

Abstract
The E6 and E7 genes of the high-risk human papillomavirus (HPV) types encode oncoproteins, and both act by interfering with the activity of cellular tumor suppressor proteins. E7 proteins act by associating with members of the retinoblastoma family, while E6 increases the turnover of p53. p53 has been implicated as a regulator of both the G1/S cell cycle checkpoint and the mitotic spindle checkpoint. When fibroblasts from p53 knockout mice are treated with the spindle inhibitor nocodazole, a rereplication of DNA occurs without transit through mitosis. We investigated whether E6 or E7 could induce a similar loss of mitotic checkpoint activity in human keratinocytes. Recombinant retroviruses expressing high-risk E6 alone, E7 alone, and E6 in combination with E7 were used to infect normal human foreskin keratinocytes (HFKs). Established cell lines were treated with nocodazole, stained with propidium iodide, and analyzed for DNA content by flow cytometry. Cells infected with high-risk E6 were found to continue to replicate DNA and accumulated an octaploid (8N) population. Surprisingly, expression of E7 alone was also able to bypass this checkpoint. Cells expressing E7 alone exhibited increased levels of p53, while those expressing E6 had significantly reduced levels. The p53 present in the E7 cells was active, as increased levels of p21 were observed. This suggested that E7 bypassed the mitotic checkpoint by a p53-independent mechanism. The levels of MDM2, a cellular oncoprotein also implicated in control of the mitotic checkpoint, were significantly elevated in the E7 cells compared to the normal HFKs. In E6-expressing cells, the levels of MDM2 were undetectable. It is possible that abrogation of Rb function by E7 or increased expression of MDM2 contributes to the loss of mitotic spindle checkpoint control in the E7 cells. These findings suggest mechanisms by which both HPV oncoproteins contribute to genomic instability at the mitotic checkpoint.
Human papillomaviruses (HPVs) are small double-stranded DNA viruses that induce hyperproliferative lesions of cutaneous and mucosal epithelia. Half of the more than 70 identified types of HPVs specifically infect the genital epithelium. These genital papillomaviruses can be divided into low-risk types, which induce only benign lesions, and high-risk types, which are associated with the development of malignant lesions (28). More than 90% of cervical cancers contain HPV DNA of the high-risk types (11, 34, 41, 58, 69). The two transforming proteins encoded by the high-risk HPVs, E6 and E7, function through their associations with the tumor suppressor proteins, p53 and Rb, respectively (reviewed in reference 60). E6 facilitates the degradation of p53 through its association with an accessory protein, E6-AP, a component of the ubiquitin proteolytic pathway (29, 55, 57, 63). E7 proteins of the high-risk types bind to Rb (14, 50), as well as to other pocket proteins, such as p107 and p130 (7, 13), leading to the altered activities of these cell cycle regulators. The E6 proteins from the low-risk viruses fail to abrogate p53 functions, while the low-risk E7 proteins bind Rb with substantially reduced affinities (reviewed in references 28 and 56). These differences are likely responsible for the lack of association of the low-risk types with malignancy.
p53 is a site-specific DNA binding protein which activates expression of genes involved in cell cycle control such as the cyclin kinase inhibitor p21 (15). In response to DNA-damaging agents, p53 levels increase by a posttranscriptional mechanism resulting in arrest via inhibition of cyclin-associated kinase activity at the G1/S interface of the cell cycle (reviewed in reference 32). Loss of p53 or expression of mutant p53 results in a failure to arrest in G1, and p53-negative cells exhibit gene amplification, a marker of genomic instability (40, 68). p53 is commonly mutated in human cancers, many of which contain amplifications and aneuploid chromosomes, consistent with its role as a “guardian of the genome” (reviewed in references 36 and 39). In this capacity, p53 has been postulated to play a role in maintaining genomic integrity. All of these functions of p53 are inhibited by HPV E6 of the high-risk types (18, 20, 27, 31, 37, 38, 46).
The product of the retinoblastoma gene, Rb, in addition to p53, plays a significant role in the regulation of the cell cycle and its checkpoints. Prior to S phase, Rb, in complex with the transcription factor E2F, is hyperphosphorylated, leading to the release of E2F, which binds to promoters of numerous genes required for DNA synthesis (reviewed in reference 62). The binding of E7 to Rb inhibits the association of Rb with E2F, resulting in constitutive activation of E2F and expression of these genes (3). Furthermore, Rb can bind p107 and p130, which also negatively regulate E2F transcription (7, 14). In this way, E7 can modulate the cell cycle by inappropriately inducing S-phase progression. Additional studies with E7 have demonstrated the ability of E7 to bypass p21-mediated G1 arrest following DNA damage (8, 9, 25, 59). The mechanism for abrogation of this checkpoint is independent of p53 and likely acts through deregulation of E2F activity (26, 48, 54).
In addition to directing the G1/S checkpoint, p53 also functions in the mitotic spindle checkpoint at G2/M. Whereas the treatment of wild-type cells with the mitotic spindle inhibitor nocodazole results in arrest at G2/M, mouse embryo fibroblasts from p53 knockout mice treated with nocodazole continue to replicate their DNA, resulting in a significant polyploid population (6). The absence of p53 results in aberrant chromosomal replication, implicating it in control of this mitotic checkpoint. The loss of p53 function in cervical cancer, through the action of E6, could, by this mechanism, result in the acquisition of numerous genetic alterations and contribute to the multistep progress of this disease. In this study, we have examined the ability of the HPV oncoproteins E6 and E7 to inhibit the mitotic spindle checkpoint. Expression of either E6 or E7 was found to independently bypass the mitotic checkpoint. We believe that E6 is able to inhibit this function via degradation of p53. E7 appears to bypass this checkpoint in the presence of high levels of p53, possibly through the loss of Rb and/or the increased expression of the cellular oncoprotein MDM2.
MATERIALS AND METHODS
Cell culture.
Human foreskin keratinocytes (HFKs) were derived from neonatal human foreskin epithelium as previously described (22) and were maintained in serum-free keratinocyte growth medium (KGM; Clonetics). Retrovirally infected cells were grown in serum-containing medium (44) with mitomycin C (Boehringer Mannheim)-treated J2 3T3 fibroblast feeders (45) kindly provided by the Howard Green laboratory.
Infection of HFKs and cell lines.
The retrovirus vector and construction of the LXSN plasmids have been described previously (21). PA317 packaging cell lines generating HPV-16 E6, -16 E7, and -16 E6E7 recombinant retroviruses were provided by Denise Galloway. For infection of HFKs, 1.5 ml of the amphotropic viral supernatant was combined with 5 ml of KGM containing Polybrene (Sigma) (9 μg/ml) and added to subconfluent HFKs in a 100-mm dish. After 6 to 8 h, 10 ml of fresh KGM was added to the dish and cells were allowed to incubate for an additional 12 to 18 h. Cells were then split 1:3 and replated in serum-containing medium in the presence of fibroblast feeders. Selection with G418 sulfate (Gibco) (100 to 200 μg/ml) began 2 days postinfection and continued for 8 days. The LKP31 cell line was generated by transfection of HFKs with HPV-31 DNA as previously described (19).
Flow cytometry analyses.
For cell cycle analyses, cells were either untreated or treated with nocodazole (Sigma) (50 ng/ml) at various time points and harvested following trypsinization. Cells grown with fibroblast feeders were treated with EDTA prior to harvesting to remove feeders as described elsewhere (45). Harvested cells (1.5 × 106) were washed with phosphate-buffered saline and centrifuged, and the cell pellet was resuspended in 0.5 ml of stain solution (0.1 mg of propidium iodide [PI] per ml, 0.5 mg of RNase A per ml, 1% PBA–Triton [1 mg of bovine serum albumin per ml in phosphate-buffered saline–10% Triton X-100], 3.34 mM sodium citrate, 30 mg of polyethylene glycol per ml). Cells were then passed through an 18-gauge needle six times and incubated at 37°C for 20 min with shaking. An equal volume of hypertonic solution (0.1 mg of PI per ml, 1% PBA–Triton, 0.356 M NaCl, 30 mg of polyethylene glycol per ml) was added, the cell extracts were sheared again, and samples were stored at 4°C for at least 6 h. Stained nuclei were analyzed on a Becton-Dickinson (Mountain View, Calif.) FACScan with Lysis II software.
Antibodies and Western blot analyses.
Antibodies directed against p53 protein (Ab-2) and MDM2 (Ab-1) were obtained from Oncogene Science, Inc. Antibodies directed against p21 protein (clone 6B6) and cyclin B1 (clone GNS-1) were obtained from PharMingen. Antibodies against cdc2 p34 (17) were obtained from Santa Cruz Biotechnology, and horseradish peroxidase-linked sheep anti-mouse secondary antibody was obtained from Amersham. Nocodazole-treated and untreated cells were harvested by centrifugation (1,100 rpm; Beckman TJ6) following trypsinization and lysed with 0.5% Nonidet P-40 lysis buffer. HFK-16E7 cells were harvested after 96 h of nocodazole treatment; all other cell populations were harvested after 48 h. Protein concentrations were determined by Bradford assays, and 100 μg of whole-cell extract from each cell type was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were then transferred to a polyvinylidene difluoride membrane (Millipore) and incubated with 5% milk solution to block nonspecific binding. Primary and secondary antibodies were incubated with the membrane for 1 h and 30 min, respectively, followed by chemiluminescence detection as described by the manufacturer (ECL; Amersham).
RESULTS
Abrogation of mitotic checkpoint by E6.
p53 has been shown to play a role in regulation of the mitotic spindle checkpoint at the end of S phase in mouse embryo fibroblasts (6). In order to investigate whether this function of p53 can be inhibited by E6 proteins from high-risk HPV types, we examined the effects of E6 expression on cell cycle progression of nocodazole-treated HFKs. HFKs were infected with recombinant retroviruses which express the HPV-16 E6 gene or the -16 E6 and E7 genes together. Following selection for neomycin resistance, colonies were pooled, expanded, and treated with nocodazole. Flow cytometry analysis of DNA content was performed on uninfected and HPV oncoprotein-infected HFKs to determine their ability to arrest at G2/M after exposure to nocodazole (Fig. 1). Normal HFKs were found to accumulate with a 4N (tetraploid) DNA content after exposure to nocodazole (Fig. 1A and B), and very few cells continued to replicate to an 8N (octaploid) population. Untreated HFK-16E6 cells exhibited a cell cycle profile identical to that of normal HFKs (Fig. 1C) but failed to arrest DNA synthesis after nocodazole treatment. Instead, the cells remained in S phase, resulting in the generation of a substantial 8N population (Fig. 1D). Moreover, HFK-16E6E7 cells (Fig. 1E and F), as well as HFKs transfected with the HPV-31 genome (LKP31 cells) (Fig. 1G and H), exhibited a similar loss of G2/M arrest upon nocodazole treatment. Infections, treatments, and analyses were repeated at least three times for normal keratinocytes and E6- and E7-expressing cell lines.
FIG. 1.
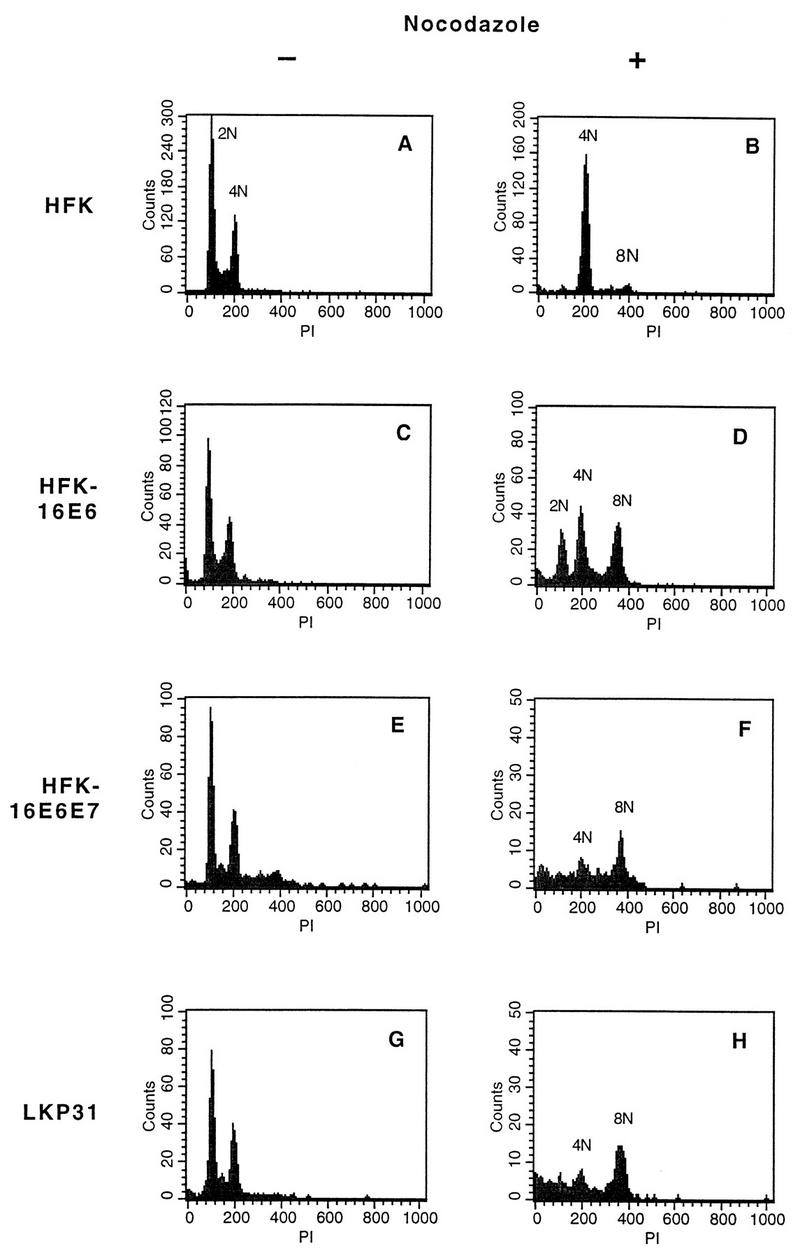
DNA content flow cytometry analysis of normal and HPV oncoprotein-expressing HFKs after nocodazole treatment. Cells were treated with nocodazole for 48 h and harvested, and nuclei were isolated and stained with PI. (A, C, E, and G) Untreated cells showing comparable DNA content profiles. 2N represents the DNA content of those cells in the G0/G1 phase of the cell cycle, while 4N indicates increased DNA content and is representative of cells in G2/M. (B) HFKs treated with nocodazole showing an accumulation of cells at a DNA content of 4N. (D, F, and H) Nocodazole-treated cells demonstrating a loss of the G2/M checkpoint and subsequent accumulation at 8N.
In order to monitor the progression of cell populations through the cell cycle after nocodazole treatment, we repeated the above analysis at successive time points up to 48 h of nocodazole treatment. As shown in Fig. 2, HFK-16E6 cells demonstrated a gradual change in DNA content from 2N into 4N and 8N populations. Analysis of HFK-16E6E7 as well as LKP31 cells showed that the two cell lines exhibited similar effects, whereas low-risk E6-alone cells did not bypass the checkpoint (data not shown). These data indicate that normal human keratinocytes contain a mitotic checkpoint that can be abrogated by high-risk E6 protein alone, E6 in combination with E7, or E6 in the context of the entire HPV-31 genome.
FIG. 2.

DNA content flow cytometry time course analysis of HFK-16E6 cells after nocodazole treatment. Cells were either left untreated (UT) or treated with nocodazole for the indicated times. DNA content profiles demonstrate a gradual, but not complete, loss of cells in the G0/G1 phase and gain of an 8N DNA content population upon nocodazole treatment.
Abrogation of mitotic checkpoint by E7.
To examine whether E7 also affected the mitotic spindle checkpoint, normal human keratinocytes were infected with retroviruses expressing HPV-16 E7 and neomycin-resistant colonies were pooled and expanded. We first observed that HFK-16E7 cells proliferated at a rate much lower than that of normal HFKs and HFK-16E6 cells. By measuring growth rates in tissue culture, we determined that HFK-16E7 cells exhibit approximately a twofold increase in doubling time compared to normal keratinocytes and other keratinocytes expressing HPV oncoproteins, which have comparable doubling times (data not shown). In order to compare effects in HFK-16E7 cells with those in HFK-16E6 cells after nocodazole treatment, it was important to examine them at similar points relative to their cycling times, since the accumulation in 8N for the E6-expressing cells occurred only after an additional cell cycle (Fig. 2). From the above data, we determined that nocodazole treatment of E7 cells for 96 h would be equivalent to 48-h treatment for the other cell lines. DNA content flow cytometry analysis was therefore performed on HFK-16E7 cells at progressive time points up to 96 h (Fig. 3). Unexpectedly, we observed that, in the presence of nocodazole, HFK-16E7 cells were able to overcome the mitotic spindle checkpoint equally as well as were HFK-16E6 cells. The accumulation of cells with an 8N DNA content can be observed in HFK-16E7 cells as early as 48 h after nocodazole treatment. Low-risk E7 cells did not overcome the spindle checkpoint following nocodazole treatment (data not shown). Furthermore, normal HFKs treated with nocodazole for 96 h still did not bypass the checkpoint and accumulated with a 4N DNA content (data not shown). This indicates that the effect of E7 on the mitotic spindle checkpoint was not due to the extended length of treatment. These data indicate a role for high-risk E7 in abrogation of the mitotic spindle checkpoint.
FIG. 3.

DNA content flow cytometry time course analysis of HFK-16E7 cells after nocodazole treatment. Cells were either untreated (UT) or treated with nocodazole at the indicated times. HFK-16E7 cells exhibit a loss of the G2/M checkpoint with no cells in G0/G1 and a gain of cells at 8N after nocodazole treatment.
A summary of the cell cycle distribution of all cells analyzed expressed as population percentages with and without nocodazole treatment is shown in Table 1. All cells expressing HPV oncoproteins exhibit obvious increases in the percentage of cells reaching a DNA content of 8N, ranging from 29% for HFK-16E7 to 46% for LKP31. Normal HFKs have less than 5% of cells falling in this population after nocodazole treatment. These data further confirm that E6 and E7 can independently abrogate the mitotic spindle checkpoint.
TABLE 1.
Summary of cell cycle distribution of normal and HPV oncoprotein-expressing HFKs after nocodazole treatment
| Cell population | Nocodazole | % of cells with ploidy:
|
||
|---|---|---|---|---|
| 2N | 4N | 8N | ||
| HFK | − | 60 | 39 | 1 |
| + | 3 | 90 | 5 | |
| HFK-16E6 | − | 60 | 37 | 1 |
| + | 23 | 41 | 32 | |
| HFK-16E7 | − | 65 | 30 | 2 |
| + | 7 | 41 | 29 | |
| HFK-16E6E7 | − | 45 | 42 | 12 |
| + | 13 | 35 | 41 | |
| LKP31 | − | 54 | 42 | 2 |
| + | 18 | 25 | 46 | |
Levels of p53 and p21 in nocodazole-treated cells.
The observation that E6 can abrogate the mitotic spindle checkpoint was not unexpected, given the presumed role of p53 and the ability of E6 to facilitate its degradation. However, it was less clear how E7 could alter this checkpoint. Either E7 could target p53, or alternatively, it could act through other, unknown components of the checkpoint. In order to investigate the mechanism by which E7 can overcome G2/M arrest, we first analyzed the levels of p53 protein before and after nocodazole treatment of both E6- and E7-expressing keratinocytes. One possibility was that nocodazole-treated E7 cells had dramatically reduced levels of p53. As shown in Fig. 4A, normal HFKs contain significant levels of p53, which are increased twofold after nocodazole treatment (lanes 1 and 2). Cells expressing E6, whether alone, in combination with E7, or in the context of the entire HPV-31 genome, have decreased levels of p53 due to the ability of E6 to facilitate its degradation (lanes 3, 4, and 7 to 10). Consistent with previous reports, HFK-16E7 cells contain higher levels of p53 compared to normal HFKs (lanes 5 and 6) (10, 54). Following nocodazole treatment, cells expressing E6 alone or in combination with E7 did not demonstrate significant changes in levels of p53 (lanes 4 and 8). However, cells expressing the entire HPV-31 genome exhibited elevated, though still low, levels of p53 after nocodazole treatment (lane 10). This is similar to the increase in p53 levels seen following treatment with actinomycin D, which arrests cells at G1/S; however, the molecular basis for it is unclear.
FIG. 4.

Western blot analysis of normal and HPV oncoprotein-expressing keratinocytes before and after nocodazole treatment for cyclin B and cdc2. One hundred micrograms of whole-cell extract was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis transferred to a polyvinylidene difluoride membrane, and Western blot analysis was performed. Odd-numbered lanes represent untreated samples, and even-numbered lanes show nocodazole-treated samples. (A) p53 protein levels. (B) p21 protein levels.
To examine whether the p53 present in the oncoprotein-expressing cells was functional, we carried out Western blotting analyses for the kinase inhibitor p21. p21 is transcriptionally regulated by p53, and the levels of p21 have been shown to parallel increases in p53 (reviewed in reference 32). As shown in Fig. 4B, the p21 protein levels directly correlate with the levels of p53, indicating that the p53 present is functional, at least in its role as a transcriptional transactivator. Most importantly, the levels of p21 increased following nocodazole treatment of E7-expressing cells. The ability of E7-containing cells to overcome this checkpoint appears to be independent of p53, since both levels and activity remain high. This finding is reminiscent of the ability of E7 to bypass the G1/S checkpoint after DNA damage. While both checkpoints are thought to be controlled by p53, E7 does not diminish the activity or levels of p53.
MDM2 protein levels in nocodazole-treated cells.
We wanted to further examine the mechanism by which HFK-16E7 cells were able to bypass the mitotic checkpoint. Recently, Lundgren et al. (42) examined the role of the MDM2 cellular oncoprotein in the cell cycle and in tumorigenesis in a p53 null background. Their data indicate that overexpression of MDM2 in mammary cells resulted in multiple rounds of S phase without intervening mitosis. The phenotype is similar in p53 wild-type and in p53 null backgrounds. Because it appeared that HFK-16E7 cells were bypassing the spindle checkpoint independently of p53, we examined the levels of MDM2 before and after nocodazole treatment to see if expression of this protein could be contributing to aneuploidy (Fig. 5). The levels of MDM2 in HFK-16E7 cells (lanes 5 and 6) were found to be significantly increased above that seen either in HFKs (lanes 1 and 2) or in E6-expressing cells (lanes 3 and 4), whose levels were very low or undetectable. Interestingly, HFK-16E6E7 and LKP31 cells also have undetectable levels of MDM2 (lanes 7, 8, 9, and 10), indicating that expression of E7 alone is not sufficient to induce overexpression of MDM2. It is likely that E7 activates factors which function synergistically with p53 to increase MDM2 levels. These studies were repeated three times with similar results. Given the previously documented role of MDM2 in mediating an abrogation of the spindle checkpoint, it is possible that its elevated expression may contribute to E7’s ability to act in this process.
FIG. 5.

Western blot analysis comparing levels of MDM2 protein in normal and oncoprotein-expressing keratinocytes after nocodazole treatment. Samples were prepared as described in Materials and Methods. Odd-numbered lanes represent untreated samples, and even-numbered lanes represent nocodazole-treated samples.
Cyclin B and cdc2 levels in nocodazole-treated cells.
Additional cell cycle regulators of the G2/M phase of the cell cycle may also be affected by the expression of the HPV oncoproteins and consequently contribute to the ability of these cells to bypass the mitotic spindle checkpoint. Therefore, we examined the levels of cyclin B and the cdc2 kinase in nocodazole-treated cells, which are required for entrance into mitosis. As shown in Fig. 6A, cyclin B levels are reduced in nocodazole-treated normal HFKs compared to untreated cells (lanes 1 and 2). All other cells tested which express the HPV oncoproteins (lanes 3 to 10) show little or no decrease in the levels of cyclin B after nocodazole treatment. Furthermore, cdc2 levels (Fig. 6B) are also decreased in normal HFKs after nocodazole treatment (lanes 1 and 2) but remain high for all other cell lines tested (lanes 3 to 10). The presence of a noncycling, quiescent population in the E6-expressing cells (see 2N population [Fig. 1D]) may account for the lower levels of cyclin B and cdc2. However, the levels of cyclins and cdc2 remain unchanged before and after nocodazole treatment (lanes 3 and 4). These data suggest that expression of the HPV oncoproteins alone, or in combination, does not lead to decreased expression of cyclin B and cdc2 following nocodazole treatment.
FIG. 6.

Western blot analysis of normal and HPV oncoprotein-expressing keratinocytes before and after nocodazole treatment for cyclin B and cdc2. Samples were prepared as described in Materials and Methods. Odd-numbered lanes represent untreated samples, and even-numbered lanes show nocodazole-treated samples. (A) Cyclin B protein levels. (B) cdc2 protein levels.
DISCUSSION
The present study demonstrates the ability of both E6 and E7 oncoproteins of the HPV high-risk types to bypass the mitotic spindle checkpoint in human keratinocytes. p53 has been previously shown to be a component of this checkpoint, and in its absence, cells inappropriately replicate their DNA without intervening mitoses when treated with a mitotic spindle inhibitor (6). E6-expressing cells most likely overcome this checkpoint through their ability to facilitate the degradation of p53, since the levels of p53 are dramatically reduced in these cells. This would be consistent with our observation that E6 proteins from low-risk types, which fail to induce p53 degradation, cannot overcome the mitotic checkpoint.
Cells expressing E7 alone can also overcome the mitotic checkpoint. Levels and activity of p53 are high in E7 cells, indicating a p53-independent mechanism. E7 functions in transformation through binding and inhibiting the activities of Rb, another tumor suppressor protein (reviewed in reference 49). We observed that cells expressing low-risk E7, which binds Rb with significantly reduced affinity compared to high-risk E7, are unable to bypass the mitotic checkpoint. This suggests that deregulation of Rb function may contribute to loss of this checkpoint. During preparation of this manuscript, studies by Wahl and coworkers (12) were published which show that loss of Rb function in Rb−/− fibroblasts resulted in loss of the mitotic spindle checkpoint. This is consistent with our findings and points to one mechanism by which E7 cells can bypass the checkpoint.
An additional mechanism which may contribute to the ability of E7-expressing cells to overcome the spindle checkpoint stems from our observation that the levels of MDM2 are elevated only in E7 cells. Previous studies have shown that overexpression of MDM2, a cellular oncogene, can lead to multiple rounds of S-phase replication without intervening mitosis in mammary epithelial cells (42). Most importantly, this phenotype is similar in p53 wild-type and p53 null backgrounds. In the E7-alone-expressing cells, high levels of functional p53 are present, as demonstrated by inducible p21 expression. Despite these high levels, E7 can overcome this checkpoint, suggesting that its action is independent of p53. It is possible that both loss of Rb and overexpression of MDM2 can act cooperatively to result in loss of the mitotic spindle checkpoint.
MDM2 was originally identified as the product of the murine double minute 2 gene from a spontaneously transformed BALB/c 3T3 cell line (2). Overexpression of MDM2 has been shown to increase the tumorigenic potential of cells in culture (16) and has been found to be amplified in human sarcomas (51). The ability of MDM2 to promote tumorigenesis is a result of its interactions with tumor suppressor proteins such as p53. The product of the mdm2 oncogene forms a complex with p53 and inhibits p53-mediated transactivation (47, 52), growth suppression (17), G1 arrest following DNA damage, and apoptosis (4, 5, 23). Interestingly, MDM2 expression is transcriptionally activated by p53 (1, 30, 65), indicating the presence of an autoregulatory loop between the two proteins. Other interactions of MDM2 include its association with Rb (66). This interaction results in the deregulation of E2F-DP1 activity, normally negatively controlled by Rb, leading to stimulation of S phase entry. Furthermore, MDM2 stimulates the transcriptional activity of two cooperating transcription factors, E2F and DP1, which enhances the stimulation of S phase (43).
It is interesting that HFK-16E6E7 cells and LKP31 cells do not express high levels of MDM2 since they, too, express E7. These cells contain low levels of p53, which may explain the lack of MDM2 protein since MDM2 is transcriptionally activated by p53. These data suggest that p53 may be required for MDM2 expression; however, our data indicate that expression of p53 alone is not sufficient to induce high levels of MDM2. For instance, normal HFKs treated with nocodazole contain levels of p53 similar to that seen in HFK-16E7 cells and yet do not have detectable levels of MDM2. Furthermore, expression of E7 alone is not sufficient to induce expression of MDM2, since HFK-16E6E7 and LKP31 cells do not have detectable levels of MDM2. We conclude that E7 may activate factors which function synergistically with p53 to increase MDM2 levels. Finally, recent work by two different groups has found that MDM2 is able to promote the degradation of p53 (24, 33) and that there is an inverse correlation between the levels of MDM2 and those of p53; high levels of MDM2 were shown to correlate with reduced levels of p53. We do not see decreased levels of p53 due to overexpression of MDM2 in the HFK-16E7 cells, demonstrating that this correlation is not true in all cases.
In addition to p53 and MDM2, p21 has been implicated in control of the mitotic spindle checkpoint. Work from the Vogelstein laboratory has shown that cells lacking p21 are able to undergo additional S phases without intervening mitoses when arrested in a G2-like state (61). Unlike those authors’ findings, our E7-alone-expressing cells have elevated levels of p21 and yet still undergo rereplication through a mechanism which appears to be independent of p53-p21 activity. Furthermore, recent reports have demonstrated that some tumor cell lines contain elevated levels of both p53 and MDM2 but lack significant levels of p21 (35). It is possible that p53 is not fully functional in these tumor lines. In our studies, E7-alone-expressing cells contain elevated levels of p53 and MDM2, as well as functional p21. E7 cells, therefore, appear to be unusual in that they express high levels of all three of these cell cycle regulators.
Previous studies by other groups have examined the role of the HPV oncoproteins in alteration of genomic stability. White et al. demonstrated that expression of E6 in human fibroblasts results in a failure to arrest in G1 and G2 following exposure to the metabolic inhibitor PALA as well as CAD gene amplification (64). In contrast, E7-expressing cells display massive cell death with the rare appearance of PALA-resistant aneuploid cells, which occurs by a p53-independent mechanism. This indicates that each of the HPV oncoproteins alters distinct pathways, resulting in different types of genomic instability. The alteration of cyclin-CDK complexes by E6, but not by E7, is implicated in this loss of genomic stability (67). Likewise, Reznikoff et al. (53) reported statistically significant differences in genomic stability between E6- and E7-immortalized human uroepithelial cells. In our study, we observe a similar loss of cell cycle control for E6 and E7 in their natural host cells, keratinocytes. Both HPV oncoproteins, through different mechanisms, are able to overcome the mitotic spindle checkpoint, leading to altered genomic integrity.
We conclude that both HPV oncoproteins contribute to genomic instability at the mitotic checkpoint: E6 through degradation of p53, a component of the checkpoint, and E7 through a p53-independent mechanism. The loss of Rb as well as the increased levels of MDM2 in the E7 cells may play a role in the loss of this checkpoint by providing the signal to undergo DNA synthesis but not allowing for a complete transition through mitosis. Both of these mechanisms afford the accumulation of genetic alterations, which may contribute to the progressive nature of cervical cancer.
ACKNOWLEDGMENTS
We thank Denise Galloway for the LXSN retroviral constructs. We gratefully acknowledge Margaret Ruesch, Neil Clipstone, Kathy Rundell, and Mary Hummel for critical review of the manuscript and the members of the Laimins laboratory for helpful discussions. We also thank Stephanie Gaillard and Robert Caldwell for technical advice.
This work was supported by the Carcinogenesis Training Grant (5T32 CA09560-12) and a Gramm Fellowship Award from Northwestern University to J.T.T. and by a grant to L.A.L. from the National Institute of Allergy and Infectious Diseases (AI31494).
REFERENCES
- 1.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cahilly-Snyder L, Yang-Feng T, Francke U, George D L. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somatic Cell Mol Genet. 1987;13:235–244. doi: 10.1007/BF01535205. [DOI] [PubMed] [Google Scholar]
- 3.Chellappan S, Hiebert S, Mudryj M, Horowitz J, Nevins J. The E2F transcription factor is a cellular target for the Rb protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 4.Chen C, Oliner J D, Zhan Q, Fornace A J, Jr, Vogelstein B, Kastan M B. Interaction between p53 and MDM2 in a mammalian cell cycle checkpoint pathway. Proc Natl Acad Sci USA. 1994;91:2684–2688. doi: 10.1073/pnas.91.7.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Wu X, Lin J, Levine A J. mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol Cell Biol. 1996;16:2445–2452. doi: 10.1128/mcb.16.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cross S M, Sanchez C A, Morgan C A, Schimke M K, Ramel S, Idzerda R L, Raskind W H, Reid B J. A p53-dependent mouse spindle checkpoint. Science. 1995;267:1353–1356. doi: 10.1126/science.7871434. [DOI] [PubMed] [Google Scholar]
- 7.Davies R, Hicks R, Crook T, Morris J, Vousden K. Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J Virol. 1993;67:2521–2528. doi: 10.1128/jvi.67.5.2521-2528.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Demers G, Foster S, Halbert C, Galloway D. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7 protein. Proc Natl Acad Sci USA. 1994;91:4382–4386. doi: 10.1073/pnas.91.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demers G W, Espling E, Harry J B, Etscheid B G, Galloway D A. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol. 1996;70:6862–6869. doi: 10.1128/jvi.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demers G W, Halbert C L, Galloway D A. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology. 1994;198:169–174. doi: 10.1006/viro.1994.1019. [DOI] [PubMed] [Google Scholar]
- 11.de Villiers E-M. Human pathogenic papillomavirus types: an update. Curr Top Microbiol Immunol. 1994;186:1–12. doi: 10.1007/978-3-642-78487-3_1. [DOI] [PubMed] [Google Scholar]
- 12.Di Leonardo A, Khan S H, Linke S P, Greco V, Seidita G, Wahl G M. DNA replication in the presence of mitotic spindle inhibitors in human and mouse fibroblasts lacking either p53 or pRb function. Cancer Res. 1997;57:1013–1019. [PubMed] [Google Scholar]
- 13.Dyson N, Guida P, Munger K, Harlow E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interactions with the same set of cellular proteins. Science. 1992;243:934–936. doi: 10.1128/jvi.66.12.6893-6902.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dyson N, Howley P, Munger K, Harlow E. The human papillomavirus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 15.El-Diery W S, Tokino T, Velculescu D B, Levy D B, Parsons J M, Trent D L, Mercer W E, Kinzler K W, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 16.Fakharzadeh S S, Trusko S P, George D L. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991;10:1565–1569. doi: 10.1002/j.1460-2075.1991.tb07676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finlay C A. The mdm-2 oncogene can overcome wild-type p53 suppression of transformed cell growth. Mol Cell Biol. 1993;13:301–306. doi: 10.1128/mcb.13.1.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foster S A, Demers G W, Etscheid B G, Galloway D A. The ability of human papillomavirus E6 proteins to target p53 for degradation in vivo correlates with their ability to abrogate actinomycin D-induced growth arrest. J Virol. 1994;68:5698–5705. doi: 10.1128/jvi.68.9.5698-5705.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frattini M G, Lim H B, Laimins L A. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc Natl Acad Sci USA. 1996;93:3062–3067. doi: 10.1073/pnas.93.7.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu Z, Pim D, Labrecque S, Banks L, Matlashewski G. DNA damage induced p53 mediated transcription is inhibited by human papillomavirus type 18 E6. Oncogene. 1994;9:629–633. [PubMed] [Google Scholar]
- 21.Halbert C L, Demers G W, Galloway D A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Halbert C L, Demers G W, Galloway D A. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J Virol. 1992;66:2125–2134. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haupt Y, Barak Y, Oren M. Cell type-specific inhibition of p53-mediated apoptosis by mdm2. EMBO J. 1996;15:1596–1606. [PMC free article] [PubMed] [Google Scholar]
- 24.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 25.Hickman E, Picksley S, Vousden K H. Cells expressing HPV 16 E7 continue cell cycle progression following DNA damage induced p53 activation. Oncogene. 1994;9:2177–2181. [PubMed] [Google Scholar]
- 26.Hickman E S, Bates S, Vousden K H. Perturbation of the p53 response by human papillomavirus type 16 E7. J Virol. 1997;71:3710–3718. doi: 10.1128/jvi.71.5.3710-3718.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoppe-Seyler F, Butz K. Repression of endogenous p53 transactivation function in HeLa cervical carcinoma cells by human papillomavirus type 16 E6, human mdm-2, and mutant p53. J Virol. 1993;67:3111–3117. doi: 10.1128/jvi.67.6.3111-3117.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howley P M. Papillomavirinae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fundamental virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 947–978. [Google Scholar]
- 29.Huibregtse J M, Scheffner M, Howley P M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10:4126–4135. doi: 10.1002/j.1460-2075.1991.tb04990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Juven T, Barak Y, Zauberman A, George D, Oren M. Wild type p53 can mediate sequence-specific transactivation of an internal promoter within the mdm2 gene. Oncogene. 1993;8:3411–3416. [PubMed] [Google Scholar]
- 31.Kessis T D, Slebos R J, Nelson W G, Kastan M B, Plunkett B S, Han S M, Lorincz A T, Hedrick L, Cho K R. Human papillomavirus 16 E6 expression disrupts the p53-mediated cellular response to DNA damage. Proc Natl Acad Sci USA. 1993;90:3988–3992. doi: 10.1073/pnas.90.9.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ko L J, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 33.Kubbutat M H G, Jones S N, Vousden K H. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 34.Laimins L A. The biology of human papillomavirus: from warts to cancer. Infect Agents Dis. 1993;2:74–86. [PubMed] [Google Scholar]
- 35.Landers J E, Cassel S L, George D L. Translational enhancement of mdm2 oncogene expression in human tumor cells containing a stabilized wild type p53 protein. Cancer Res. 1997;57:3562–3568. [PubMed] [Google Scholar]
- 36.Lane D P. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 37.Lechner M S, Laimins L A. Inhibition of p53 DNA binding by human papillomavirus E6 proteins. J Virol. 1994;68:4262–4273. doi: 10.1128/jvi.68.7.4262-4273.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lechner M S, Mack D H, Finicle A B, Crook T, Vousden K H, Laimins L A. Human papillomavirus E6 proteins bind p53 in vivo and abrogate p53-mediated repression of transcription. EMBO J. 1992;11:3045–3052. doi: 10.1002/j.1460-2075.1992.tb05375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levine A J, Momand J, Finlay C A. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 40.Livingstone L R, White A, Sprouse J, Livanos E, Jacks T, Tlsty T D. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 41.Lowy D R, Kirnbauer R, Schiller J T. Genital human papillomavirus infection. Proc Natl Acad Sci USA. 1994;91:2436–2440. doi: 10.1073/pnas.91.7.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lundgren K, Montes de Oca Luna R, McNeill Y B, Emerick E P, Spencer B, Barfield C R, Lozano G, Rosenberg M P, Finlay C A. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. 1997;11:714–725. doi: 10.1101/gad.11.6.714. [DOI] [PubMed] [Google Scholar]
- 43.Martin K, Trouche D, Hagemeier C, Sorenson T S, La Thangue N B, Kouzarides T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature. 1995;375:691–694. doi: 10.1038/375691a0. [DOI] [PubMed] [Google Scholar]
- 44.Meyers C, Frattini M G, Hudson J B, Laimins L A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science. 1992;257:971–973. doi: 10.1126/science.1323879. [DOI] [PubMed] [Google Scholar]
- 45.Meyers C, Laimins L A. In vitro systems for the study and propagation of human papillomaviruses. Curr Top Microbiol Immunol. 1994;186:199–215. doi: 10.1007/978-3-642-78487-3_11. [DOI] [PubMed] [Google Scholar]
- 46.Mietz J A, Unger T, Huibregtse J M, Howley P M. The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. EMBO J. 1992;11:5013–5020. doi: 10.1002/j.1460-2075.1992.tb05608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Momand J, Zambetti G P, Olson D C, George D, Levine A J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 48.Morozov A, Shiyanov P, Barr E, Leiden J M, Raychaudhuri P. Accumulation of human papillomavirus type 16 E7 protein bypasses G1 arrest induced by serum deprivation and by the cell cycle inhibitor p21. J Virol. 1997;71:3451–3457. doi: 10.1128/jvi.71.5.3451-3457.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Munger K, Scheffner M, Huibregtse J M, Howley P M. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 1992;12:197–217. [PubMed] [Google Scholar]
- 50.Munger K, Werness B, Dyson N, Phelps W, Harlow E, Howley P. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oliner J D, Kinzler K W, Meltzer P S, George D L, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 52.Oliner J D, Pietenpol J A, Thiagalingam S, Gyuris J, Kinzler K W, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 53.Reznikoff C A, Belair C, Savelieva E, Zhai Y, Pfeifer K, Yeager T, Thompson K J, DeVries S, Bindley C, Newton M A, Sekhon G, Waldman F. Long-term genome stability and minimal genotypic and phenotypic alterations in HPV16 E7-, but not E6-, immortalized human uroepithelial cells. Genes Dev. 1994;8:2227–2240. doi: 10.1101/gad.8.18.2227. [DOI] [PubMed] [Google Scholar]
- 54.Ruesch M N, Laimins L A. Initiation of DNA synthesis by human papillomavirus E7 oncoproteins is resistant to p21-mediated inhibition of cyclin E-cdk2 activity. J Virol. 1997;71:5570–5578. doi: 10.1128/jvi.71.7.5570-5578.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scheffner M, Huibregtse J M, Vierstra R D, Howley P M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 56.Scheffner M, Romanczuk H, Munger K, Huibregtse J M, Mietz J A, Howley P M. Functions of human papillomavirus proteins. Curr Top Microbiol Immunol. 1994;186:83–99. doi: 10.1007/978-3-642-78487-3_5. [DOI] [PubMed] [Google Scholar]
- 57.Scheffner M, Werness B A, Huibregtse J M, Levine A J, Howley P M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 58.Schiffman M H. Epidemiology of cervical human papillomavirus infections. Curr Top Microbiol Immunol. 1994;186:55–81. doi: 10.1007/978-3-642-78487-3_4. [DOI] [PubMed] [Google Scholar]
- 59.Slebos R J, Lee M H, Plunkett B S, Kessis T D, Williams B O, Jacks T, Hedrick L, Kastan M B, Cho K R. p53-dependent G1 arrest involves pRb-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proc Natl Acad Sci USA. 1994;91:5320–5324. doi: 10.1073/pnas.91.12.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vousden K H. Interactions between papillomavirus proteins and tumor suppressor gene products. Adv Cancer Res. 1994;64:1–24. doi: 10.1016/s0065-230x(08)60833-7. [DOI] [PubMed] [Google Scholar]
- 61.Waldman T, Lengauer C, Kinzler K W, Vogelstein B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature. 1996;381:713–716. doi: 10.1038/381713a0. [DOI] [PubMed] [Google Scholar]
- 62.Weinberg R A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 63.Werness B A, Levine A J, Howley P M. Association of human papillomavirus types 16 and 18 proteins with p53. Science. 1990;248:76–79. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 64.White A E, Livanos E M, Tlsty T D. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins. Genes Dev. 1994;8:666–677. doi: 10.1101/gad.8.6.666. [DOI] [PubMed] [Google Scholar]
- 65.Wu X, Bayle J H, Olson D, Levine A J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 66.Xiao Z, Chen J, Levine A J, Modjtahedi N, Xing J, Sellers W R, Livingston D M. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature. 1995;375:694–698. doi: 10.1038/375694a0. [DOI] [PubMed] [Google Scholar]
- 67.Xiong Y, Kuppuswamy D, Li Y, Livanos E M, Hixon M, White A, Beach D, Tlsty T D. Alteration of cell cycle kinase complexes in human papillomavirus E6- and E7-expressing fibroblasts precedes neoplastic transformation. J Virol. 1996;70:999–1008. doi: 10.1128/jvi.70.2.999-1008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yin Y, Tainsky M A, Bischoff F Z, Strong L C, Wahl G M. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- 69.zur Hausen H, De Villiers E. Human papillomaviruses. Annu Rev Microbiol. 1994;48:427–447. doi: 10.1146/annurev.mi.48.100194.002235. [DOI] [PubMed] [Google Scholar]