

Abstract
A recombinant retrovirus, termed MoFe2-MuLV, was constructed in which the U3 region of T-lymphomagenic Moloney murine leukemia virus (Mo-MuLV) was replaced by that of FeLV-945, a provirus of unique long terminal repeat (LTR) structure identified only in non-T-cell, non-B-cell lymphomas of the domestic cat. The LTR of FeLV-945 is unusual in that it contains only a single copy of the transcriptional enhancer followed 25 bp downstream by a 21-bp sequence in triplicate in tandem. Infectivity of MoFe2-MuLV was demonstrated in vitro in SC-1 cells and in vivo in neonatal NIH-Swiss mice. Tumors occurred in MoFe2-MuLV-infected animals following a latency period of 4 to 10 months (average, 6 months). The results of Southern blot analysis of the T-cell receptor beta locus demonstrated that all tumors were lymphomas of T-cell origin. MoFe2-MuLV LTRs were amplified by PCR from tumor DNA and were characterized by nucleotide sequence analysis. LTRs from the tumors that occurred with relatively shorter latency predominantly retained the original MoFe2-MuLV sequence intact and unaltered. Tumors that occurred with relatively longer latency contained LTRs that also retained the 21-bp sequence triplication characteristic of the original virus but had acquired various duplications of enhancer sequences. The repeated identification of enhancer duplications in late-appearing tumors suggests that the duplication affords a selective advantage, although apparently not in the efficient induction of T-cell lymphoma. Proto-oncogenes known to be targets of insertional mutagenesis in the majority of Mo-MuLV-induced tumors or in feline non-T-cell, non-B-cell lymphomas were shown not to be rearranged in any tumor examined. Mink cell focus-inducing (MCF) proviral DNA was readily detectable in some, but not all, tumors. The presence or absence of MCF did not correlate with the kinetics of tumor induction. These studies indicate that the single-enhancer, triplication-containing FeLV LTR, typical of non-T-cell, non-B-cell lymphomas in cats, is competent in the induction of T-cell lymphoma in mice. The findings suggest that the mechanism of MoFe2-MuLV-mediated lymphomagenesis may differ from that of Mo-MuLV-mediated disease, considering the possible involvement of novel oncogenes and the variable presence of MCF recombinants.
The replication-competent murine and feline leukemia viruses lack an oncogene within the genome and induce malignant disease in a tissue-specific manner. For example, Moloney murine leukemia virus (Mo-MuLV), a replication-competent nonacute retrovirus, induces a T-lymphoblastic lymphoma in virtually 100% of susceptible neonatal mice. Mo-MuLV induces T-cell lymphoma with a latency period of 3 to 4 months, an indicator that multiple steps are involved in disease development. Events characteristically associated with Mo-MuLV-induced lymphoma include (i) virus replication in target cells of the bone marrow, spleen, and thymus, (ii) the appearance of mink cell focus-inducing (MCF) viruses that arise by env gene recombination between Mo-MuLV and endogenous MuLV-related sequences, and (iii) insertional mutagenesis of proto-oncogenes via adjacent proviral integration, thus activating the malignant potential of the proto-oncogene in the target cell for transformation (reviewed in reference 19).
The major Mo-MuLV-encoded determinant of both tumorigenic potency and T-cell disease specificity resides in the long terminal repeat (LTR), particularly within the tandemly duplicated, 75-bp transcriptional enhancers present in the U3 region of the LTR (13, 14, 17, 18, 25, 29). The LTR of Mo-MuLV, like other mammalian leukemia viruses, is implicated in oncogenesis because of its influence on virus replication in relevant target tissues, on the formation and propagation of env recombinant viruses, and on the activation of cellular proto-oncogenes adjacent to sites of proviral integration (reviewed in reference 19). The Mo-MuLV LTR functions preferentially in T cells, a consequence of the recognition of protein binding sites within the transcriptional enhancers by cellular factors whose activity is restricted to specific cell types or differentiation states. The cell type-specific enhancer activity of Mo-MuLV, as of other MuLVs, has been correlated positively with the tumorigenic spectrum of the retrovirus (7, 8, 22, 30, 38–41).
The tandem repeat structure of the MuLV enhancer is considered to be important to its cell type-specific function and tumorigenic potential. For example, the oncogenic potential of Mo-MuLV, containing the tandemly repeated enhancer, has been compared with that of a Mo-MuLV mutant containing only a single copy of the enhancer. The comparison demonstrated that both the parental type and the mutant can induce neoplastic disease of the thymus but that the mutant does so with significantly prolonged latency (29). Similar observations have been reported for pathogenic AKR MCF recombinant viruses (24). It is noteworthy that the tandemly duplicated enhancer structure is also conserved in T-cell-lymphoma-derived isolates of feline leukemia virus (FeLV), a naturally occurring retrovirus associated with thymic lymphoma of T-cell origin in the domestic cat. The majority of FeLV proviruses found in naturally occurring T-cell lymphomas contain LTRs with tandem repeats of two or three copies of the enhancer. By comparison, the majority of FeLV proviruses isolated from healthy animals or from animals with nonneoplastic diseases contain only a single enhancer in the LTR. On the basis of these observations, it has been suggested that the enhancer repeats arise de novo during FeLV infection and are strongly associated with the induction of T-cell lymphoma (3, 21, 31, 32). In a recent test of this hypothesis, cats were infected experimentally with a natural isolate of FeLV containing only a single enhancer in the U3 region. Examination of proviruses derived from the resulting thymic tumors revealed that enhancer duplications had occurred in the LTR; however, FeLV LTRs from animals that did not develop tumors were observed to retain the single-enhancer structure (36).
Unlike Mo-MuLV, infection with FeLV does not uniformly induce T-cell lymphoma. In fact, natural FeLV infections are associated with a variety of malignant, proliferative, and degenerative disorders (35). In previous studies, we have extensively examined FeLV-positive, naturally occurring, non-T-cell, non-B-cell lymphomas classified clinically as multicentric, whose cell type of origin is apparently a primitive lymphoid cell or other hematopoietic progenitor (1, 2, 27). FeLV proviruses derived directly from these non-T-cell, non-B-cell lymphomas contain LTRs of unique structure. The U3 region of the FeLV LTR from these tumors is unusual in that it contains only a single copy of the enhancer, followed 25 bp downstream by a tandem triplication of a 21-bp sequence. Triplication of the 21-bp sequence has not been observed in any other isolate of FeLV or in any other mammalian retrovirus (1, 27). Our previous studies indicate that the 21-bp sequence triplication contributes enhancer function to the LTR that contains it and that it functions preferentially in a primitive hematopoietic cell line (2). The repeated identification of the single-enhancer, triplication-containing FeLV LTR uniquely in non-T-cell, non-B-cell multicentric lymphomas and its relatively high level of activity in a multipotential hematopoietic cell line suggest that it is an important determinant of the tumor type in which it is identified. On this basis, we hypothesized that replacement of the U3 region of Mo-MuLV LTR with the homologous region from the triplication-containing FeLV LTR would alter the tumorigenic spectrum of Mo-MuLV to include primitive hematopoietic targets. A recombinant retrovirus, termed MoFe2-MuLV, was developed to test this hypothesis.
MATERIALS AND METHODS
Construction of recombinant virus.
The recombinant virus MoFe2-MuLV was constructed from an infectious molecular clone of Mo-MuLV proviral DNA termed p63-2. Through a series of steps of restriction enzyme digestions and ligations (diagrammed in Fig. 1B), both LTRs of p63-2 were replaced between the EcoRV and SmaI sites with the homologous fragment from the LTR of FeLV-945 (27). The nucleotide sequence of the recombinant LTR was verified. To obtain infectious MoFe2-MuLV virus particles, MoFe2-MuLV plasmid DNA was introduced by electroporation into SC-1 cells (ATCC CRL-1404), an embryo fibroblast line derived from a feral mouse. Transfected cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum. Productive MoFe2-MuLV infection in SC-1 cells was detected after 10 days by demonstration of reverse transcriptase activity in culture supernatants as previously described (6). Culture supernatants from productively infected cells were collected, filtered to remove cellular debris, and frozen in aliquots at −70°C. The titer of virus in culture supernatants was determined by XC assay (37).
FIG. 1.
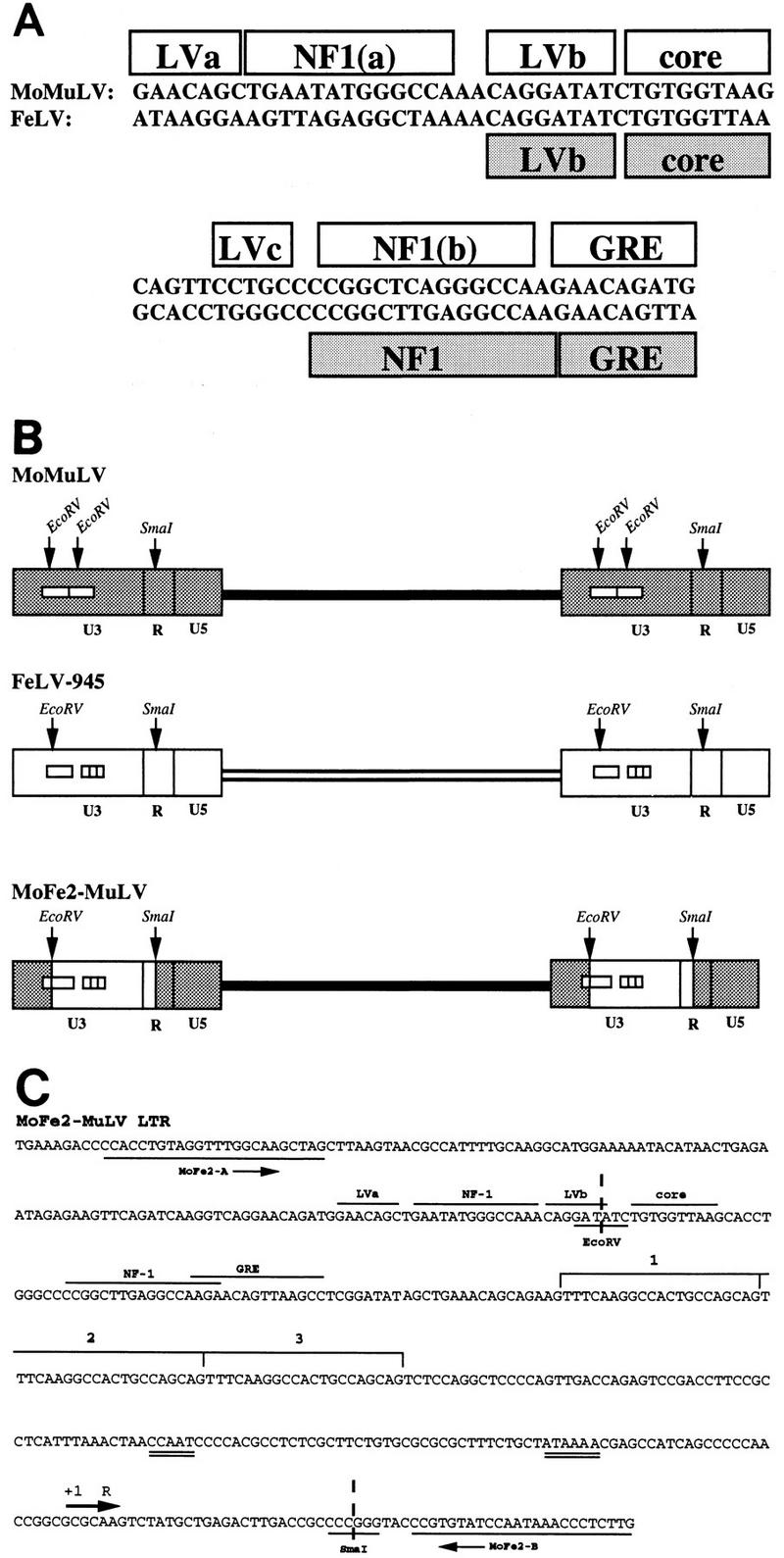
Construction of the recombinant retrovirus MoFe2-MuLV. To construct MoFe2-MuLV, the U3 region of Mo-MuLV was replaced by U3 sequences from the LTR of FeLV-945 (1) between the homologous EcoRV and SmaI sites. (A) Nucleotide sequence and protein binding sites identified in the enhancers found in the U3 region of Mo-MuLV (39) and FeLV (21) LTRs. (B) Diagrammatic representation of the proviral DNAs of Mo-MuLV, FeLV-945, and MoFe2-MuLV, indicating the number of enhancer repeats (open boxes), the 21-bp sequence triplication in the LTR of FeLV-945, and the EcoRV and SmaI sites used to construct the recombinant. (C) Nucleotide sequence of a segment of the LTR of MoFe2-MuLV, indicating a single copy of the transcriptional enhancer with predicted nuclear protein binding sites (LVa, LVb, core, NF-1, and GRE [21]), followed by a 21-bp sequence repeated three times in tandem (indicated by the brackets and numbers). CCAAT and TATA boxes in the promoter (double underline), the positions of PCR primers used for amplification as described in the text (MoFeA and MoFeB), and the EcoRV and SmaI sites that represent the junctions between MuLV- and FeLV-derived sequences are also indicated.
Inoculation of mice.
Neonatal NIH-Swiss mice (1 to 2 days old) were inoculated intraperitoneally with 0.2 ml of culture supernatant containing MoFe2-MuLV. Animals were monitored regularly until they died naturally from lymphoma or until significant morbidity required sacrifice. Necropsy was performed on all infected animals for the collection of tissues.
Southern blot analysis.
High-molecular-weight DNA was isolated from tumors, and Southern blot analysis was performed as previously described (1). The T-cell receptor beta (TCRβ) locus was examined with 86T5, a 600-bp EcoRI fragment cloned as a cDNA for the murine TCRβ locus (23). The flvi-1 locus was examined with probe A, a 1.2-kb PstI-EcoRI fragment, and probe D, a 0.9-kb BamHI-SacI fragment, cloned from feline flvi-1 flanking the domain of common retroviral insertion (26). The c-myc locus was examined with pSVcmyc1 (ATCC 41029), a clone of mouse genomic DNA representing c-myc exons 2 and 3. The pim-1 locus was examined with a 0.9-kb BamHI fragment of murine pim-1 genomic DNA (a gift of Anton Berns, Netherlands Cancer Institute). The pvt-1 locus was examined with a 2.2-kb XbaI fragment of murine pvt-1 (16). The probe for the 3′ env region of MoFe2-MuLV was a 1.1-kb BamHI-ClaI fragment isolated from p63-2, an infectious molecular clone of Mo-MuLV.
PCR amplification, cloning and sequencing of products.
Primer pair MoFe2A and MoFe2B (sequences shown in Fig. 1C) were used to amplify a portion of the LTR from integrated proviruses in tumor DNA. PCR mixtures (100-μl total volume) contained 1 μg of tumor DNA, 400 ng of each primer, 1.5 mM MgCl2, and 0.2 mM each of the four deoxyribonucleoside triphosphates, in a Taq DNA polymerase reaction buffer provided by the manufacturer (Promega). An initial denaturation step at 98°C for 5 min was followed by addition of 2.5 U of Taq DNA polymerase (Promega), annealing at 58°C for 5 min, and extension at 72°C for 1 min. This was followed by 35 cycles of PCR, with denaturation at 94°C for 30 s, annealing at 58°C for 30 s, and extension at 72°C for 1 min, with a final extension step at 72°C for 15 min. Amplification products were examined by agarose gel electrophoresis and cloned into the pGEM-T plasmid vector (Promega). Nucleotide sequence analysis was performed by using dideoxy chain termination reactions with Sequenase as described by the manufacturer (Amersham Life Sciences).
RESULTS
Construction of recombinant virus and infection of neonatal mice.
To examine the influence of the triplication-containing FeLV LTR on tumorigenesis, we developed a recombinant retrovirus, termed MoFe2-MuLV, in which the U3 region of Mo-MuLV was replaced by U3 sequences from the LTR of FeLV-945. FeLV-945 is an FeLV provirus derived from a naturally occurring, non-T-cell, non-B-cell lymphoma of a domestic cat. The LTR of FeLV-945, typical of proviruses derived from tumors of this type, contains in U3 a single copy of a transcriptional enhancer followed 25 bp downstream by a tandem triplication of a 21-bp sequence (1, 27). By comparison, the LTR of Mo-MuLV contains in U3 a tandemly duplicated direct repeat of a 75-bp enhancer. The single enhancer present in FeLV-945 contains nuclear protein binding sites homologous to some of those previously identified in the Mo-MuLV enhancer (21, 39). Of particular note are the LVb and core binding sites, previously identified as major determinants of the T-cell specificity and tumorigenic potential of Mo-MuLV (22, 29, 40). The LVb binding sites in the enhancers of Mo-MuLV and FeLV-945 are identical in sequence; however, the core binding sites differ at two residues (underlined) (TGTGGTAAG versus TGTGGTTAA [Fig. 1A]). The FeLV-945 core binding site is identical in sequence to that of the T-lymphomagenic murine leukemia virus SL3-3. In fact, recent studies of T-cell lymphoma-derived FeLV, containing a tandem repeat of this enhancer, show that it substitutes functionally for that of SL3-3 in the induction of T-cell lymphoma (34).
To construct MoFe2-MuLV, the U3 region of Mo-MuLV in both LTRs was replaced by that of FeLV-945 between conserved EcoRV and SmaI restriction sites as shown (Fig. 1B). Nucleotide sequence analysis of the recombinant MoFe2-MuLV LTR revealed a single copy of enhancer that includes LVa, NF-1, and LVb binding sites contributed by Mo-MuLV sequences followed by core, NF-1, and GRE binding sites contributed by FeLV sequences. The 21-bp sequence triplication characteristic of FeLV-945 is also evident, beginning 25 bp downstream of the enhancer (Fig. 1C). The MoFe2-MuLV recombinant virus was shown to be infectious in vitro following transfection into murine SC-1 cells, although the virus titer in culture supernatants was relatively low (1,300 XC PFU/ml). Neonatal NIH-Swiss mice (n = 14) were inoculated intraperitoneally with tissue culture supernatant of SC-1 cells productively infected with MoFe2-MuLV (0.2 ml; 260 XC PFU). Tumors occurred in 13 of 14 infected animals, with a latency period of 4 to 10 months (average, 6 months). Tumors generally involved the spleen, thymus, and occasionally the lymph nodes of diseased animals.
Cell type of origin of MoFe2-induced tumors.
Tumors induced by Mo-MuLV are invariably of T-cell origin, as detected by clonal, somatic rearrangement of the TCRβ locus in tumor DNA (9). To determine whether tumors induced by MoFe2-MuLV were of T-cell origin, Southern blot analysis of the TCRβ locus was performed. Southern blot analysis clearly demonstrated clonal, somatic rearrangement of the TCRβ locus in the DNA from all MoFe2-MuLV-induced tumors examined (11 of 14) (Fig. 2). This finding demonstrates that MoFe2-MuLV-induced tumors are lymphomas of T-cell origin and, thus, that the tumorigenic spectrum of Mo-MuLV is unchanged by the substitution of LTR sequences from non-T-cell lymphoma-derived FeLV.
FIG. 2.

Southern blot analysis of the TCRβ locus in DNA from tumors induced by MoFe2-MuLV. DNA from five MoFe2-MuLV-induced tumors (lanes a to e) and from an uninfected NIH-Swiss mouse (lane f) was digested with HpaI and hybridized to a murine TCRβ probe (23) as previously described (1). The arrows indicate the previously identified germ line HpaI fragments of 11.6 and 6.1 kb (9).
Sequence of the LTR in MoFe2-MuLV-induced tumors.
The finding that MoFe2-MuLV uniformly induces lymphomas of T-cell origin is unexpected because the LTR of MoFe2-MuLV lacks the tandemly repeated enhancer known to function as a major determinant of T-cell lymphomagenicity and contains FeLV sequence elements identified uniquely in non-T-cell lymphomas (reviewed in references 3 and 19). One possible explanation for this result is that recombination occurred in vivo in infected animals to regenerate an LTR similar in structure to that of wild-type Mo-MuLV. To test this possibility, LTRs were amplified from the DNA from MoFe2-MuLV-induced tumors, using PCR primers derived from the Mo-MuLV LTR sequence (shown in Fig. 1C). The results of PCR analysis demonstrated variation in LTR structure as a function of time after infection (Fig. 3). In relatively early appearing tumors, a predominant amplification product was observed of the size predicted from the intact MoFe2-MuLV LTR (457 bp). Nucleotide sequence analysis revealed that the predominant amplification product from early-appearing tumors retained the original MoFe2-MuLV sequence intact and unaltered. A minor, slightly larger amplification product seen in two early-appearing tumors (477-2 and 477-3) was shown to contain the 21-bp sequence in quadruplicate in tandem (Fig. 4A). A minor amplification product seen in the early-appearing tumor 493-1 was shown to contain the unaltered MoFe2-MuLV sequence, except with only a single copy of the 21-bp sequence (not shown). In contrast, in the relatively late appearing tumors, multiple amplification products were evident by PCR amplification (Fig. 3). Nucleotide sequence analysis demonstrated that LTRs from these tumors retained the 21-bp sequence triplication but acquired various duplications of enhancer during replication in vivo. The acquired duplications of enhancer were of variable length and included the FeLV core, NF-1, and GRE binding sites. The LVb/Ets site was not necessarily included in the duplication. Variable spacing was observed between the acquired duplications of enhancer and between the enhancers and the 21-bp sequence triplication (Fig. 4B).
FIG. 3.

PCR amplification of the LTRs in MoFe2-MuLV-induced tumors from two litters of mice (litters 477 and 493) infected independently. LTRs from tumor DNA were amplified by PCR with the primer pair shown in Fig. 1C. The identifying number of each animal (given after the litter number and hyphen) and the latency of tumor induction (in weeks postinoculation) are given above and below the gels, respectively. The arrows indicate the predominant amplification product (457 bp), demonstrated by subsequent sequence analysis to represent the intact MoFe2-MuLV LTR.
FIG. 4.

Diagrammatic representation of the nucleotide sequences of the MoFe2-MuLV LTR and of the predominant PCR products from MoFe2-MuLV-induced tumors. LTRs amplified from relatively early arising lymphomas (A) and relatively late arising lymphomas (B). The enhancer (stippled boxes), predicted nuclear protein binding sites within the enhancer (21), the 21-bp sequence triplication (open boxes), and the spacing between the elements are indicated. 477 and 493 represent different litters of mice inoculated independently. Multiple amplification products derived from one tumor are indicated by the letters a and b (e.g., 477-3a and 477-3b).
Proto-oncogene involvement in MoFe2-MuLV-induced tumors.
T-cell lymphomas induced by Mo-MuLV typically contain proviral insertions within at least one of three cellular loci: c-myc, pim-1, and pvt-1 (reviewed in reference 19). The tandemly repeated enhancer in the MuLV LTR has been implicated as an important determinant of its ability to activate these proto-oncogenes in T-cell lymphomas, particularly c-myc (15, 33). In contrast, in the non-T-cell, non-B-cell lymphomas like those from which FeLV-945 was identified, neither c-myc, pim-1, nor pvt-1 is targeted for insertional mutagenesis. Rather, a locus in feline DNA of unknown function, termed flvi-1, is a common target of proviral integration. Presumably, the unique structure of the FeLV-945 LTR is relevant to activation of a gene sequence within or near flvi-1 (1, 27). Considering that the MoFe2-MuLV LTR contains sequence elements typical of both Mo-MuLV and FeLV-945, we wished to examine the pattern of proto-oncogene involvement in MoFe2-MuLV-induced tumors. We tested by Southern blot analysis the organization of c-myc, pim-1, pvt-1, and flvi-1 loci in the DNA from 11 MoFe2-MuLV-induced lymphomas. Tumor DNA samples were digested with at least two different restriction enzymes in order to visualize approximately 20 kb of each proto-oncogene locus by Southern blot analysis. No rearrangements were detected in any of the loci examined (data not shown). Thus, the pattern of proto-oncogene involvement is apparently distinct from those of both Mo-MuLV and FeLV-945.
MCF viruses in MoFe2-MuLV-induced lymphomas.
Strongly associated with Mo-MuLV-mediated lymphomagenesis is the appearance of recombinant MCF viruses, so named because of their expanded host range. Moloney MCF viruses represent env gene recombinants between the infecting Mo-MuLV and endogenous polytropic provirus resident in the mouse genome (19). By virtue of distinctive restriction enzyme sites in the recombinant env gene, Moloney MCF proviruses in tumor DNA can be detected by Southern blot analysis using a 1.1-kb BamHI-ClaI fragment of the Mo-MuLV env gene as the probe (5). As previously described (5), digestion of tumor DNA with BamHI and ClaI is predicted to yield a 1.1-kb fragment generated from the ecotropic envelope of MoFe2-MuLV; however, the same digestion is predicted to yield a 1.9-kb fragment diagnostic of the MCF recombinant virus. Southern blot analysis of the DNA from 10 MoFe2-MuLV-induced tumors, digested with BamHI and ClaI and hybridized to the 1.1-kb env gene probe, revealed the 1.1-kb fragment generated from ecotropic MoFe2-MuLV in all samples examined. MCF proviral DNA, as evidenced by the diagnostic 1.9-kb fragment, was readily detectable in five tumors examined and weakly detectable or absent in five others (Fig. 5). The presence or absence of MCF virus did not correlate with the kinetics of tumor induction.
FIG. 5.

Southern blot analysis of tumor DNA to detect the presence of recombinant MCF virus. DNA from 10 MoFe2-MuLV-induced tumors and from an uninfected (uninf.) NIH-Swiss mouse was digested with BamHI and ClaI and hybridized to a 1.1-kb BamHI-ClaI fragment of the Mo-MuLV env gene as previously described (5). The 1.1-kb fragment generated from the ecotropic MoFe2-MuLV env gene, the 1.9-kb fragment diagnostic of MCF proviral DNA (arrows), the identifying number of each animal, and the latency of tumor induction (in weeks postinoculation [p.i.]) are indicated.
DISCUSSION
In this study, an infectious, recombinant retrovirus (MoFe2-MuLV) was generated by replacing the U3 region of the Mo-MuLV LTR with homologous sequences from the LTR of FeLV-945 (Fig. 1). The FeLV-945 LTR has been identified in 73% of natural feline lymphomas of non-T-cell, non-B-cell origin and is identified uniquely in those tumors (1, 27). On this basis, we hypothesized that substitution of the U3 region of FeLV-945 into Mo-MuLV would alter the tumorigenic spectrum of that virus. However, the results of this study demonstrate that neonatal mice infected with the recombinant virus MoFe2-MuLV develop lymphomas exclusively of T-cell origin (Fig. 2). Thus, the presence of the FeLV-945 LTR does not redirect the tumorigenic spectrum of T-lymphomagenic Mo-MuLV toward a more primitive hematopoietic cell.
The latency of tumor induction following MoFe2-MuLV infection is somewhat prolonged, i.e., 4 to 10 months (average, 6 months) compared to the 3- to 4-month latency associated with Mo-MuLV infection. This delay in tumor formation may be due to the relatively low inoculum (260 XC PFU of MoFe2-MuLV) compared to the 400-fold-greater inoculum typically used in studies of Mo-MuLV pathogenesis (4, 12). Alternatively, the delay in tumor formation may reflect the presence of only a single enhancer in the MoFe2-MuLV LTR. Single-enhancer mutants of Mo-MuLV have been shown by others to induce T-cell tumors with a prolonged latency that is similar to that of MoFe2-MuLV (mean, 190 days [29]). This similarity suggests that the presence of the 21-bp sequence triplication in the MoFe2-MuLV LTR may not accelerate the malignant process in T-cells compared to a single-enhancer derivative of Mo-MuLV that lacks the triplication.
It is striking that proviruses in the most rapidly arising tumors contained the input LTR of MoFe2-MuLV intact and unaltered (Fig. 3 and 4); thus, the single-enhancer, triplication-containing U3 region of FeLV-945 is competent in the induction of T-cell lymphomas in mice. In this respect, it is noteworthy that the LVb binding site in the FeLV-945 enhancer is identical to that of Mo-MuLV and that the FeLV-945 core binding site is identical to that of SL3-3. The LVb and core binding sites of Mo-MuLV and SL3-3 have been amply demonstrated to determine the tumorigenic potential and T-cell target specificity of lymphomagenesis mediated by those viruses (7, 22, 29, 30, 40). The ability of MoFe2-MuLV to induce T-cell lymphoma is also consistent with our previous studies of the function of the FeLV-945 LTR in reporter gene assays in vitro. Those studies demonstrated that the FeLV-945 LTR directs reporter gene expression in T cells to a level indistinguishable from that of the double-enhancer, T-cell-lymphoma-derived FeLV LTR (2). Thus, it might be predicted that the FeLV-945 LTR functions efficiently in T-cell lymphomagenesis. However, our previous studies also showed that the FeLV-945 LTR drives reporter gene expression in a primitive hematopoietic cell line to significantly higher levels than does the T-cell-derived, double-enhancer LTR. We correlated this high level of activity in primitive hematopoietic cells with the fact that LTRs of the FeLV-945 type are identified exclusively in non-T-cell, non-B-cell lymphomas (1, 2, 27). It is not yet clear why FeLV-945 is associated uniquely with non-T-cell, non-B-cell lymphomas, when the LTR is clearly functional in T-cell lymphomagenesis. However, the env gene of FeLV-945 is known to contain sequence elements clearly distinguishable from the highly conserved env genes of horizontally transmissible FeLV (1, 27a). One possibility is that in addition to the unique LTR, the unique env gene of FeLV-945 is an important participant in determining its tumorigenic spectrum. FeLV-945 env sequences were not present in the MoFe2-MuLV recombinant virus and thus could not exert an influence on its tumorigenic spectrum. Indeed, experiments with other murine retroviruses suggest that the nature of the envelope glycoprotein influences the type of leukemia that results. For instance, when a recombinant Mo-MuLV containing the avian v-myc oncogene is pseudotyped with an amphotropic MuLV helper, it induces both B and T lymphomas in mice (9); however, when the same virus is pseudotyped with ecotropic MuLVs, only T lymphomas result (22a).
In contrast to the tumors appearing most rapidly in MoFe2-MuLV-infected mice, late-appearing lymphomas were observed to contain LTRs in which a variety of enhancer duplications had occurred. All such LTRs retained the 21-bp sequence triplication characteristic of the FeLV-945 sequence but had acquired a complete or nearly complete duplication of the FeLV-derived enhancer element (Fig. 3 and 4). The repeated identification of enhancer duplications in late-appearing tumors suggests that the duplication may afford some selective advantage. The advantage is apparently not in the efficient induction of T-cell lymphoma, however, since the input virus is equally tumorigenic yet lacks a duplicated enhancer. An intriguing possibility is that the enhancer duplication offers a selective advantage not in the malignant process but in persistent virus propagation in the animal. Alternatively, the enhancer duplication may not afford a selective advantage; rather, enhancer duplications may occur repeatedly in late-appearing tumors simply because the probability for duplication may increase as a function of time after infection and cycles of replication. A test of these possibilities would require longitudinal studies in which the appearance, prevalence, and tissue distribution of the duplicated enhancer-containing LTR could be tracked.
The mechanism of action of the recombinant MoFe2-MuLV LTR in T-cell tumorigenesis is of interest, particularly with respect to the patterns of insertional mutagenesis of proto-oncogenes. Mo-MuLV-induced T-cell lymphomas typically contain proviral insertions within or near the cellular genes c-myc, pim-1, and pvt-1 (reviewed in reference 19). Previous studies of MuLVs indicate that the double-enhancer structure of the LTR is relevant to its ability to activate expression of the adjacent proto-oncogene in T-cell tumors, particularly in the case of c-myc. One approach to this issue has been to inoculate animals with MuLVs bearing LTR mutations and deletions. T-cell tumorigenesis is then examined as a potent selective force for LTR rearrangements, duplications, and mutations in vivo that increase the malignant potential of the virus. The results of such studies demonstrate that MuLV LTRs integrated adjacent to c-myc in tumor DNA typically contain multiple enhancers, in some cases with a distinctive sequence in the nuclear protein binding sites (15, 33). Similarly, in natural feline T-cell lymphomas, the FeLV LTR integrated adjacent to c-myc also contains multiple enhancer repeats (32). Taken together, these findings indicate that the repeated enhancer impacts on the ability of the LTR to activate proto-oncogenes in T-cell tumors. The absence of proviral insertions near c-myc, pim-1, or pvt-1 in MoFe2-MuLV-induced tumors is consistent with this hypothesis, since the MoFe2-MuLV LTR lacks a tandem repeat of enhancer. The absence of insertional mutagenesis of flvi-1 is also noteworthy, since this is a common domain of FeLV-945 integration in feline lymphomas (27). These findings suggest that the MoFe2-MuLV LTR interacts with proto-oncogenes different from those associated with either parental type; indeed, novel oncogenes may be activated by the recombinant LTR. The search to identify the targets of MoFe2-MuLV-mediated insertional mutagenesis is ongoing at present. When such targets are identified, a key issue will be analysis of the LTR structure at the commonly integrated site.
Finally, we examined the generation of env recombinant MCF viruses in MoFe2-MuLV-induced tumors, since their appearance has been established as an important preleukemic event in Mo-MuLV lymphomagenesis. Although their exact role is not yet known, MCFs may function to circumvent superinfection interference in ecotropic virus-infected cells, thus permitting increased virus replication in the target tissue. MCFs may also function more actively in the malignant process, e.g., by suppressing the growth of bone marrow stromal cells, thus inducing a compensatory extramedullary hematopoiesis that provides an actively dividing population of cells vulnerable to secondary transformation events (5, 10, 28) and/or by directly stimulating the growth of the T-cell target for transformation through mitogenic signal pathways (20). In the case of another recombinant derivative of Mo-MuLV, in which enhancer sequences of murine polyomavirus were inserted between enhancer and promoter elements of the Mo-MuLV LTR, the ability to generate MCF viruses during infection was linked strongly to the malignant potential of the virus (4, 5, 11, 12). In MoFe2-MuLV-induced lymphomas, MCF viruses could be detected in some tumors but not in others (Fig. 5). It is noteworthy that the appearance of MCF does not correlate with the kinetics of tumor induction; thus, the formation of MCF recombinants is apparently not a requirement in tumorigenesis by this chimeric virus, nor is it linked to the efficiency of tumor induction.
These findings show that the substitution into Mo-MuLV of a unique FeLV LTR, obtained exclusively from natural non-T-cell, non-B-cell lymphomas, does not alter or expand the tumorigenic spectrum of Mo-MuLV beyond the induction of T-cell lymphoma. The mechanisms of disease induction, however, may be distinct, since the patterns of proto-oncogene induction and MCF formation are different. These differences may reflect a different combination of events leading to transformation of the target T lymphocyte during infection with MoFe2-MuLV.
ACKNOWLEDGMENTS
We acknowledge with appreciation the technical assistance of Joshua Kayser.
This work was supported in part by American Cancer Society grant RPG-94-012-04-VM to L.S.L., by Development Funds of the Tulane Cancer Center, and by NIH grant CA32455 to H.F. C.R.S. has been supported by a predoctoral fellowship from the Louisiana Education Quality Support Fund. S.G. was partially supported by NIH training grant T32CA09054. The financial support of the biotechnology core facility of the UCI Cancer Center and of the UCI Cancer Research Institute is acknowledged.
REFERENCES
- 1.Athas G, Choi B, Prabhu S, Lobelle-Rich P, Levy L S. Genetic determinants of feline leukemia virus-induced multicentric lymphomas. Virology. 1995;214:431–438. doi: 10.1006/viro.1995.0053. [DOI] [PubMed] [Google Scholar]
- 2.Athas G, Lobelle-Rich P, Levy L S. Function of a unique sequence motif in the long terminal repeat of feline leukemia virus isolated from an unusual set of naturally occurring tumors. J Virol. 1995;69:3324–3332. doi: 10.1128/jvi.69.6.3324-3332.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Athas G, Starkey C, Levy L S. Retroviral determinants of leukemogenesis. Crit Rev Oncog. 1994;5:169–199. doi: 10.1615/critrevoncog.v5.i2-3.40. [DOI] [PubMed] [Google Scholar]
- 4.Belli B, Fan H. The leukemogenic potential of an enhancer variant of Moloney murine leukemia virus varies with the route of inoculation. J Virol. 1994;68:6883–6889. doi: 10.1128/jvi.68.11.6883-6889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belli B, Patel A, Fan H. Recombinant mink cell focus-inducing virus and long terminal repeat alterations accompany the increased leukemogenicity of the Mo+PyF101 variant of Moloney murine leukemia virus after intraperitoneal inoculation. J Virol. 1995;69:1037–1043. doi: 10.1128/jvi.69.2.1037-1043.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonham L, Lobelle-Rich P A, Henderson L A, Levy L S. Transforming potential of a myc-containing variant of feline leukemia virus in vitro in early-passage feline cells. J Virol. 1987;61:3072–3081. doi: 10.1128/jvi.61.10.3072-3081.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boral A L, Okenquist S A, Lenz J. Identification of the SL3-3 virus enhancer core as a T-lymphoma cell-specific element. J Virol. 1989;63:76–78. doi: 10.1128/jvi.63.1.76-84.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bösze Z, Thiesen H-J, Charnay P. A transcriptional enhancer with specificity for erythroid cells is located in the long terminal repeat of the Friend murine leukemia virus. EMBO J. 1986;5:1615–1623. doi: 10.1002/j.1460-2075.1986.tb04404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brightman B K, Chandy K G, Spencer R H, Gupta S, Pattengale P K, Fan H. Characterization of lymphoid tumors induced by a recombinant murine retrovirus carrying the avian v-myc oncogene. J Immunol. 1988;141:2844–2854. [PubMed] [Google Scholar]
- 10.Brightman B K, Davis B R, Fan H. Preleukemic hematopoietic hyperplasia induced by Moloney murine leukemia virus is an indirect consequence of viral infection. J Virol. 1990;64:4582–4584. doi: 10.1128/jvi.64.9.4582-4584.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brightman B K, Farmer C, Fan H. Escape from in vivo restriction of Moloney mink cell focus-inducing viruses driven by the Mo+PyF101 long terminal repeat (LTR) by LTR alterations. J Virol. 1993;67:7140–7148. doi: 10.1128/jvi.67.12.7140-7148.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brightman B K, Rein A, Trepp D J, Fan H. An enhancer variant of Moloney murine leukemia virus defective in leukemogenesis does not generate detectable mink cell focus-inducing virus in vivo. Proc Natl Acad Sci USA. 1991;88:2264–2268. doi: 10.1073/pnas.88.6.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chatis P A, Holland C A, Hartley J W, Rowe W P, Hopkins N. Role for the 3′ end of the genome in determining disease specificity of Friend and Moloney murine leukemia viruses. Proc Natl Acad Sci USA. 1983;80:4408–4411. doi: 10.1073/pnas.80.14.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatis P A, Holland C A, Silver J E, Frederickson T N, Hopkins N, Hartley J W. A 3′ end fragment encompassing the transcriptional enhancers of nondefective Friend virus confers erythroleukemogenicity on Moloney leukemia virus. J Virol. 1984;52:248–254. doi: 10.1128/jvi.52.1.248-254.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Yoshimura F K. Identification of a region of a murine leukemia virus long terminal repeat with novel transcriptional regulatory activities. J Virol. 1994;68:3308–3316. doi: 10.1128/jvi.68.5.3308-3316.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cory S, Graham M, Webb E, Corcoran L, Adams J M. Variant (6;15) translocations in murine plasmacytomas involve a chromosome 15 locus at least 72 kb from the c-myc oncogene. EMBO J. 1985;4:675–681. doi: 10.1002/j.1460-2075.1985.tb03682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DesGroseillers L, Jolicoeur P. The tandem direct repeats within the long terminal repeat of murine leukemia viruses are the primary determinant of their leukemogenic potential. J Virol. 1984;52:945–952. doi: 10.1128/jvi.52.3.945-952.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DesGroseillers L, Rassart E, Jolicoeur P. Thymotropism of murine leukemia virus is conferred by its long terminal repeat. Proc Natl Acad Sci USA. 1983;80:4203–4207. doi: 10.1073/pnas.80.14.4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan H. Leukemogenesis by Moloney murine leukemia virus: a multistep process. Trends Microbiol. 1997;5:74–82. doi: 10.1016/S0966-842X(96)10076-7. [DOI] [PubMed] [Google Scholar]
- 20.Flubacher M M, Bear S E, Tsichlis P N. Replacement of interleukin-2 (IL-2)-generated mitogenic signals by a mink cell focus-forming (MCF) or xenotropic virus-induced IL-9-dependent autocrine loop: implications for MCF virus-induced leukemogenesis. J Virol. 1994;68:7709–7716. doi: 10.1128/jvi.68.12.7709-7716.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fulton R, Plumb M, Shield L, Neil J C. Structural diversity and nuclear protein binding sites in the long terminal repeats of feline leukemia virus. J Virol. 1990;64:1675–1682. doi: 10.1128/jvi.64.4.1675-1682.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golemis E, Li Y, Fredrickson T N, Hartley J W, Hopkins N. Distinct segments within the enhancer region collaborate to specify the type of leukemia induced by nondefective Friend and Moloney viruses. J Virol. 1989;63:328–337. doi: 10.1128/jvi.63.1.328-337.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22a.Granger, S., B. K. Brightman, and H. Fan. Unpublished data.
- 23.Hedrick S M, Cohen D I, Nielsen E A, Davis M M. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature. 1984;308:149–153. doi: 10.1038/308149a0. [DOI] [PubMed] [Google Scholar]
- 24.Holland C A, Thomas C Y, Chattopadhyay S K, Koehne C, O’Donnell P V. Influence of enhancer sequences on thymotropism and leukemogenicity of mink cell focus-forming viruses. J Virol. 1989;63:1284–1292. doi: 10.1128/jvi.63.3.1284-1292.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lenz J, Celander D, Crowther R L, Patarca R, Perkins D W, Haseltine W A. Determination of the leukaemogenicity of a murine retrovirus by sequences within the long terminal repeat. Nature. 1984;308:467–470. doi: 10.1038/308467a0. [DOI] [PubMed] [Google Scholar]
- 26.Levesque K, Mattei M-G, Levy L S. Evolutionary conservation and chromosomal localization of flvi-1. Oncogene. 1991;6:1377–1379. [PubMed] [Google Scholar]
- 27.Levesque K S, Bonham L, Levy L S. flvi-1, a common integration domain of feline leukemia virus in naturally occurring lymphomas of a particular type. J Virol. 1990;64:3455–3462. doi: 10.1128/jvi.64.7.3455-3462.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27a.Levy, L. S. Unpublished observations.
- 28.Li Q X, Fan H. Combined infection by Moloney murine leukemia virus and a mink cell focus-forming virus recombinant induces cytopathic effects in fibroblasts or in long-term bone marrow cultures from preleukemic mice. J Virol. 1990;64:3701–3711. doi: 10.1128/jvi.64.8.3701-3711.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Golemis E, Hartley J W, Hopkins N. Disease specificity of nondefective Friend and Moloney murine leukemia viruses is controlled by a small number of nucleotides. J Virol. 1987;61:693–700. doi: 10.1128/jvi.61.3.693-700.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LoSardo J E, Boral A L, Lenz J. Relative importance of elements within the SL3-3 virus enhancer for T-cell specificity. J Virol. 1990;64:1756–1763. doi: 10.1128/jvi.64.4.1756-1763.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsumoto Y, Momoi Y, Watari T, Goitsuka R, Tsujimoto H, Hasegawa A. Detection of enhancer repeats in the long terminal repeats of feline leukemia viruses from cats with spontaneous neoplastic and nonneoplastic diseases. Virology. 1992;189:745–749. doi: 10.1016/0042-6822(92)90598-j. [DOI] [PubMed] [Google Scholar]
- 32.Miura T, Shibuya M, Tsujimoto H, Fukasawa M, Hayami M. Molecular cloning of a feline leukemia provirus integrated adjacent to the c-myc gene in a feline T-cell leukemia cell line and the unique structure of its long terminal repeat. Virology. 1989;169:458–461. doi: 10.1016/0042-6822(89)90172-4. [DOI] [PubMed] [Google Scholar]
- 33.Morrison H L, Soni B, Lenz J. Long terminal repeat enhancer core sequences in proviruses adjacent to c-myc in T-cell lymphomas induced by a murine retrovirus. J Virol. 1995;69:446–455. doi: 10.1128/jvi.69.1.446-455.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pantginis J, Beaty R M, Levy L S, Lenz J. The feline leukemia virus long terminal repeat contains a potent genetic determinant of T-cell lymphomagenicity. J Virol. 1997;71:9786–9791. doi: 10.1128/jvi.71.12.9786-9791.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rezanka L J, Rojko J L, Neil J C. Feline leukemia virus: pathogenesis of neoplastic disease. Cancer Invest. 1992;10:371–389. doi: 10.3109/07357909209024796. [DOI] [PubMed] [Google Scholar]
- 36.Rohn J L, Overbaugh J. In vivo selection of long terminal repeat alterations in feline leukemia virus-induced thymic lymphomas. Virology. 1995;206:661–665. doi: 10.1016/s0042-6822(95)80085-9. [DOI] [PubMed] [Google Scholar]
- 37.Rowe W P, Pugh W E, Hartley J. Plaque assay techniques for murine leukemia viruses. Virology. 1970;42:1136–1139. doi: 10.1016/0042-6822(70)90362-4. [DOI] [PubMed] [Google Scholar]
- 38.Short M K, Okenquist S A, Lenz J. Correlation of leukemogenic potential of murine retroviruses with transcriptional tissue preference of the viral long terminal repeat. J Virol. 1987;61:1067–1072. doi: 10.1128/jvi.61.4.1067-1072.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Speck N A, Baltimore D. Six distinct nuclear factors interact with the 75-base-pair repeat of the Moloney murine leukemia virus enhancer. Mol Cell Biol. 1987;7:1101–1110. doi: 10.1128/mcb.7.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Speck N A, Renjifo B, Golemis E, Fredrickson T N, Hartley J W, Hopkins N. Mutation of the core or adjacent LVb elements of the Moloney murine leukemia virus enhancer alters disease specificity. Genes Dev. 1990;4:233–242. doi: 10.1101/gad.4.2.233. [DOI] [PubMed] [Google Scholar]
- 41.Yoshimura F K, Davison B, Chaffin K. Murine leukemia virus long terminal repeat sequences can enhance gene activity in a cell-type-specific manner. Mol Cell Biol. 1985;5:2832–2835. doi: 10.1128/mcb.5.10.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]