the UL6, UL15, UL25, UL28, UL32, and UL33 genes have been shown to be required for processing and packaging of viral DNA (1, 2, 4, 6, 28, 29, 37, 40, 44, 45, 49, 55, 56). In addition, the product of the UL12 gene has been shown to play an important but not essential role in cleavage and packaging. Mutants which fail to express the UL12 protein retain the ability to cleave and package DNA, but release of DNA-containing capsids from the nucleus is defective (33). No functions have been described for the six essential cleavage/packaging genes, although the UL15 gene shares homology with the ATP binding protein (gp17) of bacteriophage T4, a protein required for DNA cleavage and packaging of T4 DNA, suggesting that UL15 protein may perform the same function for HSV DNA packaging (55). The UL12 gene encodes an alkaline nuclease that appears to be required to resolve recombination intermediates generated during viral DNA replication (33). Homologs for the six essential HSV-1 cleavage/packaging genes are found in other herpesviruses (34), suggesting that the mechanism of DNA cleavage and packaging is the same for most herpesviruses.
The UL25 gene is predicted to encode a 580-amino-acid (60-kDa) protein (34). Ali et al. (3) demonstrated that rabbit antisera prepared against a UL25–glutathione S-transferase fusion protein expressed in Escherichia coli immunoprecipitated a 60-kDa protein from both HSV-1- and HSV-2-infected cells. In addition, they showed that the UL25 protein was found to be associated with purified HSV-1 virions. Studies with two temperature-sensitive (ts) mutants (ts1204 and ts1208) bearing lesions which map to the vicinity of the UL25 open reading frame suggested that the UL25 gene is a virion component which functions in both virus entry and capsid assembly (1). To further investigate the role of the UL6, UL15, UL25, UL28, UL32, and UL33 genes in cleavage and encapsidation of viral DNA, we have set out to construct mutants with more defined mutations in these genes. In this report, we describe the isolation and characterization of an HSV-1 UL25 mutant (KUL25NS) that was constructed by insertion of an in-frame stop codon in the UL25 open reading frame. The mutant was propagated on a complementing Vero cell line, and the phenotype of the UL25 mutant was characterized. The results of studies with this mutant demonstrate that the product of the UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. This is the first description of an HSV-1 mutant which cleaves DNA in the absence of packaging and demonstrates that the UL25 protein is essential for retaining DNA in capsids.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
Previously described procedures were used for growth and maintenance of African green monkey kidney cells (Vero; ATCC CCL 81 [26]). Vero-derived C1 (49), F3 (19), D6 (11), and 3A6 cells were grown in Dulbecco modified Eagle medium plus 10% fetal bovine serum and 0.5 mg of G418 (Geneticin; GIBCO-BRL) per ml. The KOS strain of HSV-1 was used as the wild-type strain. The HSV-1 UL28 deletion mutant GCB (49) was grown on UL28-complementing cell line C1. The UL26 and UL27 null viruses, KUL26ΔZ (19) and K082 (11), were grown on 3A6 and D6 cells, respectively. Baculovirus Autographa californica nuclear polyhedrosis virus was grown in Spodoptera frugiperda Sf9 cells (ATCC CRL 1711) as previously described (50). Recombinant baculoviruses expressing the HSV UL18, UL19, UL26, UL26.5, UL35, and UL38 genes have been described elsewhere (50). Monoclonal antibody MCA406 (Serotec Inc.) was used to analyze the UL26 and UL26.5 proteins (50). The UL19, UL38, and UL18 proteins were analyzed by using rabbit polyclonal antisera NC1 (UL19), NC2 (UL38), and NC5 (UL18) provided by R. Eisenberg and G. Cohen, University of Pennsylvania (14). A UL9-specific monoclonal antibody was obtained from S. Weller, University of Connecticut, Farmington (32).
Plasmids.
pRSV-neo contains the bacterial gene for neomycin resistance under control of the Rous sarcoma virus promoter (22). pKEF-B5 contains a 12.4-kb DNA fragment (nucleotides 47986 to 60362) that contains the UL25 through UL28 genes (19). The 6.5-kb NotI fragment derived from pKEF-B5 was subcloned into pBluescript II KS(−) to generate pKEF-NotI as previously described (19). The 6,282-bp EcoRI-SnaBI fragment (nucleotides 47986 to 52588) which contains the entire open reading frame of the UL25 gene (nucleotides 48813 to 50553) was subcloned from pKEF-B5 into pUC18 to generate pKUL25. The single NotI site (nucleotide 49126) present in this fragment was converted to an SpeI site by insertion of a 14-bp linker containing stop codons in all three reading frames to generate pKUL25NS.
The open reading frames for the UL25, UL6, and UL9 genes were cloned into the BamHI site of the baculovirus transfer vector pVL941 as follows. pAC-UL25 was constructed by digesting pSG18 (21) with BsrI, the ends were made blunt, and BamHI linkers were ligated. The sample was then digested with BamHI and EcoRV, and a 1,616-bp fragment containing the UL25 gene was gel purified. The EcoRV site (nucleotide 49049) is located within the UL25 gene, and the BsrI site (nucleotide 50665) is located 3′ to the UL25 stop codon. PCR primers were used to amplify sequences starting 5′ to the UL25 translation start codon to a region 3′ of the EcoRV site. The 5′ primer was designed to insert a BamHI site 13 bp 5′ to the UL25 ATG. The 250-bp fragment generated by PCR was digested with BamHI and EcoRV and used in a three-way ligation with the UL25 fragment (nucleotides 49049 to 50665) generated in the first step along with BamHI-digested pVL941. Clones which contained BamHI inserts in the proper orientation with respect to the baculovirus polyhedrin promoter were sequenced to verify that the region amplified by PCR was correct. pAC-UL6 (open reading frame nucleotides 15132 to 17160) was constructed by first digesting pSG10 (21) with SalI, and a 1,060-bp SalI fragment (nucleotides 16263 to 17323) that contains the 3′ end of the UL6 gene was cloned into the SalI site of pUC18; isolates were screened for the proper orientation relative to the BamHI site in pUC18. The resulting clone was digested with KpnI (nucleotide 16273) and BamHI, and the 1,050-bp fragment generated by this digest was isolated. The 5′ end of the UL6 gene was cloned by digesting pSG10 with BfaI, the ends were made blunt, and BamHI linkers were ligated. The sample was digested with BamHI and KpnI (nucleotide 16273), and a 1,170-bp fragment containing the UL6 gene was gel purified and ligated into the BamHI/KpnI site of pVL941. The BfaI site (nucleotide 15103) is located 100 bp 5′ of the UL6 ATG codon. The resulting clone was digested with KpnI and EcoRI, and the vector fragment (6,700 bp) generated by this digest was isolated and ligated with the 1,050-bp KpnI/BamHI UL25 fragment along with a 3,100-bp EcoRI/BamHI fragment that contains the remainder of the pVL941 vector. The resulting clone contained the UL6 open reading frame under the control of the polyhedrin promoter. pAC-UL9 (open reading frame nucleotides 23261 to 20708) was constructed by digesting pSG10 with NruI, and a 5-kbp fragment (nucleotides 20395 to 25412) was isolated and blunt-end ligated into the SmaI site of pUC19 to generate pUC-UL9. pUC-UL9 was digested with NarI (nucleotide 23540) and EcoRV (nucleotide 20463); following the addition of BamHI linkers, this fragment was ligated into the BamHI site of pVL941. The resulting clones were screened for proper orientation of the UL9 open reading frame with regard to the polyhedrin promoter.
Plasmid pAPV-UL25 contains the UL25 gene expressed from the HSV-1 ICP6 promoter. The pAPV vector was obtained from S. Weller (29). pAPV was generated by ligating the HindIII/XhoI fragment containing the ICP6 promoter into pUC118 digested with HindIII/SalI. A BamHI-to-BclI fragment from simian virus 40 containing the poly(A) site was then inserted at the BamHI site. This vector contains the inducible ICP6 promoter and the simian virus 40 polyadenylation sequence on either side of a BamHI site into which the BamHI UL25 containing fragment of pAC-UL25 was cloned.
Plasmid pMAL-UL25 was constructed by digesting pAC-UL25 with BamHI and filling in the ends with Klenow enzyme, followed by ligation of a 12-bp XbaI linker and digestion with XbaI. The 1,885-bp UL25-containing fragment was purified and ligated into the XbaI site of pMAL-cR1. Plasmid pMAL-UL6 was constructed by digesting pAC-UL6 with BamHI, followed by ligation of the UL6-containing BamHI fragment into the BamHI site of pMAL-cRI. The resulting clones contained in-frame fusions of the E. coli maltose binding protein (MBP) with the entire UL25 or UL6 open reading frame.
Generation of polyclonal and monoclonal antisera.
Rabbit polyclonal UL25 and UL6 antisera and mouse monoclonal antisera to the UL25 protein were produced by immunizing animals with E. coli-expressed MBP-UL25 or MBP-UL6 fusion protein. The fusion proteins was purified as instructed by the manufacturer (New England Biolabs). Following immunization of rabbits with the fusion protein (100 μg on days 1, 14, and 21) in Freund’s complete adjuvant, serum was collected by standard protocols and used as described in Results. Spleens from mice immunized with the MBP-UL25 fusion protein (100 μg on days 1, 17, and 24) in Freund’s complete adjuvant were used to isolate cell lines expressing monoclonal UL25-specific antibodies following standard protocols, and the antibodies were used as described in Results.
Isolation of UL25 mutant.
Marker transfer of mammalian cells with HSV-1 genomic DNA and cloned DNA fragments was done as described previously (49). Briefly, infectious KUL26ΔZ viral DNA was mixed with a fivefold molar excess of pKUL25NS and transfected into exponentially growing F3 cells. The resulting virus stocks were plated on Vero, F3, and 3A6 cells. Isolates that formed plaques on F3 but not Vero or 3A6 cells were replated on F3 cells in the presence of the chromogenic substrate 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-Gal), and plaques which failed to turn blue were selected and plaque purified three times on F3 cells before stocks were made.
Isolation of UL25-complementing cell line (8-1 cells).
One day prior to transfection, approximately 2 × 106 Vero cells were seeded in 100-mm-diameter culture dishes. Cells were cotransformed with 2 μg of pAPV-UL25 and 0.5 μg of pRSV-neo as described previously (49) and grown in medium containing 1 mg of G418 per ml. After 14 to 21 days, individual G418-resistant colonies were isolated and screened for the ability to support growth of KUL25NS.
Capsid purification.
Vero cells or 8-1 cells were infected with KOS or KUL25NS at a multiplicity of infection (MOI) of 5 PFU per cell; at 24 h postinfection, cells were harvested. Suspension cultures (100 ml) of Sf9 cells were infected with baculovirus recombinants at an MOI of 5 PFU per cell (each virus); at 64 h postinfection, the cells were harvested. Capsid or capsid structures were purified by banding on 20 to 60% sucrose gradients as previously described (50, 51).
Extraction of capsids with GuHCl.
The procedure followed that of Newcomb and Brown (36), with the following modifications. Sucrose gradient-purified capsids were resuspended in TNE buffer (0.5 M NaCl, 1 mM EDTA, 20 mM Tris HCl [pH 7.5]) at a concentration of 1.0 mg/ml at 4°C. guanidine HCl (GuHCl; 6.0 M) in TNE was slowly added to the capsid sample until the final concentration was 2.0 M GuHCl. After incubation for 1 h at 4°C, the extracted capsids were recovered by centrifugation through 200 μl of a 25% sucrose–2 M GuHCl solution made in TNE buffer. Centrifugation was for 1 h at 20,000 rpm in a Beckman SW55 rotor. The pelleted capsids were resuspended in 0.5 ml of TNE and then rebanded on a 20 to 60% sucrose gradient. Capsid bands were identified by light scattering and removed from the gradient with a Pasteur pipette, the sample was diluted 10-fold with TNE, and capsids were pelleted by centrifugation (1 h 20,000 rpm in a Beckman SW40.1 rotor).
Western immunoblotting and transmission EM.
Sf9 cells infected with recombinant baculoviruses were prepared for Western blot analysis or for transmission electron microscopy (EM) (thin sections) as described previously (49).
PFGE.
Pulsed-field gel electrophoresis (PFGE) was performed on a Bio-Rad (Melville, N.Y.) CHEF mapper. One 60-mm-diameter dish of Vero cells or 8-1 cells infected at an MOI of 2 PFU per cell were harvested at various times postinfection. The medium was removed; the cells were washed with phosphate-buffered saline (PBS), scraped into PBS, and pelleted. The cell pellet was resuspended in approximately 300 μl of 55°C 1.0% low-melting-temperature agarose (Bio-Rad) in PBS (without CaCl2 and MgCl2) and cast in three blocks of approximately 100 μl each in casting blocks provided by Bio-Rad. The blocks were removed from the mold and stored at 4°C in 50 mM EDTA (pH 8.0).
Prior to electrophoresis, the blocks were incubated for 20 to 24 h at 37°C in 1.0% laurylsarcosine–0.4 M EDTA (pH 9.0)–protease K (1 mg/ml) at 37°C. Blocks were then washed five times for 15 min each in 50 mM Tris-HCl (pH 7)–1 mM EDTA (TE) at 45°C. The plugs were sealed into the wells of a 1% agarose gel made in 0.5× TBE (1× TBE is 0.089 M Tris [pH 8.0], 0.089 M boric acid, and 0.002 M EDTA [pH 8.0]). The gels were run at 6 V/cm for 22 h at 14°C; the angle was 120° with a pulse time of 50 to 90 s with 0 ramping factor. The gel was stained with ethidium bromide at 0.5 mg/ml for 1 h at room temperature and photographed. The gel was then soaked in acid (0.25 M HCl) for 45 min, denatured (0.6 M NaCl, 0.4 M NaOH) for 30 min, and finally neutralized (1.5 M NaCl, 0.5 M Tris [pH 7.5]) for 30 min. The DNA was transferred to GeneScreen Plus in 10× SSC (1.5 M NaCl, 0.15 M sodium citrate [pH 7.0]) for 16 to 20 h. The blot was prehybridized and hybridized with 32P-labeled BamHI K fragment as described previously (49).
DNase digestion.
Vero cells or 8-1 cells infected at an MOI of 2 PFU per cell for 18 h at 37°C were harvested and embedded in agarose. The agarose plugs were treated in a solution of 150 mM NaCl, 10 mM Tris (pH 7.5), 1.5 mM MgCl2, and 0.2% Nonidet P-40 for 2 to 4 h at 4°C. The plugs were rinsed in reticulocyte standard buffer (RSB; 10 mM Tris-HCl [pH 7.4], 10 mM KCl, 1.5 mM MgCl2) four times at 45°C for 15 min each, and 0.5 ml of RSB was added to each tube along with 5 to 10 μl of DNase I (100 U/μl; Gibco-BRL catalog no. 18047-019) and allowed to digest for 2 to 4 h at 37°C. The RSB solution was removed, and the plugs were treated with cell lysis solution and processed for PFGE as described above.
RESULTS
Isolation of a mutant with an in-frame stop codon in the open reading frame of the UL25 gene.
The 580-amino-acid open reading frame for the UL25 gene extends from nucleotides 48813 (ATG) to 50553 (TAG) in the HSV-1 genome (Fig. 1). Five mRNAs are expressed as 3′-coterminal transcripts from this region of the HSV-1 genome; the three largest transcripts (5.6, 5.4, and 4.2 kb) contain the entire UL25 open reading frame (25). The products of the UL26 and UL26.5 genes are expressed from two smaller transcripts of 2.4 and 1.4 kb, respectively (31). The 6,282-bp EcoRI-SnaBI fragment that encodes the UL25 gene was cloned into a vector and the single NotI site present in this fragment was converted to an SpeI site by insertion of a 14-bp linker (Fig. 1). The SpeI linker contains stop codons in all three reading frames. The NotI site is located at codon 104 of the UL25 open reading frame; therefore, insertion of the SpeI linker would terminate translation of UL25 at this point. The resulting plasmid was linearized and used with KUL26ΔZ viral DNA to cotransfect F3 cells. The F3 cell line harbors an HSV-1 DNA fragment that specifies genes UL25 to UL28, and this cell line has previously been shown to complement the growth of UL26, UL27, and UL28 null viruses (19). The KUL26ΔZ virus is an HSV-1 UL26 null mutant (19) that was constructed by replacing DNA sequences specifying codons 41 through 593 of the UL26 gene with sequences coding for a lacZ reporter gene (Fig. 1). Progeny virus isolated from the transfection of KUL26ΔZ DNA with pKUL25NS were plated on Vero, F3, and 3A6 cells (the latter a cell line that expresses only the UL26 gene). Because pKUL25NS contains all of the sequences deleted from the UL26 gene, homologous recombination between the plasmid and viral sequences would result in the introduction of the SpeI linker into the UL25 gene and replacement of the lacZ sequences with the wild-type UL26 gene. The desired recombinant virus was subsequently screened for its ability to form plaques on F3 cells but not on Vero or 3A6 cells and for the loss of blue plaques when the monolayer was overlaid with Bluo-gal, indicating that the lacZ sequences were gone from the virus. The recombinant virus isolated (designated KUL25NS) was plaque purified three times, and stocks were prepared in F3 cells.
FIG. 1.
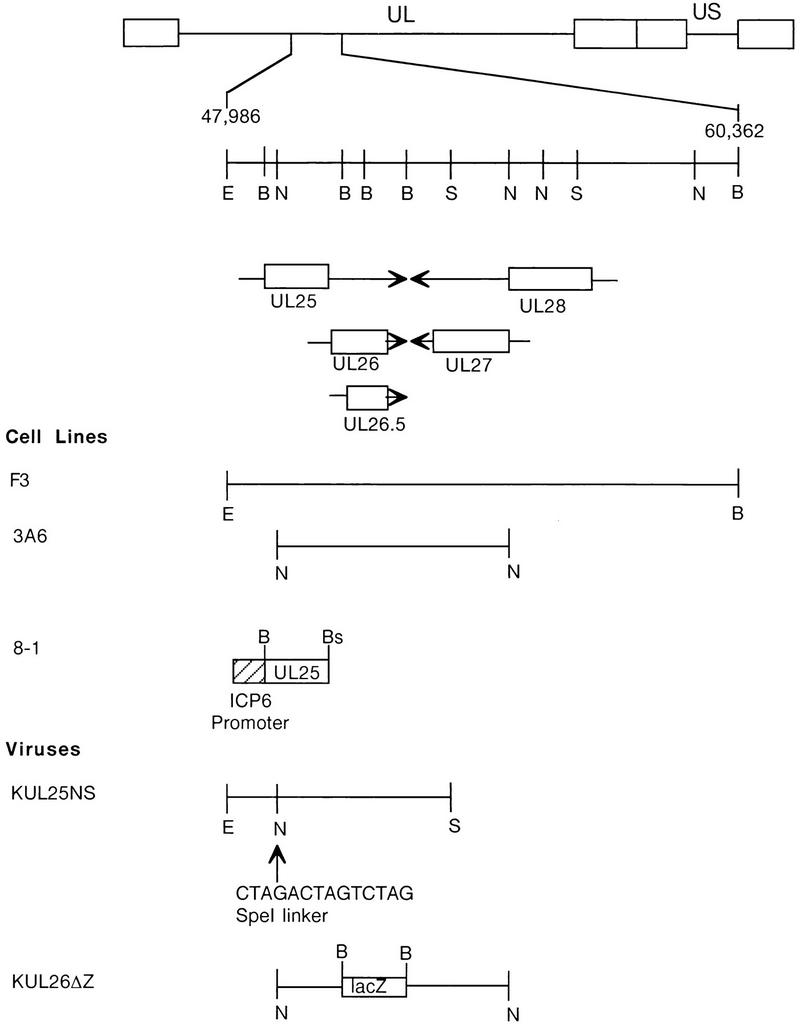
Regions of the HSV-1 genome that were used to isolate transformed cells and recombinant viruses. The HSV-1 genome is shown at the top; UL and US refer to the long and short unique region sequences. On the next line, the EcoRI-to-BamHI region located between nucleotides 47986 and 60362 of the HSV-1 genome is expanded. The locations and directions of transcription of the UL25, UL26, UL26.5, UL27, and UL28 genes are indicated in the next three lines, with the boxed regions representing the open reading frames for these genes. (Cell lines) Regions of the HSV-1 genome contained in recombinant plasmids (see Materials and Methods) used to isolate transformed Vero cell lines F3 (pKEF-B5), 3A6 (pKEF-NotI), and 8-1 (pAPV-UL25). (Viruses) Plasmid pKUL25NS, which contains an SpeI oligomer inserted within the UL25 open reading frame encoding termination codons in all three reading frames, was used to isolate the UL25 mutant KUL25NS. KUL26ΔZ is a UL26 mutant in which a lacZ cassette was inserted in place of UL26 codons 41 through 593 (19). Restriction endonuclease sites: E, EcoRI; B, BamHI; N, NotI; S, SpeI; Bs, BsrI.
To confirm that the KUL25NS virus harbored the SpeI linker, viral DNA was prepared from Vero cells infected with KOS or from Vero cells separately infected with two independent isolates of the KUL25NS mutant. The DNA samples were digested with BamHI and SpeI and subjected to gel electrophoresis and Southern blot analysis (Fig. 2). The blot was probed with a 2,314-bp BamHI U fragment (nucleotides 48634 to 50928) that contains the entire UL25 open reading frame. The presence of the SpeI linker in the UL25 gene of the mutant would result in cleavage of the 2,314-bp BamHI fragment into 1.8- and 0.5-kb fragments. The digests demonstrated that DNA isolated from cells infected with KUL25NS contained the UL25 gene harboring the SpeI linker (Fig. 2; compare lane 1 to lanes 2 and 3).
FIG. 2.
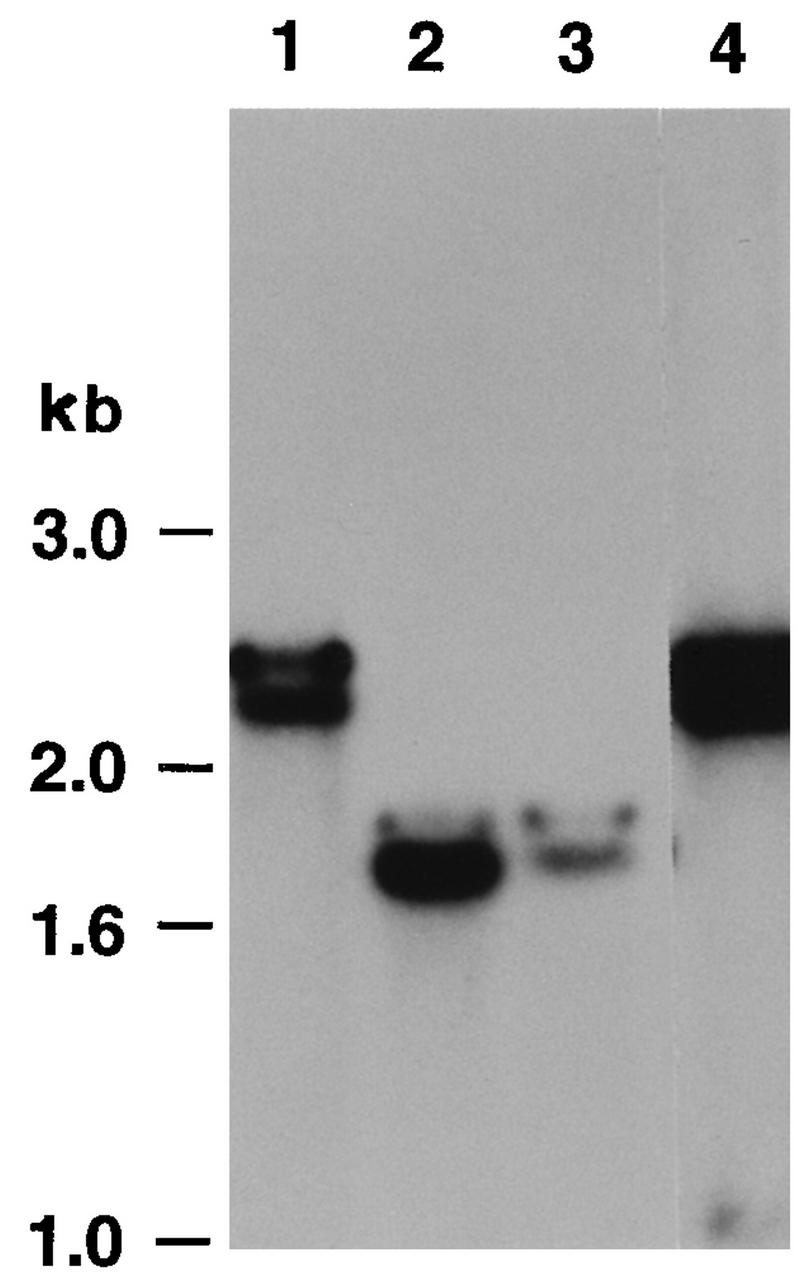
Southern blot analysis of the UL25 null mutant. DNA was extracted from Vero cells infected with KOS (lanes 1), two independent isolates of the KUL25NS mutant (lanes 2 and 3), or a plaque isolate of the KUL25NS mutant that grew on Vero cells (lanes 4). The DNA was digested with BamHI and SpeI, and the resulting restriction fragments were separated on a 0.8% agarose gel and transferred to GeneScreen Plus. The filters were hybridized with a UL25-specific probe (BamHI fragment, nucleotides 48634 to 50791). The 0.5-kb UL25 fragment (lanes 2 and 3) ran off the bottom of the gel.
Phenotypic analysis of KUL25NS.
The plating efficiencies of the KUL25NS and KOS viruses were tested on Vero, 3A6, and F3 cells. The results of this assay are shown in Table 1. As expected, KUL25NS gave rise to plaques on F3 cells but not on Vero or 3A6 cells, while KOS formed plaques on all three cell lines. The low levels of wild-type virus in the mutant stocks is due to recombination that occurs between homologous sequences present in the mutant viral genome and the wild-type sequences in the transformed cell line. To demonstrate that the KUL25NS virus that plaques on Vero cells is due to wild-type virus, we picked a plaque from Vero cells and prepared viral DNA. The DNA was digested with BamHI and SpeI and analyzed by Southern blot analysis. The blot demonstrated that the isolate had lost the SpeI site that had been inserted into the UL25 gene (Fig. 2, lane 4).
TABLE 1.
Plaquing efficiencies of KOS, KUL25NS, KUL26ΔZ, KO82, and GCB on Vero, F3, 8-1, and 3A6 cells
| Cell line | Integrated HSV gene(s) | Virus (PFU/ml)
|
||||
|---|---|---|---|---|---|---|
| KOS (109) | KUL25NS | KUL26ΔZ | KO82 | GCB | ||
| Vero | None | 3.2 | 1.6 × 105 | 2.0 × 105 | 1.5 × 103 | <50 |
| F3 | UL25–UL28 | 1.7 | 1.0 × 108 | 1.3 × 109 | 2.5 × 109 | 4 × 107 |
| 8-1 | UL25 | 4.2 | 5.2 × 108 | 2.0 × 105 | 1.0 × 103 | <50 |
| 3A6 | UL26 | 2.9 | 1.6 × 105 | 1.3 × 109 | 2.0 × 103 | <50 |
As already mentioned, the F3 cell line is capable of complementing UL26, UL27, and UL28 null viruses. To demonstrate that the KUL25NS virus did not contain mutations outside of the UL25 gene, we constructed a second cell line that expressed just the UL25 open reading frame. Vero cells were transformed with pSV2neo (22) and a plasmid, pAPV-UL25, that contains the 1,865-bp UL25 open reading frame under control of the HSV ICP6 promoter. Colonies that were resistant to the drug G418 were harvested and tested for the ability to complement growth of KUL25NS. Cell line 8-1 gave a plaquing efficiency for KUL25NS that was similar to that of the F3 cell line (Table 1). In contrast to F3 cells, 8-1 cells failed to support growth of the UL26 (KUL26ΔZ) null mutant. In addition, 8-1 cells failed to support growth of a UL27 (KO82) or UL28 (GCB) null virus (Table 1). These results demonstrate that KUL25NS fails to grow on Vero cells because of a mutation in the UL25 gene.
To identify the protein product of the HSV-1 UL25 gene, an anti-UL25 rabbit polyclonal antiserum and a mouse monoclonal antibody (2D9) were raised against an E. coli-expressed MBP-UL25 fusion protein (see Materials and Methods). The specificities of the two antisera were tested by Western blot analysis, following sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), of total cell extracts of HSV-infected Vero cells or insect cells infected with a recombinant baculovirus (BAC-UL25) expressing the UL25 open reading frame (Fig. 3). A single immunoreactive band of 60 kDa was readily detected both in HSV-infected Vero cells and in insect cells infected with BAC-UL25. Lower-molecular-weight proteins which probably represented breakdown of the full-length UL25, since neither antisera reacted with proteins from mock-infected cells, were also detected in the BAC-UL25 extracts (Fig. 3B, lane mock). The open reading frame of the UL25 gene predicts a protein of approximately 62 kDa, which is in good agreement with the size of the protein detected with the two antisera. In addition, the molecular mass of the UL25 protein agrees with what Ali et al. have shown in assays using antisera prepared against a UL25–glutathione S-transferase fusion protein (3).
FIG. 3.
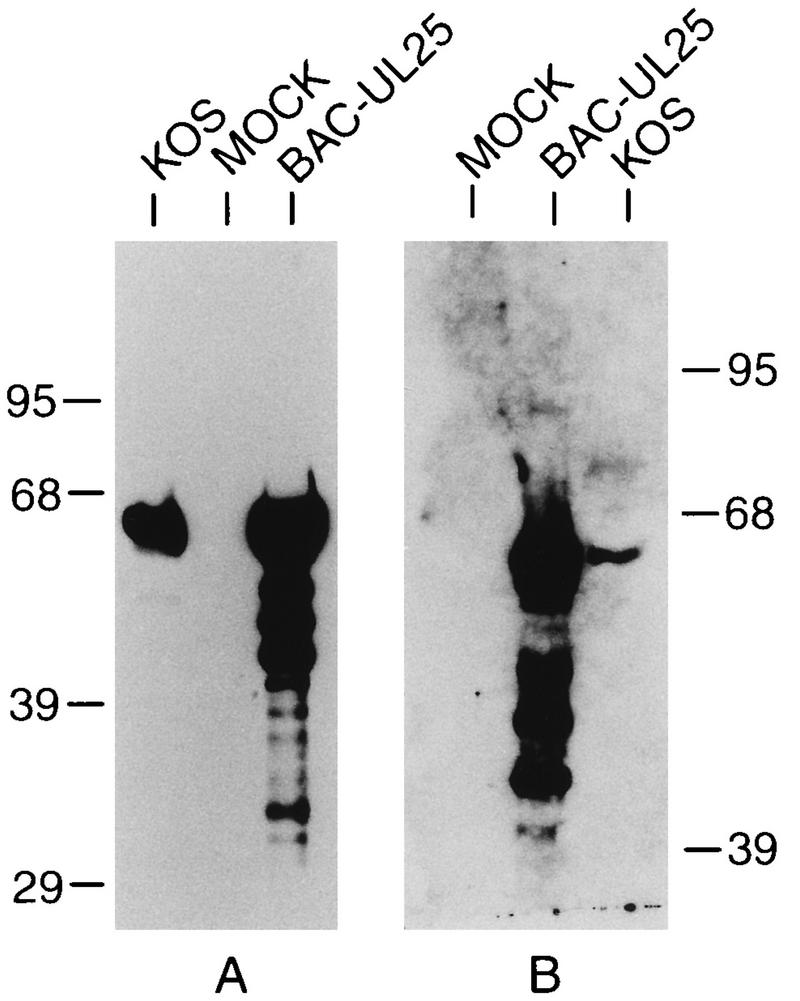
Specificity of UL25 polyclonal and monoclonal antibodies. Lysates of Vero cells infected with HSV-1 (KOS) or insect cells infected with BAC-UL25 were prepared, separated by SDS-PAGE, and immunoblotted with mouse 2D9 (A) or rabbit UL25 (B) antiserum. Mock lysates are from Vero cells (A) and from insect cells (B). The migration of protein size markers (in kilodaltons) is shown at the side of each panel.
Total cell extracts from KOS- or KUL25NS-infected cells were prepared at various times postinfection, and expression of the UL25 gene product was analyzed by using monoclonal antibody 2D9 (Fig. 4). In KOS-infected Vero cells, the 60-kDa UL25 protein was first detected at 6 h postinfection, and the intensity of this band steadily increased at later times postinfection (Fig. 4B). The 60-kDa UL25 protein was not detected in KUL25NS-infected Vero cells out to 20 h postinfection (Fig. 4A). KUL25NS is predicted to encode a 102-amino-acid (12-kDa) protein; however, a band of this size was not detected (data not shown), indicating that either this protein is not recognized by our antisera or the truncated protein is not stable. The time courses of expression of the HSV UL26 and UL26.5 proteins (Fig. 4C and D) were similar in KOS- and KUL25NS-infected cells, demonstrating that the UL25 mutant virus expresses late viral proteins (25).
FIG. 4.

Expression of the UL25 protein in KOS- and KUL25NS-infected Vero cells. Vero cells were infected with KOS (B and D) or KUL25NS (A and C) at an MOI of 5 PFU per cell. At the indicated times (hours) postinfection, cell lysates were prepared, separated by SDS-PAGE, and immunoblotted. Expression of the UL25 protein was detected by using mouse monoclonal antibody 2D9 (A and B). The UL26 and UL26.5 proteins were detected by using mouse monoclonal antibody MCA406 (C and D). The positions of protein standards in order of decreasing molecular mass (200, 95, 68, 39, and 29 kDa) are marked to the left of each panel.
KUL25NS is defective in DNA packaging but still able to cleave replicated viral DNA into unit-length monomers.
Previous studies with two ts mutants (ts1204 and ts1208) whose mutations map to the UL25 gene showed that when these mutants were grown at the nonpermissive temperature they produced few capsids, and the capsids that were made failed to package viral DNA (1). HSV mutants which are defective in DNA packaging also fail to cleave replicated HSV DNA into unit-length monomers (reviewed in reference 27). To determine if the UL25 gene product is required for viral DNA cleavage, cells infected with KUL25NS were examined for the presence of free genomic termini. Total DNA was isolated from Vero or 8-1 cells infected with KOS or KUL25NS at several times postinfection. The DNA was digested with BamHI, and the presence of genomic termini was monitored by Southern blot hybridization using the joint-spanning BamHI K fragment as a probe (Fig. 5). Cleaved viral DNA with free ends will give rise to the terminal BamHI Q and S fragments, whereas concatemeric DNA gives rise only to the joint-spanning BamHI K fragment (Fig. 5). The junction fragment in KOS- or KUL25NS-infected Vero cells was detected as early as 6 h postinfection, and the amount of this fragment steadily increased at later times postinfection, confirming that the UL25 mutant was capable of near wild-type levels of DNA replication (Fig. 5). The Q and S terminal fragments were also detected in KOS- and KUL25NS-infected Vero cells and in KUL25NS-infected 8-1 cells, indicating that the UL25 gene product was not required for cleavage of viral DNA concatemers to generate free viral genomic termini. It should be noted that there appears to be slightly less terminal fragments relative to junction fragments in KUL25NS-infected Vero cells than in KUL25NS-infected 8-1 cells or KOS-infected Vero cells, suggesting that cleavage may be reduced in the absence of the UL25 protein.
FIG. 5.

Processing of viral DNA. Vero cells or 8-1 cells were infected with the indicated virus at an MOI of 5 PFU per cell. At the indicated times (hours) postinfection, total infected cell DNA was isolated, digested with BamHI, and subjected to Southern blot analysis using the BamHI K joint fragment (32P labeled) as a probe. Scanned images of the autoradiograph obtained from the Southern blots are shown. The location of the BamHI K joint fragment and the two end fragments, BamHI-Q and -S, in the HSV-1 genome are shown at the bottom.
The structure of HSV DNA was examined by PFGE to demonstrate that the genomic termini found in KUL25NS-infected Vero cells resulted from the cleavage of DNA concatemers into mature monomeric genomes. PFGE of DNA from HSV-infected cells results in the separation of viral DNA into two bands, one which fails to enter the gel (well DNA) and a second which migrates as 152-kbp genome-length DNA (7, 43, 58). The well DNA represents replicated, concatemeric HSV DNA that contains branched structures that probably arise by recombination during DNA replication (7, 43, 58). Total DNA isolated at 0, 6, 8, 12, 16, 20, and 24 h postinfection from Vero or 8-1 cells infected with either KOS or KUL25NS was subjected to PFGE and subsequently analyzed by Southern blot analysis using the BamHI K fragment as a probe. DNA from KOS-infected Vero cells and either KUL25NS-infected Vero or 8-1 cells contained both well DNA and genome-size HSV DNA (Fig. 6A, C, and E). Both the well DNA and 152-kbp HSV DNA were detected at either 8 h (Fig. 6C) or 12 h (Fig. 6E) postinfection, and the intensity of both bands increased at later times postinfection. The comparable increase in the amount of the 152-kbp HSV DNA detected in KOS-infected Vero cells and both KUL25NS-infected Vero and 8-1 cells demonstrated that the free genomic termini detected in Fig. 5 were the result of cleavage of replicated viral DNA into monomer-size HSV genomes and that cleavage was nearly as efficient as in wild-type-infected cells. These results support the conclusion that the UL25 gene is not required for DNA cleavage.
FIG. 6.
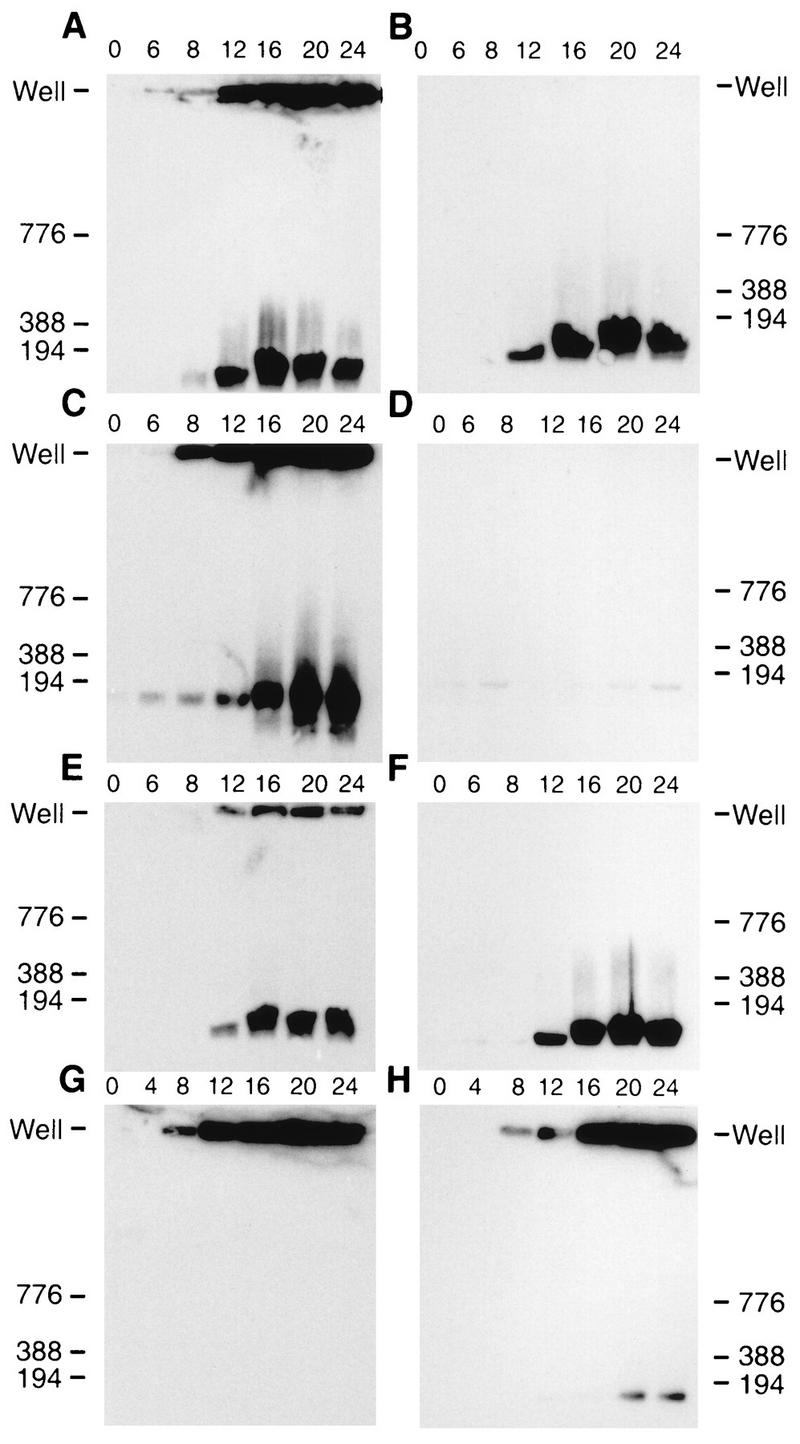
HSV-1 DNA analyzed by PFGE. Infected cells were harvested at the indicated times (hours) postinfection and treated as described in Materials and Methods. Following PFGE, the DNA was transferred to GeneScreen Plus and hybridized by using the HSV-1 BamHI K fragment (32P labeled) as a probe. Scanned images of the autoradiograph obtained from the Southern blots are shown. (A and B) Vero cells infected with KOS; (C and D) Vero cells infected with KUL25NS; (E and F) 8-1 cells infected with KUL25NS; (G) Vero cells infected with GCB; (H) C1 cells infected with GCB. (B, D, and F) Samples were treated with DNase prior to PFGE as described in Materials and Methods. Numbers next to each blot refer to molecular weight markers (kilobase pairs); the position of well DNA is marked for each blot.
An example of a mutant which fails to cleave replicated viral DNA is shown in Fig. 6G and H. DNA from Vero cells infected with a null mutant (GCB) which does not express the product of the HSV UL28 gene (49) contains only well DNA (Fig. 6G). When this virus was grown on a cell line (C1) that expresses the UL28 protein, monomer-size HSV DNA was detected at late times postinfection (Fig. 6H).
To determine if the cleaved DNA generated in KUL25NS-infected cells was being packaged, the agarose plugs used for PFGE were treated with DNase I prior to electrophoresis. DNase I treatment would be expected to degrade all DNA in infected cells except for encapsidated viral DNA. DNase treatment of either KOS-infected Vero cells (Fig. 6B) or KUL25NS-infected 8-1 cells (Fig. 6F) resulted in the elimination of well bands with little if any change in the amount of genome-size viral DNA, indicating that viral monomeric DNA was encapsidated. In contrast, both well and monomeric DNA were degraded when KUL25NS-infected Vero cells were treated with DNase (Fig. 6D). These results demonstrated that the UL25 gene is essential for packaging but not cleavage of viral DNA. This phenotype is unique among previously reported cleavage/packaging mutants and indicates that the defect in KUL25NS is at a stage after cleavage of viral DNA.
Analysis of capsid formation in KUL25NS-infected cells.
Thin sections of KUL25NS-infected Vero and 8-1 cells were examined by transmission EM to determine the presence of A, B, and C capsids. Previous reports have shown that only B capsids were found in Vero cells infected with mutants defective in cleavage and packaging (reviewed in reference 27). Since both A and C capsids result when DNA packaging is completed (C capsid) or aborted (A capsid), the absence of these two capsid forms suggested that DNA packaging does not take place with these mutants. In Vero cells infected with KUL25NS, large numbers of both A and B capsids were detected within infected cell nuclei, while no C capsids were observed either in the nucleus or in the cytoplasm (Fig. 7A and B). When KUL25NS was grown on 8-1 cells, A and B capsids were again seen in the nucleus, but in contrast to Vero cells, enveloped A and C capsids were observed both inside and outside the nucleus, and these enveloped structures were usually associated with membranes (Fig. 7C). The large numbers of A capsids, along with the absence of C capsids, in the nucleus of KUL25NS-infected Vero cells suggests that abortive packaging events had occurred.
FIG. 7.
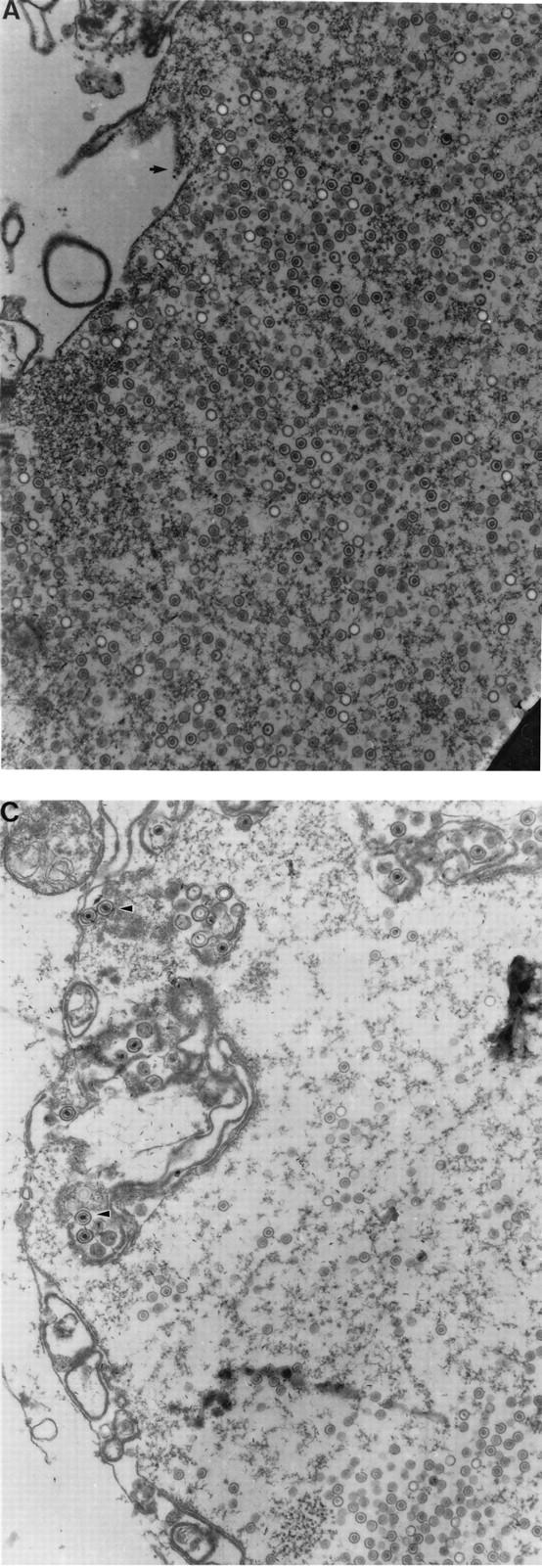
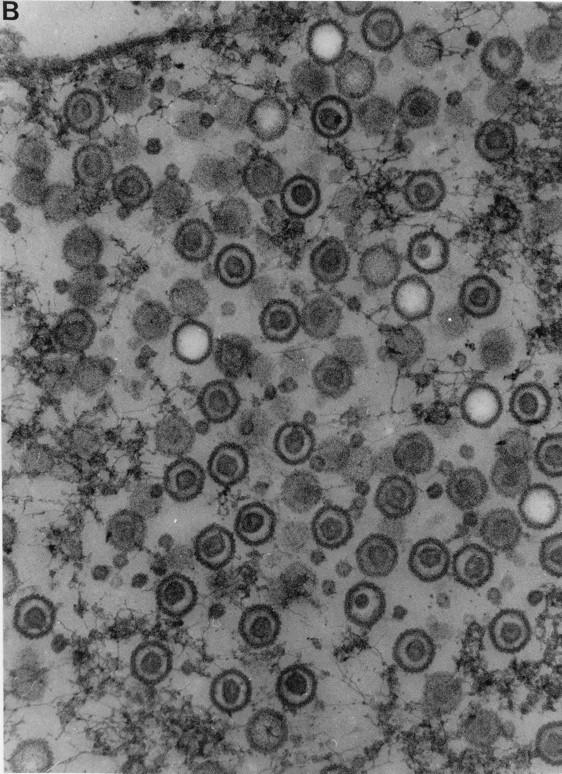
Transmission electron micrographs of thin section preparations of virus-infected cells. Vero cells (A and B) or 8-1 cells infected with KUL25NS were fixed at 16 h postinfection, and thin sections were prepared for EM analysis. In panel A, the arrow points to the region that is enlarged in panel B; in panel C, the arrowheads point to enveloped C capsids. Magnifications: (A and C) ×32,500; (B) ×107,250.
To further examine the different capsid forms made in KUL25NS-infected cells, lysates of Vero or 8-1 cells infected with KOS or KUL25NS were subjected to 20 to 60% sucrose gradient centrifugation, and capsids were viewed as visible light-scattering bands. In KOS-infected Vero cells, all three capsid forms were observed, with the B and C bands being the most prominent (Fig. 8). The A-capsid band is present but is difficult to observe. In lysates of KUL25NS-infected Vero cells, large numbers of A and B capsids were seen in approximately equal ratios and no C capsids were observed (Fig. 8). In KOS- and KUL25NS-infected 8-1 cells, the capsid pattern resembled what was found in KOS-infected Vero cells, with few if any A capsids, large numbers of B capsids, and a small but observable C-capsid band (Fig. 8). The high proportion of A capsids found in KUL25NS-infected Vero cells along with the absence of C capsids supports what was observed by EM analysis and suggests that in the absence of the UL25 protein, abortive packaging takes place.
FIG. 8.

Rate-velocity sedimentation of capsids from Vero or 8-1 cells infected with KOS or KUL25NS. Cells (two T225 flasks) infected with the indicated virus at an MOI of 5 PFU per cell were harvested at 24 h postinfection, and lysates were layered onto 20 to 60% sucrose gradients and centrifuged at 24,000 rpm (SW41 rotor) for 1 h. The positions of A-, B-, and C-capsid bands are indicated.
The UL25 protein is associated with purified capsids.
The fact that the UL25 protein is important for retaining cleaved DNA in capsids suggests that this protein may interact with capsids in vivo. It has been reported that UL25 is associated with purified virions (3). To examine if UL25 is associated with capsids, the sucrose gradients shown in Fig. 8 were fractionated (0.5 ml/fraction), regions which corresponded to where A, B, and C capsids band were isolated, and the capsids were concentrated by centrifugation. The samples were taken up in 50 μl of gel loading buffer, and 10 to 20 μl of each sample was analyzed by Western blot analysis. Capsid proteins VP5, VP19C, and VP23 were detected by using a pool of rabbit polyclonal antisera against the three proteins (Fig. 9A), and separate blots were probed with our UL25 rabbit polyclonal antisera (Fig. 9B). The UL25 protein was found associated with B and C capsids isolated from KOS-infected Vero cells. Surprisingly, even though there were few A capsids, as evidenced by the fact that only small amounts of VP5, VP19C, and VP23 were present in the A-capsid sample isolated from KOS-infected Vero cells (Fig. 9A), a strong UL25 protein band was detected in this sample (Fig. 9B). We attribute this result to the stronger reactivity of the UL25 antisera than of the VP5, VP19C, and VP23 antibodies. UL25 was not found to be associated with A or B capsids isolated from KUL25NS-infected Vero cells, and since C capsids were not made in these cells, no capsid proteins or UL25 was detected in this region of the gradient. The B and C capsids isolated from KUL25NS-infected 8-1 cells contained UL25, demonstrating that the UL25 protein expressed from these cells was incorporated into capsids (Fig. 9). Western analysis of the A-capsid region isolated from the gradient of KUL25NS-infected 8-1 cells showed that no capsid proteins (VP5, VP19C, and VP23) or UL25 were present (data not shown).
FIG. 9.

Analysis of virus capsids. A, B, and C capsids isolated from the sucrose gradients shown in Fig. 8 were run on SDS–12.5% polyacrylamide gels and immunoblotted with antisera NC1, NC2, and NC5 (A), which detect capsid proteins VP5, VP19C, and VP23, respectively, or with a UL25 polyclonal antiserum (B). Positions of the capsid proteins (A) and the UL25 protein (B) are indicated. The migration of protein size markers (in kilodaltons) is shown at the side of each panel.
The above results indicated that UL25 was a minor capsid protein, since detection of this capsid-associated protein required Western blot analysis. This contrasts with capsid proteins such as VP5, VP19C, and VP23, which can be detected on gels by Coomassie blue staining. Therefore, the above experiment could not rule out that the small quantity of UL25 found in purified capsid preparations was due to this protein smearing through the sucrose gradient as has been observed with the tegument protein VP16 (38). A model for HSV-1 capsid assembly has previously been described for insect cells coinfected with recombinant baculoviruses expressing the six HSV-1 capsid proteins (48, 50, 51). This system was used to determine if UL25 would associate with capsids in the absence of tegument proteins. Sf9 cells were infected with a mixture of recombinant baculoviruses expressing the UL18, UL19, UL26, UL26.5, UL35, and UL38 genes alone or along with baculoviruses expressing the HSV UL25, UL6, and UL9 genes. Cells were harvested at 64 h postinfection, and cell extracts were layered onto 20 to 60% sucrose gradients. Following sedimentation, the B-capsid band was harvested from the sucrose gradient. The protein composition of the banded B capsid was then determined by Western blot analysis (Fig. 10). Separate blots were probed for capsid proteins VP5, VP19C, and VP23 (Fig. 10A), scaffold proteins VP21 and VP22a (Fig. 10B), UL25 (Fig. 10C), UL6 (Fig. 10D), and UL9 (Fig. 10E), using antisera specific for each protein. Baculoviruses expressing the UL6 and UL9 proteins were included in these experiments as positive (UL6) and negative (UL9) controls since UL6 has previously been shown to be a minor capsid protein (3) whereas UL9 should not be found associated with capsids. Both UL25 (Fig. 10C, lanes 6 and 9) and UL6 (Fig. 10D, lanes 6 and 9) were detected in B capsids isolated from KOS-infected Vero cells and in capsids assembled in insect cells. In contrast, UL9 (Fig. 10E, lanes 6 and 9) was not found in capsids assembled in either Vero cells or insect cells. Surprisingly, UL25 (Fig. 10C, lane 7) and UL6 (Fig. 10D, lane 7) but not UL9 (Fig. 10E, lane 7) were found to band in the same region of the sucrose gradient as B capsids when these proteins were expressed in insect cells in the absence of HSV capsid proteins. This unexpected observation suggested that UL25 and UL6 form insoluble aggregates when overexpressed in insect cells and that these aggregates purify with B capsids. It should be noted that we have analyzed the distribution of UL25 across a sucrose gradient of HSV-infected Vero cells and found that as with the baculovirus samples, the UL25 protein is found from the top to the bottom of the gradient, with slightly more UL25 associated with the region where capsid bands are found (data not shown).
FIG. 10.

Protein composition of HSV particles made in KOS-infected Vero cells or Sf9 cells infected with recombinant baculoviruses expressing HSV proteins. Cells were harvested at 12 h (Vero cells) or 64 h (Sf9 cells) postinfection, and cell extracts were loaded onto 20 to 60% sucrose gradients. Following centrifugation, B capsids were harvested. If there was no visible band on the gradient, then the region of the gradient where B capsids should band was harvested. A portion of each sample was removed for Western analysis, and the remainder of the sample was treated with 2 M GuHCl as described in Materials and Methods. Following guanidine treatment, particles were purified by banding on a 20 to 60% sucrose gradients. Purified particles before (lanes 6 to 10) and after (lanes 1 to 5) GuHCl treatment were separated on SDS–12.5% polyacrylamide gels and immunoblotted. The blots were probed with antisera NC1, NC2, and NC5 (A), antiserum MCA406 (B), UL25 polyclonal antibody (C), UL6 polyclonal antibody (D), and UL9 monoclonal antibody (E). Samples include particles isolated from Vero cells infected with KOS (lanes 1 and 6), particles isolated from Sf9 cells infected with a mixture of recombinant baculoviruses expressing the six HSV-1 capsid proteins alone (lanes 5 and 10) or along with recombinant baculoviruses expressing the UL6, UL9, and UL25 proteins (lanes 4 and 9), and particles isolated from Sf9 cells infected with a single recombinant baculovirus expressing UL25 (A to C and E, lanes 2 and 7), UL9 (A to E, lanes 3 and 8), or UL6 (D, lanes 2 and 7). (C to E) Lane E contains total cell lysate of Sf9 cells infected with recombinant baculovirus expressing UL25 (C), UL6 (D), and UL9 (E). The position of the capsid proteins VP5, VP19C, and VP23 (A), scaffold proteins VP21 and VP22a (B), UL25 protein (C), UL6 protein (D), and UL9 protein (E) are indicated.
To determine whether UL25 and UL6 were stably incorporated into capsids made in insect cells or if they were simply aggregates that copurified with B capsids, we treated the samples with GuHCl (see Materials and Methods). Treatment of purified capsids with 2 M GuHCl has been shown to result in selective removal of some capsid proteins without altering the icosahedral capsid shell (36). Purified HSV B capsids treated with 2 M GuHCl remove VP21, VP22a, VP24, and VP26 (36). Patel et al. (38) have shown that UL6 remains stably associated with HSV B capsids treated with 2 M GuHCl. As shown in Fig. 10B, proteins VP21 and VP22a were completely removed from B capsids made in Vero (lane 1) or insect (lanes 4 and 5) cells, while proteins VP5, VP19C, and VP23 remained associated with capsids (Fig. 10A, lanes 1, 4, and 5). Gradient-purified B capsids isolated from Vero cells (Fig. 10C and D, lanes 1) or from insect cells (Fig. 10C and D, lanes 4) that had been extracted with 2 M GuHCl were found to retain UL25 and UL6 but not UL9 (Fig. 10E, lane 4). In contrast, treating the region that corresponded to where B capsids would band that was isolated from gradients of insect cells infected with baculoviruses expressing UL25 or UL6 alone eliminated these proteins (Fig. 10C and D, lanes 2). These data indicate that the UL25 and UL6 proteins are tightly associated with capsids and that their presence is not the result of protein aggregates that happen to purify with B capsids on sucrose gradients. In addition, these results demonstrate that both UL25 and UL6 will associate with capsids in the absence of tegument or envelope proteins.
DISCUSSION
Studies with ts mutants whose mutations map within HSV-1 genes UL6, UL15, UL25, UL28, UL32, and UL33 have shown that these six genes are essential for DNA cleavage and packaging and in production of A and C capsids. To further investigate the role of these six genes in cleavage and packaging, additional mutants with defined lesions in these genes have been constructed. To isolate these mutants, complementing cell lines that could be used to propagate viruses containing lethal mutations in these genes were constructed. HSV-1 mutants which fail to express the UL6, UL15, and UL28 proteins have been described elsewhere (6, 37, 49, 56). The results of studies with these null mutants support what was found with ts mutants, since these null viruses did not cleave or package replicated viral DNA and only B capsids were found in cells infected with these mutants. These results are consistent with the hypothesis that the DNA cleavage/packaging complex contains both intact capsids plus the cleavage/packaging proteins. Studies with mutants that fail to assemble capsids due to the deletion of an essential capsid protein support this model, since these mutants fail to cleave replicated viral DNA (18).
Expression of UL25 protein was first detected at 6 h postinfection, and the expression levels were found to increase out to 20 h postinfection (Fig. 4B). The pattern of expression of UL25 was similar to that of the UL26 and UL26.5 proteins (Fig. 4C and D), indicating that UL25 is expressed with late kinetics (Fig. 4). The presence of nearly identical patterns of expression of the UL26 and UL26.5 proteins in KUL25NS-infected Vero cells (Fig. 4C) and in KOS-infected Vero cells (Fig. 4D) demonstrates that the UL25 mutant enters cells and replicates with near wild-type kinetics. Therefore, it does not appear that the UL25 protein is required for virus penetration as was found with the UL25 ts mutant (1).
Ali et al. (3) have shown that the UL25 protein is associated with purified HSV virions. In this study, we show that the UL25 protein is stably associated with all three types of capsids. To discount the possibility that the UL25 protein was a tegument protein that nonspecifically associates with capsids, we showed that treatment of HSV-1 B capsids with 2 M GuHCl did not remove UL25. In addition, the UL25 protein was found to be stably associated with B capsids made in insect cells, and 2 M GuHCl treatment of these capsids did not remove the UL25 protein. Taken, together these observations strongly indicate that the UL25 protein is a minor component of HSV-1 capsids.
The one surprising observation that resulted from these studies was that cleaved/unpackaged genome-size DNA was present in Vero cells infected with the UL25 null mutant. As described above, this is a novel phenotype since all previously described mutants within this group fail to cleave and package DNA into preformed B capsids. The presence of nearly equal amounts of both A and B capsids and no C capsids in Vero cell infected with the UL25 null virus suggested that the cleaved DNA may have resulted from an abortive packaging event. In Vero cells infected with the UL25 null virus, replicated DNA is cleaved into genome-size molecules but the DNA fails to be packaged in capsids in the absence of the UL25 protein. The abortive packaging event resulted in the loss of the scaffold protein and generation of an empty A capsid. The cleavage of viral DNA in KUL25NS-infected Vero cells appears to be nearly as efficient as in KOS-infected Vero cells or KUL25NS-infected 8-1 cells, as evidenced by similar levels of genome-size DNA (Fig. 6). Therefore, cleavage and packaging occur at near wild-type levels with the UL25 null virus. The absence of DNase-resistant DNA and the abundance of A capsids suggest that UL25 may function to retain DNA in the capsid following the cleavage event.
A defect in the release of DNA-containing capsids from the nucleus while retaining the ability to cleave DNA has been shown with a UL12 null virus (33). As mentioned earlier, the UL12 gene is not essential, but the titers of mutants lacking this protein are reduced over 100-fold. Mutants which fail to express the UL12 protein make an overabundance of A capsids. Significant amounts of DNase-resistant DNA are found in the nucleus, but very little if any is found in the cytoplasm. Therefore, it appears that the DNA-containing capsids made in the absence of the UL12 protein are unstable. In contrast, no DNA-containing capsids are found in the absence of the UL25 protein, suggesting that the DNA is only transiently associated with capsids and further supporting a role for UL25 in maintaining DNA in capsids following cleavage.
Since cleavage and packaging of HSV DNA require fully formed capsids, some cleavage/packaging proteins may interact with capsids. It has been reported that UL6 is found in all three capsid forms whereas the UL15 protein is present in B capsids (38, 57). In this report, we show that UL25 is stably associated with A, B, and C capsids. Site-specific cleavage of the concatemers is found at unique sites within a sequences which are present at both genomic termini and at the internal repeated sequences (47, 53, 56). The a sequences appear to specify when a genome equivalent has entered the capsid and where the viral DNA should be cleaved. The a sequences could serve as a site to which cleavage/packaging proteins such as UL25 bind in retaining DNA in the capsid.
The process of HSV-1 capsid assembly and DNA packaging appears to be analogous to the pathway found with double-stranded DNA (dsDNA) bacteriophages such as T4, P22, and lambda (8, 9, 12, 13, 23, 24, 41, 42). The pathway in both HSV-1 and dsDNA bacteriophages initially involves the assembly of a spherical, unstable procapsid which then matures to the stable angular capsid (13, 23, 35, 41, 52). The bacteriophage and HSV procapsid lack DNA but contain scaffolding protein. As with HSV, bacteriophage DNA is synthesized as head-to-tail concatemers that are cleaved to monomers and packaged into capsids, with the loss of the scaffolding protein. The prohead is the capsid in which DNA packaging is initiated in the dsDNA bacteriophages. Transfer of DNA into bacteriophage proheads requires a protein complex consisting of the terminase protein, the portal protein, and accessory proteins. It has been speculated that the UL15 protein may function as a terminase, based on its homology with the large subunit of T4 terminase (16). In bacteriophages, the terminase is only transiently associated with the capsid, while the other proteins required for packaging (including the portal protein) are mainly structural components of the capsid. The substrates for DNA packaging are replication-generated concatemers, and the DNA is cleaved into genome-size molecules at the time of packaging. The portal protein is found at one vertex (site where phage tail is attached) of the capsid arranged in a single, ring-shaped dodecamer (9, 41). The DNA has been postulated to enter the procapsid through the hole in the center of the portal protein dodecamer. Procapsids missing the portal protein do not package DNA. There is a mechanism which determines when the procapsid is filled with DNA and activates the nucleolytic cleavage and releases the internalized DNA from the concatemer. Genetic studies have linked the portal protein to this cleavage event (12). Following cleavage, the DNA is unstably packaged and unless additional proteins are added to the portal vertex the DNA can come back out of the capsid (30). Although there is no obvious equivalent of the portal vertex in the HSV procapsid, some of the minor capsid proteins (such as the UL6 protein) may serve this function. The function of the UL25 protein appears to be similar to that of the proteins added after DNA is packaged in the dsDNA bacteriophages that aid in retaining the DNA in the capsid.
In summary, we have described a system for studying the role of the UL25 gene product in HSV-1 morphogenesis. UL25-transformed Vero cell lines which allow for the isolation and preparation of mutant virus stocks have been established. The availability of the complementing cell lines will allow for the isolation of additional UL25 mutants containing both deletion and nonsense mutations. Characterization of these mutants should be useful in understanding the role of the UL25 protein in packaging of viral DNA.
REFERENCES
- 1.Addison C, Rixon F J, Palfreyman J W, O’Hara M, Preston V G. Characterization of a herpes simplex virus type 1 mutant which has a temperature-sensitive defect in penetration of cells and assembly of capsids. Virology. 1984;138:246–259. doi: 10.1016/0042-6822(84)90349-0. [DOI] [PubMed] [Google Scholar]
- 2.Addison C, Rixon F J, Preston V G. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J Gen Virol. 1990;71:2377–2384. doi: 10.1099/0022-1317-71-10-2377. [DOI] [PubMed] [Google Scholar]
- 3.Ali M A, Forghani B, Cantin E M. Characterization of an essential HSV-1 protein encoded by the UL25 gene reported to be involved in virus penetration and capsid assembly. Virology. 1996;216:278–283. doi: 10.1006/viro.1996.0061. [DOI] [PubMed] [Google Scholar]
- 4.Al-Kobaisi M F, Rixon F J, McDougall I, Preston V G. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology. 1991;180:380–388. doi: 10.1016/0042-6822(91)90043-b. [DOI] [PubMed] [Google Scholar]
- 5.Baines J D, Poon A P W, Rovnak J, Roizman B. The herpes simplex virus 1 UL15 gene encodes two proteins and is required for cleavage of genomic viral DNA. J Virol. 1994;68:8118–8124. doi: 10.1128/jvi.68.12.8118-8124.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baines J D, Cunningham C, Nalwanga D, Davison A. The UL15 gene of herpes simplex virus type 1 contains within its second exon a novel open reading frame that is translated in frame with the UL15 gene product. J Virol. 1997;71:2666–2673. doi: 10.1128/jvi.71.4.2666-2673.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bataille D, Epstein A. Herpes simplex virus replicative concatemers contain L components in inverted orientation. Virology. 1994;203:384–388. doi: 10.1006/viro.1994.1498. [DOI] [PubMed] [Google Scholar]
- 8.Bjornsti M, Reilly B E, Anderson D L. Morphogenesis of bacteriophage phi-29 of Bacillus subtilis: oriented and quantized in vitro packaging of DNA protein gp3. J Virol. 1983;45:383–396. doi: 10.1128/jvi.45.1.383-396.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black L W. DNA packaging in dsDNA bacteriophages. In: Calendar R, editor. The bacteriophages. Vol. 2. New York, N.Y: Plenum Press; 1988. pp. 321–373. [Google Scholar]
- 10.Bronstein J C, Weller S K, Weber P C. The product of the UL12.5 gene of herpes simplex virus type 1 is a capsid-associated nuclease. J Virol. 1997;71:3039–3047. doi: 10.1128/jvi.71.4.3039-3047.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai W, Person S, Warner S C, Zhou J, DeLuca N A. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J Virol. 1987;61:714–721. doi: 10.1128/jvi.61.3.714-721.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casjens S, Hatfull G, Hendrix R. Evolution of dsDNA tailed-bacteriophage genomes. Semin Virol. 1992;3:383–397. [Google Scholar]
- 13.Casjens S, Hendrix R. Control mechanisms in dsDNA bacteriophage assembly. In: Calendar R, editor. The bacteriophages. Vol. 2. New York, N.Y: Plenum Press; 1988. pp. 15–91. [Google Scholar]
- 14.Cohen G H, Ponce de Leon M, Diggelmann H, Lawrence W C, Vernon S K, Eisenberg R J. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J Virol. 1980;34:521–531. doi: 10.1128/jvi.34.2.521-531.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dargan D J. The structure and assembly of herpes-viruses. In: Harris J R, Horne R W, editors. Electron microscopy of proteins. London, England: Academic Press; 1986. pp. 359–437. [Google Scholar]
- 16.Davison A J. Channel catfish virus: a new type of herpesvirus. Virology. 1992;186:9–14. doi: 10.1016/0042-6822(92)90056-u. [DOI] [PubMed] [Google Scholar]
- 17.Deiss L P, Chou J, Frenkel N. Functional domains within the a sequence involved in the cleavage-packaging of herpes simplex virus DNA. J Virol. 1986;59:605–618. doi: 10.1128/jvi.59.3.605-618.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desai P, Deluca N A, Glorioso J C, Person S. Mutations of herpes simplex type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J Virol. 1993;67:1357–1364. doi: 10.1128/jvi.67.3.1357-1364.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desai P, Watkins S C, Person S. The size and symmetry of B capsids of herpes simplex virus type 1 are determined by the gene products of the UL26 open reading frame. J Virol. 1994;68:5365–5374. doi: 10.1128/jvi.68.9.5365-5374.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson W, Roizman B. Proteins specified by herpes simplex virus. VIII. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J Virol. 1972;10:1044–1052. doi: 10.1128/jvi.10.5.1044-1052.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldin A L, Sandri-Goldin R M, Levine M, Glorioso J C. Cloning of herpes simplex virus type 1 sequences representing the whole genome. J Virol. 1981;38:50–58. doi: 10.1128/jvi.38.1.50-58.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gorman C R, Padmanabhan R, Howard B H. High efficiency DNA-mediated transformation of primate cells. Science. 1983;221:551–553. doi: 10.1126/science.6306768. [DOI] [PubMed] [Google Scholar]
- 23.Hendrix R W. Shape determination in virus assembly: the bacteriophage example. In: Casjens S, editor. Virus structure and assembly. Boston, Mass: Jones and Bartlett Publishers, Inc.; 1985. pp. 169–203. [Google Scholar]
- 24.Hohn B. DNA sequences necessary for packaging of bacteriophage lambda DNA. Proc Natl Acad Sci USA. 1983;80:7456–7460. doi: 10.1073/pnas.80.24.7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holland L E, Sandri-Goldin R M, Goldin A L, Glorioso J C, Levine M. Transcriptional and genetic analysis of the herpes simplex virus type 1 genome: coordinates 0.29 to 0.45. J Virol. 1984;49:947–959. doi: 10.1128/jvi.49.3.947-959.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holland T C, Marlin S D, Levine M, Glorioso J. Antigenic variants of herpes simplex virus selected with glycoprotein-specific monoclonal antibodies. J Virol. 1983;45:672–682. doi: 10.1128/jvi.45.2.672-682.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Homa F L, Brown J C. Capsid assembly and DNA packaging in herpes simplex virus. Rev Med Virol. 1997;7:107–122. doi: 10.1002/(sici)1099-1654(199707)7:2<107::aid-rmv191>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 28.Lamberti C, Weller S K. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology. 1996;226:403–407. doi: 10.1006/viro.1996.0668. [DOI] [PubMed] [Google Scholar]
- 29.Lamberti, C., and S. K. Weller. The herpes simplex virus type 1 UL32 protein is involved in capsid localization to replication compartments. Submitted for publication. [DOI] [PMC free article] [PubMed]
- 30.Lenk E, Casjens S, Weeks J, King J. Intracellular visualization of precursor capsids in phage P22 mutant infected cells. Virology. 1975;68:182–199. doi: 10.1016/0042-6822(75)90160-9. [DOI] [PubMed] [Google Scholar]
- 31.Liu F, Roizman B. The herpes simplex virus 1 gene encoding a protease also contains within its coding domain the gene encoding the more abundant substrate. J Virol. 1991;65:5149–5156. doi: 10.1128/jvi.65.10.5149-5156.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malik A K, Shao L, Shanley J D, Weller S K. Intracellular localization of the herpes simplex virus type-1 origin binding protein, UL9. Virology. 1996;224:380–389. doi: 10.1006/viro.1996.0545. [DOI] [PubMed] [Google Scholar]
- 33.Martinez R, Sarisky R T, Weber P C, Weller S K. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J Virol. 1996;70:2075–2085. doi: 10.1128/jvi.70.4.2075-2085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGeoch D J, Dalrymple M A, Davison A J, Dolan A, Frame M C, McNab D, Perry L J, Scott J E, Taylor P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol. 1988;69:1531–1574. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- 35.Newcomb W W, Homa F L, Thomsen D R, Booy F P, Trus B L, Steven A C, Spencer J V, Brown J C. Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid assembly. J Mol Biol. 1996;263:432–446. doi: 10.1006/jmbi.1996.0587. [DOI] [PubMed] [Google Scholar]
- 36.Newcomb W W, Brown J C. Structure of the herpes simplex virus capsid: effects of extraction with guanidine hydrochloride and partial reconstitution of extracted capsids. J Virol. 1991;65:613–620. doi: 10.1128/jvi.65.2.613-620.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel A H, Rixon F J, Cunningham C, Davison A J. Isolation and characterization of herpes simplex virus type-1 mutants defective in the UL6 gene. Virology. 1996;217:111–123. doi: 10.1006/viro.1996.0098. [DOI] [PubMed] [Google Scholar]
- 38.Patel A H, Maclean J B. The product of the UL6 gene of herpes simplex virus type 1 is associated with virus capsids. Virology. 1995;206:465–478. doi: 10.1016/s0042-6822(95)80062-x. [DOI] [PubMed] [Google Scholar]
- 39.Perdue M L, Cohen J C, Randall C C, O’Callaghan D J. Biochemical studies of the maturation of herpes virus nucleocapsid species. Virology. 1976;74:194–208. [PubMed] [Google Scholar]
- 40.Poon A P, Roizman B. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J Virol. 1993;67:4497–4503. doi: 10.1128/jvi.67.8.4497-4503.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prevelige P E, King J. Assembly of bacteriophage P22: a model for ds-DNA virus assembly. Prog Med Virol. 1993;40:206–221. [PubMed] [Google Scholar]
- 42.Prevelige P E, Thomas D, King J. Nucleation and growth phases in the polymerization of coat and scaffolding subunits into icosahedral procapsid shells. Biophys J. 1993;64:824–835. doi: 10.1016/S0006-3495(93)81443-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Severini A A, Morgan R, Tovell D R, Tyrrell D L J. Study of the structure of replicative intermediates of HSV-1 DNA by pulsed-field gel electrophoresis. Virology. 1994;200:428–435. doi: 10.1006/viro.1994.1206. [DOI] [PubMed] [Google Scholar]
- 44.Sherman G, Bachenheimer S L. DNA processing in temperature-sensitive morphogenic mutants of HSV-1. Virology. 1987;158:427–430. doi: 10.1016/0042-6822(87)90214-5. [DOI] [PubMed] [Google Scholar]
- 45.Sherman G, Bachenheimer S L. Characterization of intranuclear capsids made by ts morphogenic mutants of HSV-1. Virology. 1988;163:471–480. doi: 10.1016/0042-6822(88)90288-7. [DOI] [PubMed] [Google Scholar]
- 46.Steven A C, Spear P G. Herpesvirus capsid assembly and envelopment. In: Chiu W, Burnett R M, Garcea R L, editors. Structural biology of viruses. Oxford, England: Oxford University Press; 1997. pp. 312–351. [Google Scholar]
- 47.Stow N D, McMonagle E C, Davison A J. Fragments from both termini of the herpes simplex virus type 1 genome contain signals required for the encapsidation of viral DNA. Nucleic Acids Res. 1983;11:8205–8220. doi: 10.1093/nar/11.23.8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tatman J D, Preston V G, Nicholson P, Elliott R M, Rixon F J. Assembly of herpes simplex virus type 1 capsids using a panel of recombinant baculoviruses. J Gen Virol. 1994;75:1101–1113. doi: 10.1099/0022-1317-75-5-1101. [DOI] [PubMed] [Google Scholar]
- 49.Tengelsen L A, Pedersen N E, Shaver P A, Wathen M W, Homa F L. Herpes simplex virus type 1 DNA cleavage and encapsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J Virol. 1993;67:3470–3480. doi: 10.1128/jvi.67.6.3470-3480.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomsen D R, Roof L L, Homa F L. Assembly of herpes simplex virus (HSV) intermediate capsids in insect cells infected with recombinant baculoviruses expressing HSV capsid proteins. J Virol. 1994;68:2442–2457. doi: 10.1128/jvi.68.4.2442-2457.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomsen D R, Newcomb W W, Brown J C, Homa F L. Assembly of the herpes simplex virus capsid: requirement for the carboxyl-terminal twenty-five amino acids of the proteins encoded by the UL26 and UL26.5 genes. J Virol. 1995;69:3690–3703. doi: 10.1128/jvi.69.6.3690-3703.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trus B L, Booy F B, Newcomb W W, Brown J C, Homa F L, Thomsen D R, Steven A C. The herpes simplex virus procapsid: structure, conformational changes upon maturation, and roles of the triplex proteins VP19C and VP23 in assembly. J Mol Biol. 1996;263:447–462. doi: 10.1016/s0022-2836(96)80018-0. [DOI] [PubMed] [Google Scholar]
- 53.Varmuza S L, Smiley J R. Signals for site-specific cleavage of HSV DNA: maturation involves two separate cleavage events at sites distal to the recognition sequences. Cell. 1985;41:793–802. doi: 10.1016/s0092-8674(85)80060-x. [DOI] [PubMed] [Google Scholar]
- 54.Vlazny D A, Kwong K, Frenkel N. Site-specific cleavage/packaging of herpes simplex virus DNA and the selective maturation of nucleocapsids containing full-length viral DNA. Proc Natl Acad Sci USA. 1982;79:1423–1427. doi: 10.1073/pnas.79.5.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weller S K, Carmichael E P, Aschman D P, Goldstein D J, Schaffer P A. Genetic and phenotypic characterization of mutants in four essential genes that map to the left half of HSV-1 UL DNA. Virology. 1987;161:198–210. doi: 10.1016/0042-6822(87)90186-3. [DOI] [PubMed] [Google Scholar]
- 56.Yu D, Sheaffer A K, Tenney D, Weller S K. Characterization of ICP6::lacZ insertion mutants of the UL15 gene of herpes simplex virus type 1 reveals two proteins translated independently. J Virol. 1997;71:2656–2665. doi: 10.1128/jvi.71.4.2656-2665.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu, D., and S. K. Weller. Personal communication.
- 58.Zhang X, Efstathiou S, Simmons A. Identification of novel herpes simplex virus replicative intermediates by field inversion gel electrophoresis: implications for viral DNA amplification strategies. Virology. 1994;202:530–539. doi: 10.1006/viro.1994.1375. [DOI] [PubMed] [Google Scholar]