

Abstract
We report here on stable prepackaging cell lines which can be converted into packaging cell lines for high-titer vesicular stomatitis virus G protein (VSV-G)-pseudotyped retrovirus vectors by the introduction of Cre recombinase-expressing adenovirus. The generated prepackaging cell lines constitutively express the gag-pol genes and contain an inducible transcriptional unit for the VSV-G gene. From this unit, the introduced Cre recombinase excised both a neomycin resistance (Neor) gene and a poly(A) signal flanked by a tandem pair of loxP sequences and induced transcription of the VSV-G gene from the same promoter as had been used for Neor expression. By inserting an mRNA-destabilizing signal into the 3′ untranslated region of the Neor gene to reduce the amount of Neor transcript, we were able efficiently to select the clones capable of inducing VSV-G at high levels. Without the introduction of Cre recombinase, these cell lines produce neither VSV-G nor any detectable infectious virus at all, even after the transduction of a murine leukemia virus-based retrovirus vector encoding β-galactosidase. They reproducibly produced high-titer virus stocks of VSV-G-pseudotyped retrovirus (1.0 × 106 infectious units/ml) from 3 days after the introduction of Cre recombinase. We also present evidence that VSV-G-producing cells are still fully susceptible to transduction by VSV-G pseudotypes. However, in this vector-producing system, which regulates VSV-G pseudotype production in an all-or-none manner, the integration of vector DNA into packaging cell lines would be minimized. We further show that heparin significantly inhibits retransduction of VSV-G pseudotypes in the culture fluids of packaging cell lines, leading to a two- to fourfold increase in the yield of the pseudotypes after induction. This vector-producing system was very stable and should be advantageous in human gene therapy.
Vectors based on murine leukemia virus (MLV) have been developed and used as powerful tools for gene transfer in basic research as well as human gene therapy. However, some problems remain, such as relatively low titers, a poor transducibility into some kinds of cells, and the inability to transfer genes into nondividing cells (17, 19, 20, 23). When this retrovirus vector is pseudotyped with the G protein of vesicular stomatitis virus (VSV-G) (34), it has a much broader host range than the vectors having the conventional amphotropic Env, and the titers of the virus stocks can be concentrated about 1,000 times by ultracentrifugation (4, 9, 37). Expression of VSV-G protein, however, is cytotoxic for most mammalian cells and imposes a significant growth disadvantage. Pseudotyped vector titers of 105 to 106 infectious units (IU)/ml have been recovered after the transient expression of the VSV-G gene by DNA transfection into cell lines which were constitutively expressing the gag and pol genes, but these systems are not suitable for the reproducible preparation of certified vectors on a large scale. Because of the cytotoxicity of VSV-G, generation of stable packaging cell lines for the production of such pseudotyped vectors has been difficult, despite their potential advantages for gene transfer. To overcome this problem, some stable packaging cell lines (6, 22) or virus-producing cell lines (36) for VSV-G pseudotypes have been developed by using tetracycline-modulated promoters for the VSV-G expression. These cell lines have been reported to induce high titers of pseudotypes (106 to 107 IU/ml) by the removal of tetracycline.
However, these stable cell lines were reported to produce low titers (101 to 102 IU/ml [6] or 103 to 104 IU/ml [22]) of pseudotypes even in the presence of transcriptional repressors such as tetracycline. Since the receptor for VSV-G is reported to include ubiquitous anionic phospholipids such as phosphatidylserine (18, 28), this leaky virus production by these packaging cell lines before the induction of the VSV-G gene could potentially cause virus reentry into the cell culture and accumulation of the vector DNA in the chromosomes during the process of selection and subsequent passages of the packaging cell lines harboring the virus vector.
To overcome this problem, we present here a new system that is suitable for the production of VSV-G pseudotypes on a large scale. In this system, we first constructed prepackaging cell lines which express the gag-pol genes and harbor a completely silent VSV-G gene. These prepackaging cell lines were designed to be converted into packaging cell lines producing VSV-G at a high level. This conversion was carried out via loxP-specific recombination by Cre recombinase introduced by a replication-defective adenovirus vector. We show here that the prepackaging cell lines harboring a virus vector produce no detectable transducing particles but produce high titers (∼106 IU/ml) of VSV-G pseudotypes after the introduction of Cre recombinase. We also present evidence that cells producing VSV-G proteins are fully susceptible to the VSV-G pseudotypes, which confirms the advantage of our pseudotype production system, which regulates production in an all-or-none manner, over other inducible cell lines.
MATERIALS AND METHODS
Plasmid construction.
The entire VSV-G (Indiana serotype)-coding fragment (24) was excised from pSVGL (25) by EcoRI digestion, blunt ended by Klenow treatment, and inserted into the unique SwaI site in pCALNLw (15, 16) to generate pCALNLG (Fig. 1A). The fragment encoding chicken c-fos was excised from chicken c-fos genomic λ clone no. 7 (11) by BglII digestion, blunt ended by Klenow treatment, and further digested with ClaI to excise a 0.4-kb fragment containing an mRNA-destabilizing signal. pCALNLw was digested at the single ClaI site, blunt ended by Klenow treatment, and self-ligated to create an NruI site. The generated plasmid was completely digested with both NspV and NruI and ligated with the 0.4-kb fragment containing the mRNA-destabilizing signal in the 3′ untranslated region of the Neor gene (2) to generate pCALNdLw. The fragment containing the Neor gene with the mRNA-destabilizing signal was excised from pCALNdLw by MluI digestion and was inserted into the MluI site of pCALNLG digested with MluI to replace the conventional Neor gene, generating pCALNdLG (Fig. 1A).
FIG. 1.
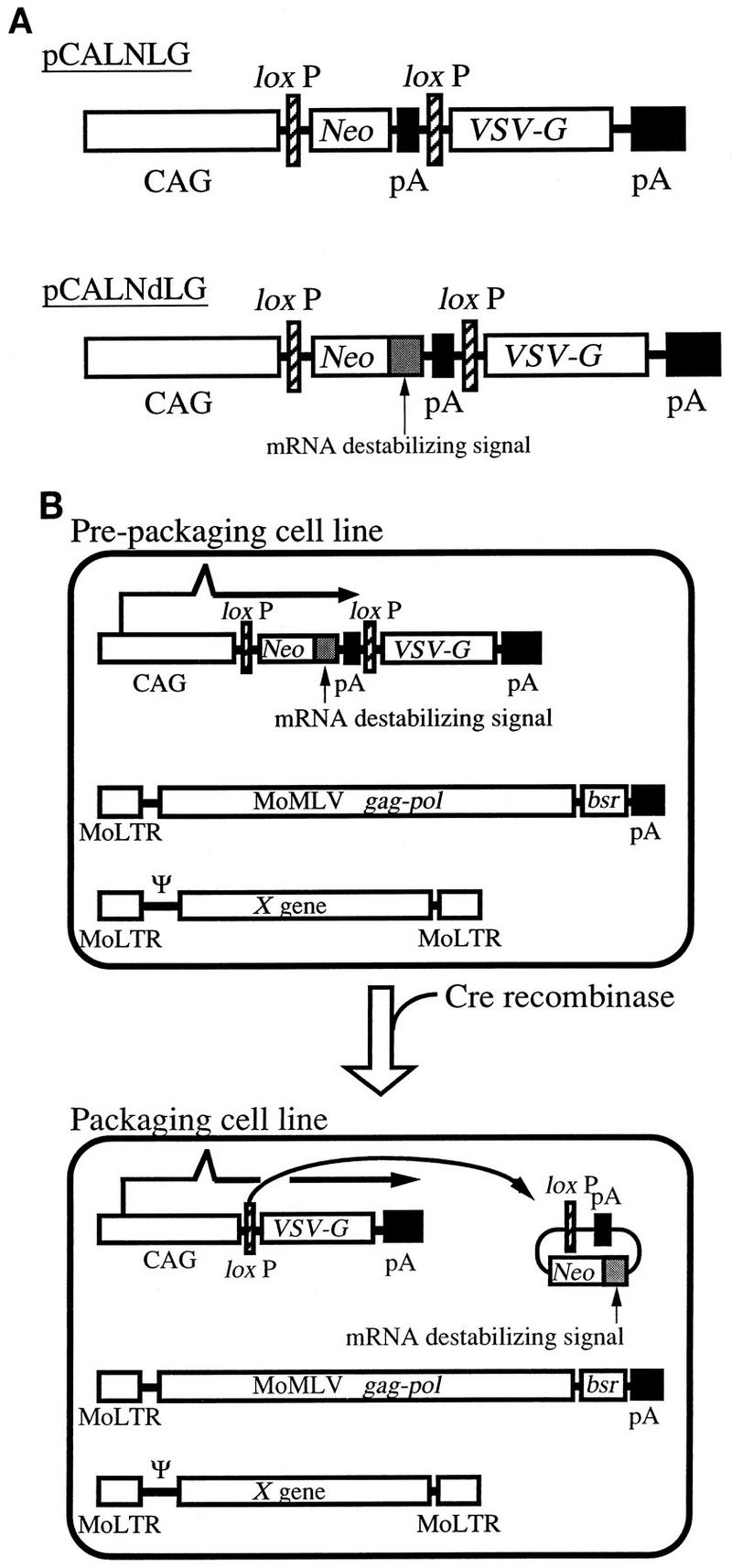
Plasmid structure for the generation of prepackaging cell lines and system for Cre-mediated pseudotyped retrovirus production. (A) Structures of pCALNLG and pCALNdLG. Two loxP sequences are tandemly located in each plasmid. CAG, CAG promoter; Neo, neomycin resistance gene; pA, polyadenylation signal; VSV-G, VSV-G (Indiana serotype)-coding sequence. The mRNA-destabilizing signal was derived from the 3′ untranslated region of chicken c-fos. (B) Schematic presentation of the conversion from the prepackaging cell line to the packaging cell line. In the prepackaging cell line generated by the transfection of pCALNdLG into FLY, the Neor gene is transcribed from a CAG promoter, while the VSV-G gene is completely silent because the RNA transcript terminates before its coding sequence. Arrows indicate the predicted transcript. The Neor transcript is expected to be unstable because it contains the mRNA-destabilizing signal. Cre recombinase excises the Neor gene, the mRNA-destabilizing signal, and the poly(A) signal by site-specific recombination between the two loxP sequences and thus converts the prepackaging cell line to the packaging cell line. In the packaging cell line, the VSV-G gene is now transcribed by the same promoter that was used for the Neor expression. MoLTR, MoMLV long terminal repeat; MoMLV gag-pol, MoMLV gag and pol genes; bsr, blasticidin resistance gene; pA, polyadenylation signal; Ψ, packaging signal of retrovirus vector; X gene, an arbitrary gene (here we used the gene encoding β-galactosidase with a nuclear localization signal).
Cell lines and drug selection.
FLY cells (8) (a derivative of human fibrosarcoma cell line HT 1080, which carries the gag and pol genes from Moloney MLV [MoMLV]), 3YI (rat fibroblast) cells, and 293 (human embryonal kidney) cells were maintained in Dulbecco’s modified Eagle’s medium (high glucose) supplemented with 10% fetal calf serum and kept at 37°C. FLY cells and derivatives were always grown in the presence of 4 μg of blasticidin S (Funakoshi) per ml, and drug selections of transfected FLY cells were performed with 1.0 mg of G418 (Gibco/BRL) per ml. Mus dunni tail fibroblast cells and PG-4 S+L− cells (Moloney sarcoma virus-infected G355 cat cells) were grown in McCoy’s 5A medium supplemented with 10% fetal calf serum.
DNA transfection and cloning of the transfectants.
FLY cells (5 × 105 cells/100-mm-diameter dish) were seeded at 1 day before transfection and transfected with pCALNdLG or pCALNLG (10 to 30 μg) by the calcium phosphate method (5). Two days after transfection, the cell cultures were split at several ratios, and G418 (final concentration, 1 mg/ml) was added to the medium 3 days after transfection for the selection of stable transformants. G418-resistant colonies were picked up with cloning cylinders and transferred for growth.
Retrovirus transduction, concentration, and titration.
To transduce an MLV-based retrovirus vector encoding β-galactosidase with a nuclear localization signal (MFGnlslacZ) into selected clones, stocks of the amphotropic virus vector were collected from FLYA4lacZ3 cells (8). Transduction was carried out in the presence of Polybrene (Sigma) (8 μg/ml) 1 day after passage. For the titration of amphotropic MFGnlslacZ or VSV-G-pseudotyped MFGnlslacZ, 3Y1 cells were plated at 1.5 × 103 cells/well in 96-well plates 1 day before transduction and incubated for 3 days with serial dilutions of virus supernatants containing 8 μg of Polybrene per ml. Transduced 3Y1 cells were fixed with 1.25% glutaraldehyde and stained with 5 mM K4[Fe(CN)6]–5 mM K3[Fe(CN)6]–2 mM MgCl2–1 mg of X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (Wako) per ml for more than 4 h, and then the numbers of cell clones with blue-stained nuclei were counted.
Adenovirus vector infection for pseudotyped retrovirus production.
Prepackaging cells were seeded at 3 × 104 cells/well in 24-well plates; 1 day later, they were infected with AxCANCre at various multiplicities of infection (MOIs) for 1 h in 100 μl of the culture medium to promote infection and then were cultured in a volume of 500 μl as usual. G418 was removed from the culture medium just after the AxCANCre infection. In some experiments, the culture temperature was changed to 32°C 2 days after infection. The medium was changed every day, and at each medium change the cells were washed three times with the medium to minimize contamination of the unabsorbed adenovirus vector. Southern blotting analysis was performed as follows. Total chromosome DNA was prepared by standard techniques (27) from PtG-S2 and PtG-L1 before or 4 days after AxCANCre infection. The digested DNA was transferred to a nylon membrane (Hybond N+; Amersham) by the capillary transfer method. The probe was labeled with 32P, and hybridized DNA was detected by autoradiography.
Protein analysis.
Expression of VSV-G protein was evaluated by immunocytochemical staining with murine monoclonal anti-VSV-G immunoglobulin G (IgG) (P5D4; Sigma). Cells were fixed with phosphate-buffered saline containing 3% paraformaldehyde and 0.1% Triton X-100 at 4°C for 15 min, and then a 1:3,000 dilution of monoclonal anti-VSV-G IgG was added. VSV-G-producing cells were visualized by using biotinylated anti-mouse IgG and a Vectastain ABC kit (Vector). For Western blot analysis, cellular lysates were prepared under denaturing conditions and separated by sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis. The gels were transferred onto polyvinylidene difluoride membranes (Immobilon; Millipore) with a semidry electroblotter. Filters were immunoblotted with monoclonal anti-VSV-G (Sigma) IgG or anti-Neor (5Prime to 3Prime) and then biotinylated anti-mouse IgG. Protein bands were detected with an ECL kit (Amersham).
RESULTS
Strategy for the generation and activation of the prepackaging cell lines.
For the generation of prepackaging cell lines, we made use of a human cell line, FLY, which expresses the gag and pol genes of MoMLV at a high level. It was previously shown that retroviruses produced from human-derived cell lines are much more resistant to human serum than those produced by conventional murine-derived packaging cell lines, due to the humanized glycosylation pattern of the virus envelope protein (8, 31–33). For the efficient screening of prepackaging cell lines capable of inducing high levels of VSV-G after the introduction of Cre recombinase, we designed a plasmid, pCALNdLG (Fig. 1A), for the generation of prepackaging cell lines from FLY cells. In pCALNdLG, the VSV-G gene is preceded by the Neor gene flanked by a tandem pair of loxP sequences. The transcription of the Neor gene is designed to be driven from a CAG promoter (the chicken β-actin gene promoter connected with the cytomegalovirus immediate-early promoter [21]) present upstream of the 5′ loxP sequence and to be terminated by a poly(A) signal located upstream of the 3′ loxP sequence. Therefore, the VSV-G gene should not be transcribed from the CAG promoter or any other promoter in G418-resistant clones. Since the 3′ untranslated region of the Neor transcripts from pCALNdLG includes an mRNA-destabilizing sequence originated from the 3′ noncoding sequence of the chicken c-fos gene, the relative amount of the Neor transcripts would be reduced. When FLY cells are exposed to G418 after transfection with pCALNdLG, we can efficiently select cell lines in which the synthesis of the transcripts from the CAG promoters is sufficiently active to keep the unstable Neor transcript at high levels adequate for survival.
After site-specific recombination between the pair of loxP sequences by Cre recombinase, the Neor gene, the mRNA-destabilizing sequence, and the poly(A) signal would be removed from the chromosomal DNA as a closed circular molecule and the CAG promoter would be available for the active transcription of the VSV-G gene (Fig. 1B) to produce VSV-G pseudotypes. To assess the validity of this design, we also constructed pCALNLG (Fig. 1A), in which the mRNA-destabilizing signal was deleted from pCALNdLG, and compared the selection efficiencies.
For the introduction of Cre recombinase into the prepackaging cell lines to convert them into packaging cell lines, we chose to use a replication-defective adenovirus vector, AxCANCre, which encodes Cre recombinase tagged with a nuclear localization signal (12, 15, 16). This vector has been shown to be efficiently introduced into human cells, to induce this exogenous gene at a very high level within a short time after the infection, and to cause efficient excision between a pair of loxP sequences located on another adenovirus genome and on a cellular chromosome.
Cloning of the prepackaging cell line.
By using the calcium phosphate method, either pCALNdLG or pCALNLG was transfected into a pair of FLY cultures. One day after the transfection, the cultures were split and stable transformants were selected with G418 for 2 weeks; surviving colonies were stained with crystal violet. The colony number of pCALNdLG transfectants was about one-third of that of the pCALNLG transfectants, suggesting that the Neor transcript from pCALNdLG is less stable, as expected (data not shown). In preliminary experiments, several clones were isolated from both of the transfectants and parallel cultures of each clone were infected with the adenovirus vectors at an MOI of 10 to determine the inducibility of VSV-G protein expression by immunocytochemical staining. The appearance of clones capable of VSV-G induction at a high level was more frequent in pCALNdLG transfectants (3 of 11 transfectants) than in pCALNLG transfectants (1 of 25). In each positive clone, almost the entire population was shown to express VSV-G protein, indicating that the introduction of Cre recombinase is very efficient. These results indicate that pCALNdLG is effective for isolation of clones that produce high levels of VSV-G protein after site-specific recombination and support the feasibility of screening with pCALNdLG. For these reasons, we chose pCALNdLG for the transfection experiments for large-scale screening. We selected 26 clones that express high levels of VSV-G protein after induction as candidates for the prepackaging cell lines. The only clone among the pCALNLG transfectants (PtG-L1) which produced VSV-G at a comparable level was also used for further experiments, for comparison.
Isolation of prepackaging cell lines capable of inducing high-titer VSV-G pseudotypes.
Parallel cultures of the above-described clones (27 in all) were transduced with amphotropic MFGnlslacZ virus at an MOI of 3. This virus vector carries the nlslacZ gene, which encodes β-galactosidase with a nuclear localization signal (nlsLacZ) as a marker enzyme. The efficiency of transduction was high as judged from cytochemical staining for the LacZ product (more than 85%). These transduced cultures were infected with AxCANCre at an MOI of 10. Culture fluids were collected, and the virus titers were determined with 3Y1 rat fibroblasts as the indicator. All the clones produced virus with various titers. Two clones produced more than 5 × 105 IU/ml, and 13 clones produced more than 5 × 104 IU/ml. We selected one clone (PtG-S2) among 26 clones of pCALNdLG transfectants, because it produced the highest virus titers. Virus induction by PtG-L1 was modest (2 × 104 IU/ml). When the virus stocks obtained from these two cell lines were treated with anti-VSV-G (Indiana serotype) antiserum, the virus was neutralized completely (unpublished observations).
Efficient and precise site-specific recombination in the PtG-S2 chromosome after the introduction of AxCANCre.
Southern blot analysis of PtG-S2 (or PtG-L1) chromosomal DNA was performed to test whether the loxP-specific recombination occurred as expected after the introduction of Cre recombinase. Chromosomal DNA was isolated before or 4 days after the AxCANCre introduction and digested with NcoI, which recognizes three sites in pCALNdLG (or pCALNLG), and DNA fragments containing the VSV-G gene were detected (Fig. 2A). A single 1.5-kb (or 1.2-kb) band was detected in prepackaging cells, but it mostly disappeared and a new 2.0-kb band appeared instead after the Cre recombinase introduction (Fig. 2B). This change in the fragment size is consistent with the idea that Cre recombinase excised the DNA sequence between the two loxP sequences by recombination. By comparing the densities of the control plasmid DNA charged at various amounts, PtG-L1 was estimated to contain a single copy of pCALNLG in the diploid cell, while PtG-S2 harbored three copies of pCALNdLG. This result indicates that the higher VSV-G expression in PtG-S2 is partly supported by the gene dosage effects.
FIG. 2.
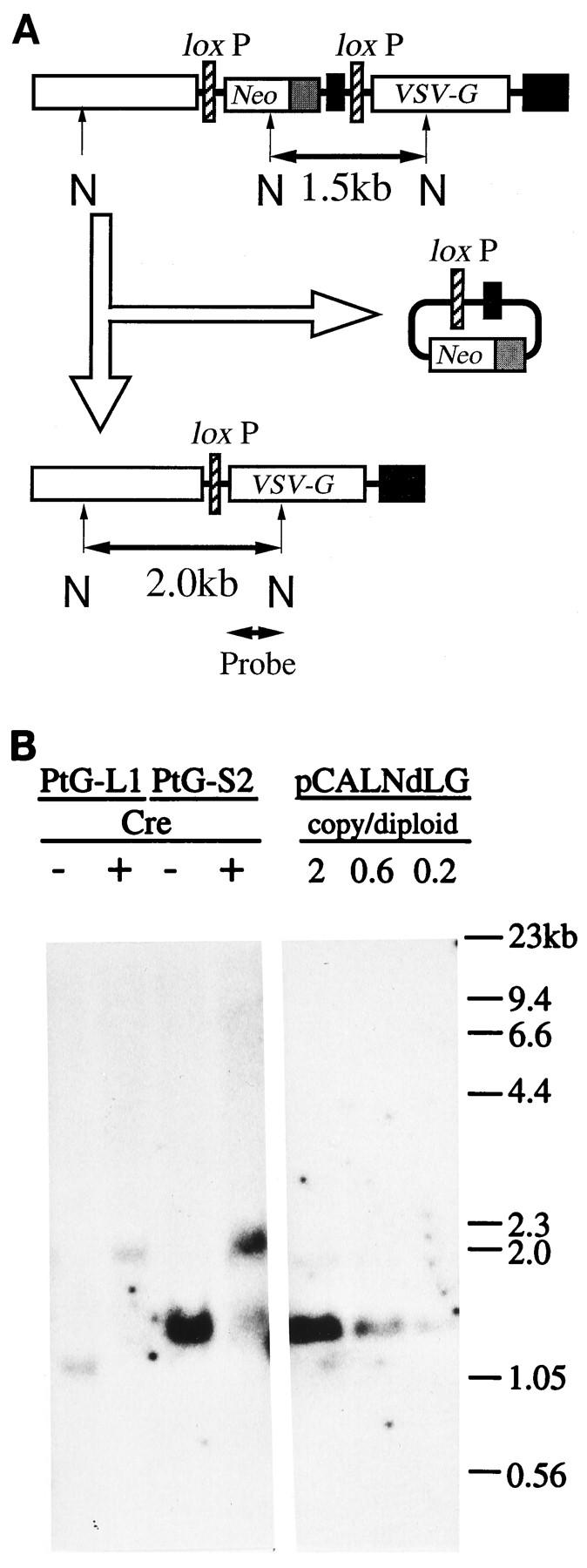
Southern blot analysis of genomic DNA. (A) Physical maps and predicted structural changes in pCALNdLG after the introduction of Cre recombinase. In the case of pCALNLG, the predicted NcoI fragment is 1.2 kb in size because of the lack of the mRNA-destabilizing signal. The 0.7-kb VSV-G probe was isolated from pCALNdLG by MluI and NcoI digestion. N, NcoI sites. (B) Autoradiogram of Southern blotting of NcoI digests of genomic DNA (15 μg per lane) from PtG-L1 and PtG-S2 before (−) and 4 days after (+) the introduction of Cre recombinase (right) and of plasmid pCALNdLG DNA for quantitation (left).
When VSV-G expression levels were analyzed by Western blotting, both PtG-S2 and PtG-L1 were found to produce no detectable VSV-G before Cre recombinase introduction (Fig. 3). Four days after AxCANCre introduction, both cell lines clearly produced VSV-G. However, the VSV-G expression level in PtG-S2 was much higher (about 10- to 15-fold) than that in PtG-L1, which is consistent with the observation that PtG-S2 is a much more efficient virus producer. Since the transcripts for the VSV-G gene from the CAG promoter in PtG-L1 and in PtG-S2 are expected to have the same molecular structure after the introduction of Cre recombinase, these results indicate that the total synthetic rate of the VSV-G transcript is more than 10-fold higher in PtG-S2. This higher synthetic rate can be partly explained by the gene dosage effect (threefold), as mentioned above, but also arises partly from the enhanced frequency of transcription initiation from the CAG promoter in PtG-S2 owing to the integration sites of the plasmid.
FIG. 3.

Protein analysis of VSV-G and the Neor gene product in PtG-S2 and PtG-L1 cells before and 4 days after AxCANCre infection. Lysates of PtG-S2 and PtG-L1 cells as well as the parent FLY cells were prepared under denaturing conditions, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (20 μg per each lane), and detected by Western blotting. Sequential dilutions (10, 5, 2.5, and 1.25 μg per lane) of PtG-L1 before induction or of PtG-S2 after induction were analyzed in parallel for quantitation.
We next analyzed the levels of expression of the Neor gene products by Western blotting with the same cell lysates (Fig. 3). In both PtG-S2 and PtG-L1, the Neor gene products were detectable before Cre recombinase introduction but were marginal 4 days after the introduction, in accordance with the observation that most of these cell lines at this stage had lost resistance to G418 (data not shown). It should be pointed out that the Neor expression level was about threefold higher in PtG-L1 than in PtG-S2, while the CAG promoter in PtG-S2 was expected to be stronger than that in PtG-L1 (as judged from the VSV-G expression levels), suggesting that the transcript for the Neor gene in PtG-S2 is much less stable than that in PtG-L1, as expected. All of these results indicate that the mRNA-destabilizing sequence in pCALNdLG was helpful for the selection of cell lines with a high total synthetic rate from the CAG promoter, as desired.
Conditions for optimum virus production.
PtG-S2 and PtG-L1 were transduced with amphotropic MFGnlslacZ virus, and these mixed populations were further used for screening to determine the optimum conditions for the induction of virus production from these prepackaging cell lines. Each AxCANCre should be introduced into every cell for the loxP-dependent recombination, and the efficiency of the recombination would be increased by dosage effects. A high dose of adenovirus, however, would cause cell damage due to the toxicity of adenovirus itself. Therefore, we next determined the time course of virus production from PtG-S2 after AxCANCre infection at several MOIs. We found the optimum range of MOIs of adenovirus for the highest induction of retrovirus to be around 10 to 30 (Table 1). Since the growth of PtG-S2 cells was significantly inhibited at an MOI of more than 30, we infected PtG-S2 with the adenovirus vector at an MOI of 10 in further studies. The titer of the virus recovered from PtG-L1 was roughly 1% of that from PtG-S2 under all of these conditions (data not shown).
TABLE 1.
Effect of MOI of AxCANCre on virus production by PtG-S2 cells
| AxCANCre MOI | Retrovirus titer (IU/ml) on the following day after Cre recombinase introduction:
|
||
|---|---|---|---|
| 2 | 4 | 6 | |
| 0 | NDa | ND | ND |
| 1 | 4.8 × 104 | 1.2 × 105 | 2.1 × 104 |
| 3 | 1.9 × 105 | 9.8 × 105 | 9.6 × 104 |
| 10 | 5.0 × 105 | 1.0 × 106 | 1.1 × 105 |
| 30 | 7.3 × 105 | 1.3 × 106 | 7.5 × 104 |
| 100 | 3.1 × 104 | 9.4 × 104 | 3.5 × 104 |
ND, undetectable.
We next determined the time course of virus production at either 37 or 32°C after adenovirus infection at an MOI of 10 (Fig. 4). The highest titer for PtG-S2 was about 1.2 × 106 IU/ml at either temperature, and the induction kinetics were rather similar at both temperatures. Growth of the AxCANCre-infected PtG-S2 cells was strongly retarded after 4 days postinfection, and some cytopathic effects were observed thereafter. To test the genetic stability of the prepackaging cell line, the production of pseudotyped retrovirus was compared for PtG-S2 freshly grown from the cellular stocks kept in liquid N2 and the same cell line that had been cultured continuously for more than 3 months in the presence of blasticidin S and G418. The kinetics of virus induction after AxCANCre introduction remained unchanged during the passage of this cell line, and similar results were obtained when PtG-L1 was used instead. These results indicate that the prepackaging cell lines established here are quite stable during cellular proliferation.
FIG. 4.
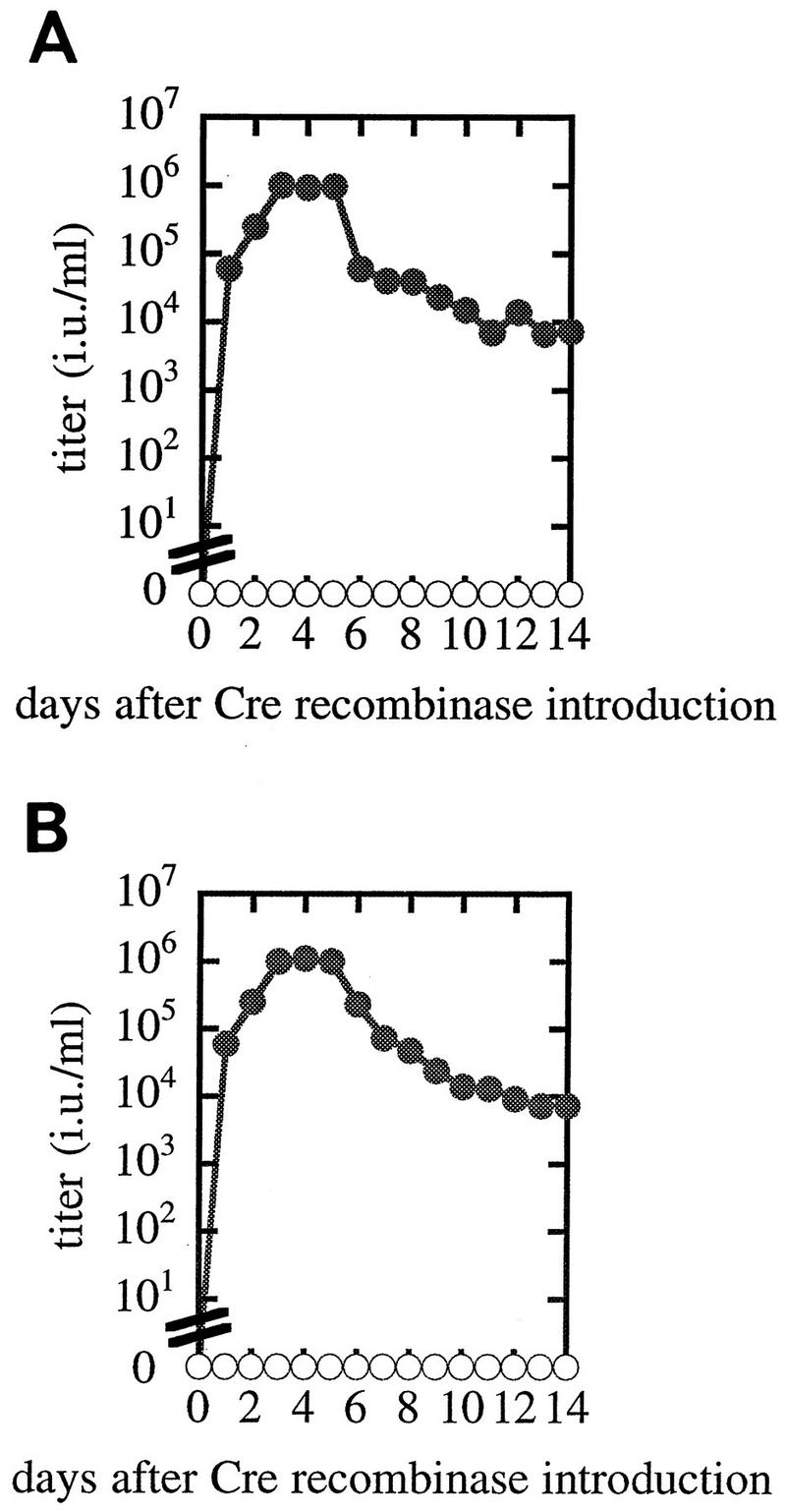
Time course of pseudotyped retrovirus production after transduction of Cre recombinase. Cre recombinase was transduced into each clone harboring MFGnlslacZ to induce VSV-G. Virus titers produced from PtG-S2 with (closed circles) or without (open circles) transduction with AxCANCre are shown. Cells were cultured at 37°C continuously (A) or cultured at 37°C until 2 days after transduction and then transferred to 32°C (B).
Characterization of VSV-G-pseudotyped retrovirus produced by prepackaging cell lines.
Virus stocks collected from induced PtG-S2 were concentrated by ultracentrifugation and titrated. These stocks could be concentrated to up to 109 IU/ml, with a yield of more than 80% (data not shown). We also observed that these virus stocks were more stable in human serum (data not shown), as expected from a previous report (33). To check whether the produced virus contains replication-competent retrovirus (1, 13), we incubated M. dunni cells with the pseudotyped virus stocks containing 5.0 × 106 IU and passaged them three times. The recovered culture medium was added to the indicator cell line, PG-4 S+L−, and kept for 5 days. No foci were detected in the culture, indicating that this virus stock of 5.0 × 106 IU contains less than one particle of replication-competent retrovirus.
We next tried to detect the adenovirus vector in pseudotyped retrovirus stocks by using a method reported previously (14). The pseudotyped virus stocks were collected every day from 3 to 5 days after adenovirus infection. These virus stocks were accumulated (total titer, 2.0 × 106 IU) and used to infect 293 cells, which can support the replication of AxCANCre. These cells were kept for 12 days but showed no cytopathic effects, eliminating the possibility of adenovirus contamination in the stock.
VSV-G-producing cells are fully susceptible to VSV-G-pseudotyped retroviruses.
The receptor for VSV-G has been reported to include anionic phospholipids, such as phosphatidylserine (18, 28). We were next interested in whether cells producing VSV-G could acquire resistance to VSV-G-pseudotyped retrovirus, as is observed in natural retroviruses (known as interference). When PtG-S2 or PtG-L1 without any vector genome was infected with MFGnlslacZ pseudotyped with VSV-G before or 4 days after AxCANCre introduction, almost the entire cell populations were infected with VSV-G-pseudotyped virus as judged from lacZ expression, independently of the AxCANCre infection (Fig. 5). We also observed that the diluted VSV-G pseudotypes (102 to 103 IU/ml) were able to infect both VSV-G-producing and nonproducing cells at similar infection efficiencies (data not shown). This result indicates that cells producing VSV-G, whether at low or high levels, are almost fully susceptible to the infection by VSV-G-pseudotyped retroviruses and further indicate that the VSV-G receptors were not saturated, unlike other natural receptors for retroviruses.
FIG. 5.
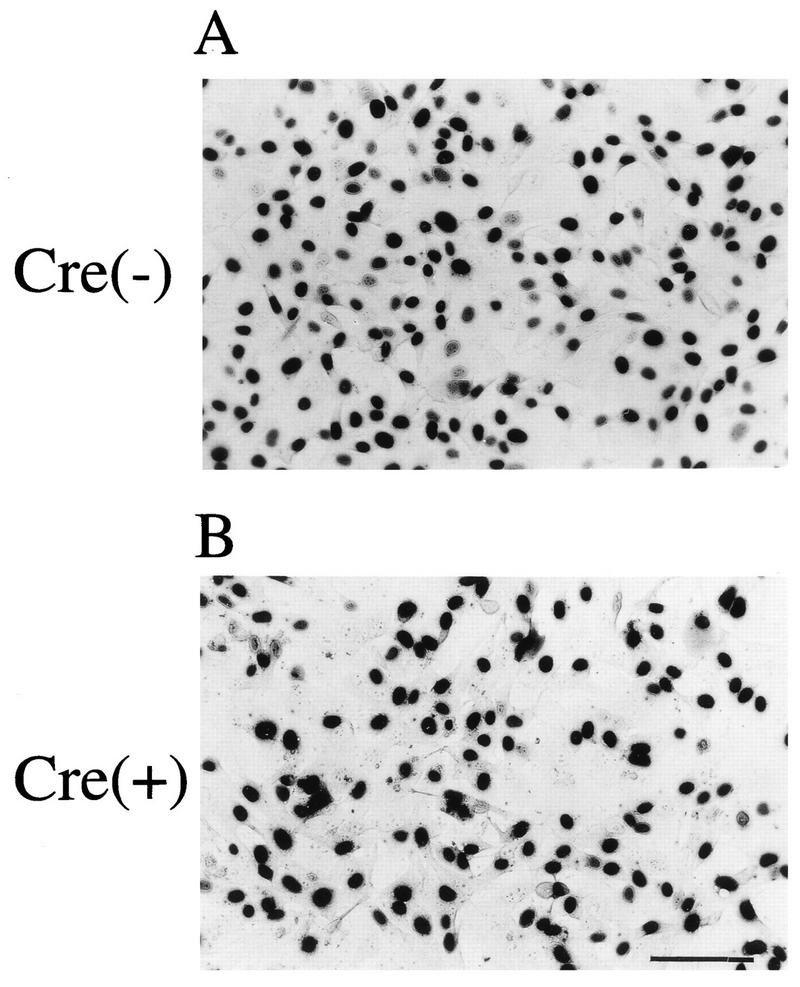
Transduction of VSV-G-pseudotyped retrovirus into PtG-S2 cells expressing VSV-G. A PtG-S2 culture, which was left uninfected (A) or was infected with AxCANCre at an MOI of 10 (B), was grown for 3 days longer and transduced with VSV-G-pseudotyped MFGnlslacZ. Two days after the transduction, the cells were fixed and cytochemically stained with X-Gal. Bar, 100 μm.
This property of VSV-G-producing cells potentially has two disadvantages for high-quality or high-titer virus production. The packaging cell line would accumulate vector DNA in their chromosomes by a “self-ping-pong” mechanism, causing their genetic instability, and recovery of the VSV-G pseudotype retrovirus from the culture medium would be reduced by its retransduction into the packaging cell lines which produced it. We have screened several reagents for the ability to suppress the transduction of the produced packaging cell lines and found that heparin (8 U/ml) inhibits the infection of the VSV-G pseudotype virus-producing cells drastically (reduced to less than 15% in the absence of Polybrene and to less than 2% in the presence of Polybrene). Immediately after this heparin-containing virus stock was diluted 10-fold with the medium, virus infectivity was fully recovered, indicating that this suppression is reversible. Indeed, addition of heparin to the PtG-S2 cultures 2 days after AxCANCre infection caused a two- to fourfold increase in the yield of VSV-G pseudotypes (data not shown). The heparin were able to be efficiently removed by subsequent ultracentrifugation.
DISCUSSION
We have presented here a unique system for the production of retrovirus vectors pseudotyped by VSV-G, the expression of which is cytotoxic for most mammalian cells, including FLY derivatives. In this study, the retrovirus vector was introduced into the prepackaging human cell line PtG-S2 by virus transduction, but DNA transfection would be one alternative. Since the prepackaging cell line contains an inducible transcriptional unit for the VSV-G gene with use of the Cre recombinase-loxP system (3, 7, 10, 26, 35), which is reported to switch the expression in an all-or-none manner (15, 16), we can select cell lines harboring the virus vector without any leaky production of transducible particles which might cause genetic instability of the cell lines during the selection and subsequent passaging procedures. The prepackaging cell line harboring the virus vector can be stocked in liquid N2 or kept in culture without any loss of virus-producing activity after the introduction of Cre recombinase. This genetic stability of the prepackaging cell lines is at least partly derived from the fact that the Neor and VSV-G genes are under the control of the same promoter. When Cre recombinase is introduced, the prepackaging cell lines are efficiently converted into packaging cell lines producing retrovirus pseudotyped by VSV-G at titers of more than 1.0 × 106 IU/ml. For the rapid and efficient introduction of Cre recombinase, we used a replication-defective adenovirus vector. DNA transfection would be an alternative for the introduction of the Cre recombinase gene. The fact that the prepackaging cell line originated from human cells has two advantages: the produced pseudotypes are resistant to human serum (our unpublished results), and the efficiency of adenovirus infection was probably higher than that with mouse cell lines.
For the isolation of prepackaging cell lines that express VSV-G at high levels after the introduction of Cre recombinase, we designed a system in which the same promoter is used for the Neor gene for the selection of prepackaging cell lines and for the VSV-G gene. Four days after Cre recombinase introduction, the Neor gene was precisely and almost completely excised from the chromosomal DNA (Fig. 2), resulting in a stringent expressional switch from the Neor gene to the VSV-G gene (Fig. 1B) as judged by protein analysis (Fig. 3). To select transfectants that efficiently transcribe the Neor gene at high levels, the Neor transcript was made very unstable by inserting an mRNA-destabilizing signal (29, 30). We showed here that this cloning design worked as expected, by comparing this construct and that without the mRNA-destabilizing element. Compared with PtG-L1, which has no mRNA-destabilizing sequence, PtG-S2 produced a smaller amount of Neor product, while PtG-S2 was converted into a producer cell line expressing much higher VSV-G levels after Cre recombinase introduction.
We also showed that cells expressing either a low or a high level of VSV-G proteins are fully susceptible to infection with VSV-G pseudotypes, consistent with the report that the VSV-G receptor contains ubiquitous anionic phospholipids. Interference would not be necessary for a lytic virus such as VSV. This means that a pseudotype can theoretically infect the same cell that produced it and implies that the packaging cell lines can accumulate virus vector during cell growth by a “self-ping-pong” mechanism. Long, leaky production of the pseudotypes would increase the possibility of genetic changes of the inducible packaging cell lines before virus induction. We believe that the pseudotype-producing system presented here is highly advantageous, in that we can control the VSV-G expression very stringently in PtG-S2 cells and can suppress the reentry of pseudotype retrovirus into the PtG-S2 cells by adding heparin. This system should be suitable for the large-scale production of pseudotypes for human gene therapy.
ACKNOWLEDGMENTS
We are grateful to Hidesaburo Hanafusa, Shih-Hui Liong, Satoshi Okazaki, and Takashi Kameda for helpful discussions. We thank Etsuko Endo and Michiru Tsukada for assistance in the preparation of the manuscript.
This work was supported in part by grants and endowments from Eisai Co., Ltd., and by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science and Culture, Japan.
REFERENCES
- 1.Bassin R H, Ruscetti S, Ali I, Haapala D K, Rein A. Normal DBA/2 mouse cells synthesize a glycoprotein which interferes with MCF virus infection. Virology. 1982;123:139–151. doi: 10.1016/0042-6822(82)90301-4. [DOI] [PubMed] [Google Scholar]
- 2.Beck E, Ludwig G, Auerswald E A, Reiss B, Schaller H. Nucleotide sequence and exact localization of the neomycin phosphotransferase gene from transposon Tn5. Gene. 1982;19:327–336. doi: 10.1016/0378-1119(82)90023-3. [DOI] [PubMed] [Google Scholar]
- 3.Bergemann J, Kühlcke K, Fehse B, Ratz I, Ostertag W, Lother H. Excision of specific DNA-sequences from integrated retroviral vectors via site-specific recombination. Nucleic Acids Res. 1995;23:4451–4456. doi: 10.1093/nar/23.21.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burns J C, Friedmann T, Driever W, Burrascano M, Yee J K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci USA. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen S T, Iida A, Guo L, Friedmann T, Yee J K. Generation of packaging cell lines for pseudotyped retroviral vectors of the G protein of vesicular stomatitis virus by using a modified tetracycline inducible system. Proc Natl Acad Sci USA. 1996;93:10057–10062. doi: 10.1073/pnas.93.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choulika A, Guyot V, Nicolas J F. Transfer of single gene-containing long terminal repeats into the genome of mammalian cells by a retroviral vector carrying the cre gene and the loxP site. J Virol. 1996;70:1792–1798. doi: 10.1128/jvi.70.3.1792-1798.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cosset F L, Takeuchi Y, Battini J L, Weiss R A, Collins M K L. High-titer packaging cells producing recombinant retroviruses resistant to human serum. J Virol. 1995;69:7430–7436. doi: 10.1128/jvi.69.12.7430-7436.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Emi N, Friedmann T, Yee J K. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J Virol. 1991;65:1202–1207. doi: 10.1128/jvi.65.3.1202-1207.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flowers C C, Woffendin C, Petryniak J, Yang S, Nabel G. Inhibition of recombinant human immunodeficiency virus type 1 replication by a site-specific recombinase. J Virol. 1997;71:2685–2692. doi: 10.1128/jvi.71.4.2685-2692.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujiwara K T, Ashida K, Nishina H, Iba H, Miyajima N, Nishizawa M, Kawai S. The chicken c-fos gene: cloning and nucleotide sequence analysis. J Virol. 1987;61:4012–4018. doi: 10.1128/jvi.61.12.4012-4018.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu H, Zou Y, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- 13.Haapala D K, Robey W G, Oroszlan S D, Tsai W P. Isolation from cats of an endogenous type C virus with a novel envelope glycoprotein. J Virol. 1985;53:827–833. doi: 10.1128/jvi.53.3.827-833.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanegae Y, Makimura M, Saito I. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn J Med Sci Biol. 1994;47:157–166. doi: 10.7883/yoken1952.47.157. [DOI] [PubMed] [Google Scholar]
- 15.Kanegae Y, Lee G, Sato Y, Tanaka M, Nakai M, Sakaki T, Sugano S, Saito I. Efficient gene activation in mammalian cells by using recombinant adenovirus expressing site-specific Cre recombinase. Nucleic Acids Res. 1995;23:3816–3821. doi: 10.1093/nar/23.19.3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanegae Y, Takamori K, Sato Y, Lee G, Nakai M, Saito I. Efficient gene activation system on mammalian cell chromosomes using recombinant adenovirus producing Cre recombinase. Gene. 1996;181:207–212. doi: 10.1016/s0378-1119(96)00516-1. [DOI] [PubMed] [Google Scholar]
- 17.Marshall E. Gene therapy’s growing pains. Science. 1995;269:1050–1055. doi: 10.1126/science.7652552. [DOI] [PubMed] [Google Scholar]
- 18.Mastromarino P, Conti C, Goldoni P, Hauttecoeur B, Orsi N. Characterization of membrane components of the erythrocyte involved in vesicular stomatitis virus attachment and fusion at acidic pH. J Gen Virol. 1987;68:2359–2369. doi: 10.1099/0022-1317-68-9-2359. [DOI] [PubMed] [Google Scholar]
- 19.Miller A D. Cell-surface receptors for retroviruses and implications for gene transfer. Proc Natl Acad Sci USA. 1996;93:11407–11413. doi: 10.1073/pnas.93.21.11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller D G, Adam M A, Miller A D. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 22.Ory D S, Neugeboren B A, Mulligan R C. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci USA. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roe T, Reynolds T C, Yu G, Brown P O. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993;12:2099–2108. doi: 10.1002/j.1460-2075.1993.tb05858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rose J K, Gallione C J. Nucleotide sequences of the mRNAs encoding the vesicular stomatitis virus G and M proteins determined from cDNA clones containing the complete coding regions. J Virol. 1981;39:519–528. doi: 10.1128/jvi.39.2.519-528.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rose J K, Bergmann J E. Expression from cloned cDNA of cell-surface secreted forms of the glycoprotein of vesicular stomatitis virus in eucaryotic cells. Cell. 1982;30:753–762. doi: 10.1016/0092-8674(82)90280-x. [DOI] [PubMed] [Google Scholar]
- 26.Russ A P, Friedel C, Grez M, von Melchner H. Self-deleting retrovirus vectors for gene therapy. J Virol. 1996;70:4927–4932. doi: 10.1128/jvi.70.8.4927-4932.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 28.Schlegel R, Tralka T S, Willingham M C, Pastan I. Inhibition of VSV binding and infectivity by phosphatidylserine: is phosphatidylserine a VSV-binding site? Cell. 1983;32:639–646. doi: 10.1016/0092-8674(83)90483-x. [DOI] [PubMed] [Google Scholar]
- 29.Shyu A B, Greenberg M E, Belasco J G. The c-fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes Dev. 1989;3:60–72. doi: 10.1101/gad.3.1.60. [DOI] [PubMed] [Google Scholar]
- 30.Shyu A B, Belasco J G, Greenberg M E. Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes Dev. 1991;5:221–231. doi: 10.1101/gad.5.2.221. [DOI] [PubMed] [Google Scholar]
- 31.Takeuchi Y, Cosset F L, Lachmann P J, Okada H, Weiss R A, Collins M K L. Type C retrovirus inactivation by human complement is determined by both the viral genome and producer cell. J Virol. 1994;68:8001–8007. doi: 10.1128/jvi.68.12.8001-8007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeuchi Y, Porter C D, Strahan K M, Preece A F, Gustafsson K, Cosset F L, Weiss R A, Collins M K. Sensitization of cells and retroviruses to human serum by (α1-3) galactosyltransferase. Nature (London) 1996;379:85–88. doi: 10.1038/379085a0. [DOI] [PubMed] [Google Scholar]
- 33.Takeuchi Y, Liong S-H, Bieniasz P D, Jäger U, Porter C D, Friedmann T, McClure M O, Weiss R A. Sensitization of rhabdo-, lenti-, and spumaviruses to human serum by galactosyl (α1-3) galactosylation. J Virol. 1997;71:6174–6178. doi: 10.1128/jvi.71.8.6174-6178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiss R A, Boettiger D, Murphy H M. Pseudotypes of avian sarcoma viruses with the envelope properties of vesicular stomatitis virus. Virology. 1977;76:808–825. doi: 10.1016/0042-6822(77)90261-6. [DOI] [PubMed] [Google Scholar]
- 35.Westerman K A, Leboulch P. Reversible immortalization of mammalian cells mediated by retroviral transfer and site-specific recombination. Proc Natl Acad Sci USA. 1996;93:8971–8976. doi: 10.1073/pnas.93.17.8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Y, Vanin E F, Whitt M A, Fornerod M, Zwart R, Schneiderman R D, Grosveld G, Nienhuis A W. Inducible, high-level production of infectious murine leukemia retroviral vector particles pseudotyped with vesicular stomatitis virus G envelope protein. Hum Gene Ther. 1995;6:1203–1213. doi: 10.1089/hum.1995.6.9-1203. [DOI] [PubMed] [Google Scholar]
- 37.Yee J K, Miyanohara A, LaPorte P, Bouic K, Burns J C, Friedmann T. A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proc Natl Acad Sci USA. 1994;91:9564–9568. doi: 10.1073/pnas.91.20.9564. [DOI] [PMC free article] [PubMed] [Google Scholar]