

Abstract
Activation of RNase L by 2′,5′-linked oligoadenylates (2-5A) is one of the antiviral pathways of interferon action. To determine the involvement of the 2-5A system in the control of human immunodeficiency virus type 1 (HIV-1) replication, a segment of the HIV-1 nef gene was replaced with human RNase L cDNA. HIV-1 provirus containing sense orientation RNase L cDNA caused increased expression of RNase L and 500- to 1,000-fold inhibition of virus replication in Jurkat cells for a period of about 2 weeks. Subsequently, a partial deletion of the RNase L cDNA which coincided with increases in virus production occurred. The anti-HIV activity of RNase L correlated with decreases in HIV-1 RNA and with an acceleration in cell death accompanied by DNA fragmentation. Replication of HIV-1 encoding RNase L was also transiently suppressed in peripheral blood lymphocytes (PBL). In contrast, recombinant HIV containing reverse orientation RNase L cDNA caused decreased levels of RNase L, increases in HIV yields, and reductions in the anti-HIV effect of alpha interferon in PBL and in Jurkat cells. To obtain constitutive and continuous expression of RNase L cDNA, Jurkat cells were cotransfected with HIV-1 proviral DNA and with plasmid containing a cytomegalovirus promoter driving expression of RNase L cDNA. The RNase L plasmid suppressed HIV-1 replication by eightfold, while an antisense RNase L construct enhanced virus production by twofold. These findings demonstrate that RNase L can severely impair HIV replication and suggest involvement of the 2-5A system in the anti-HIV effect of alpha interferon.
The interferons are a family of cytokines that suppress replication of a wide range of viruses through multiple antiviral pathways (40). Accordingly, human immunodeficiency virus (HIV) is susceptible to inhibition by interferons in primary and immortalized human T cells and monocytes/macrophages (25). Several different steps in the HIV life cycle have been reported to be affected by interferon treatment. For example, alpha interferon treatment of chronically infected cell lines was often due to defective assembly and release of virus particles (8, 16, 26). In this regard, the effect of interferons on HIV replication were similar to that previously observed for murine retroviruses (14). However, interferons have also been reported to inhibit early stages prior to production and integration of proviral DNA (15, 23), perhaps at the level of reverse transcription (33). In addition, translation of HIV mRNAs was specifically inhibited by alpha interferon in chronically infected, U937 promonocytic cells and in synchronously infected CEM T cells (8).
The 2′,5′-linked oligoadenylate (2-5A) system is an interferon-induced, RNA decay pathway implicated in some of the antiviral mechanisms of interferon action (34). Interferon treatment of cells induces several isozymes of 2-5A synthetase and a single species of the 2-5A-dependent RNase L. The synthetases require double-stranded RNA (dsRNA) to produce 2-5A [pxA(2′p5′A)y, where x = 1 to 3 and y ≥ 2] from ATP (18). 2-5A binds with high affinity to RNase L, converting the inactive monomeric protein to the activated homodimer (9, 11). Many viruses produce dsRNA molecules that can activate 2-5A synthetases, resulting in accumulation of 2-5A. Accordingly, 2-5A or related material has been observed in interferon-treated cells infected with encephalomyocarditis virus (EMCV), vaccinia virus, reovirus, herpes simplex virus, and SV40 simian virus 40 (34). Furthermore, the leader region of HIV type 1 (HIV-1) RNA is sufficiently structured to activate 2-5A synthetase activity in vitro (32). Indeed, a chemically synthesized segment of HIV-1 RNA corresponding to the 5′-terminal, 57-nucleotide TAR region was capable of activating 2-5A synthetase (20). In two studies, however, alpha interferon treatment failed to induce enhanced degradation of either HIV RNA or rRNA (8, 33). Perhaps the lack of RNase L activity was due to inhibition of TAR-mediated activation of 2-5A synthetase by the HIV-1 TAT protein (30). In contrast, a transient increase in 2-5A synthetase and RNase L activity was observed in extracts of HIV-infected H9 cells (31). In addition, several studies have suggested that direct expression or activation of 2-5A system enzymes can lead to suppression of HIV replication. For instance, expression of 2-5A synthetase from an HIV long-terminal repeat (LTR) inhibited HIV-1 replication in HeLa T4+ cells (29) and phosphorothioate-phosphodiester 2-5A derivatives inhibited HIV-induced syncytium formation (36). Also, 2-5A derivatives inhibited HIV-1 reverse transcriptase by preventing primer complex formation (35).
Here, we have taken a novel approach to investigating the potential of the 2-5A system for controlling HIV infections. The availability of cDNA encoding RNase L allowed manipulation of the enzyme which catalyzes the biological effects of the 2-5A system (45). Accordingly, we show that HIV-1 replication can be suppressed by the overexpression of RNase L from either an infectious HIV-1 molecular clone or from an expression plasmid. In contrast, decreasing endogenous levels of RNase L with a reverse orientation cDNA enhanced HIV replication and reduced the antiviral effect of interferon. Our findings thus demonstrate that the 2-5A system is able to regulate HIV replication in control and interferon-treated human cells.
MATERIALS AND METHODS
Construction of plasmids.
HIV provirus, pNL4-3Δnef, containing a 272-bp deletion in the nef coding sequence flanked by two unique restriction sites, XbaI and XhoI, was a generous gift of H. Kestler (Cleveland, Ohio) (1, 5). Human RNase L cDNA, obtained by digesting plasmid pZC5-2 with XbaI and XhoI, was purified and subcloned in both orientations into the XbaI/XhoI sites of pNL4-3Δnef (see Fig. 1). The RNase L cDNA was also cloned into pcDNAneo (Invitrogen) at the HindIII site in both orientations (see Fig. 8A) (47).
FIG. 1.

Construction of recombinant HIV containing RNase L cDNA in the sense and antisense orientations. The plasmids constructed or used for this study included NL4-3 (wild-type HIV-1 proviral DNA), NL4-3Δnef (272 bases deleted from nef), NL4-3/sRL (forward orientation RNase L cDNA cloned in place of a 272-bp segment of the nef gene), and NL4-3/aRL (reverse orientation RNase L cDNA cloned in place of a 272-bp segment of the nef gene).
FIG. 8.

Control of HIV replication by RNase L expression plasmids in Jurkat cells. (A) Plasmid maps for pcDNAneo, pcDNAneo/sRL, and pcDNAneo/aRL; (B) plasmids cotransfected with NL4-3 in Jurkat cells. p24 antigen was measured as a function of time. NL4-3 proviral DNA cotransfected with salmon sperm DNA (○), pcDNAneo (▵), pcDNAneo/aRL (▴), or pcDNAneo/sRL (•). d, days; SV40, simian virus 40; RSV, Rous sarcoma virus.
Cell culture and viruses.
Jurkat cells were cultured in RPMI 1640 medium–10% fetal bovine serum (FBS) (Gibco/BRL). Peripheral blood mononuclear cells from healthy donors were isolated from heparinized venous blood by density gradient centrifugation on Ficoll-Hypaque (Organon). The cells were washed twice in phosphate-buffered saline (PBS), resuspended to 2 × 106 cells per ml in RPMI 1640 medium–l-glutamine–antibiotics–HEPES (pH 7.2), and plated overnight in tissue culture flasks, and the nonadherent cells consisting of primary blood lymphocytes (PBL) depleted of monocytes were collected and activated by treating with 10 U of interleukin 2 (Chiron) per ml and 4 μg of phytohemagglutinin-P (PHA-P) (Sigma) per ml.
Transfections and infections of cells.
Plasmid DNAs (2.5 to 5 μg) were incubated with 10 to 15 μl of Lipofectamine (Gibco/BRL) in 100 μl of OPTI-MEM medium at room temperature for 25 to 40 min prior to transfection of PBL or Jurkat cells. The DNA mixtures were incubated with cells (5 × 105 to 10 × 105) in 1.0 ml of OPTI-MEM medium for 5 h, and then 4 ml of RPMI 1640 medium–10% FBS was added.
Virus stocks were prepared by introducing 5 μg of proviral DNA into Jurkat cells with Lipofectamine. Virus titers and 50% tissue culture infective doses (TCID50) on about 105 cells were measured as described elsewhere (17). Prior to infections, PBL were incubated for 48 h, washed in PBS, and resuspended in medium lacking PHA-P. Stimulated PBL or Jurkat cells (5 × 105 per ml) were infected with 20 TCID50 of virus in serum-free RPMI 1640 medium for 2 h at 37°C. The cells were washed four times with PBS to remove unabsorbed virus particles, and then 5 ml of RPMI 1640 medium–10% FBS was added.
Interferon treatment of cells.
Jurkat cells or stimulated PBL (1 × 105 to 2 × 105 cells per ml) were treated with 5,000 U of recombinant human interferon α-2b (Intron A; Schering) per ml for 18 to 24 h followed by infection with 10 to 20 TCID50 of virus in 1 ml of serum-free medium for 2 h. The cells were washed three times with PBS and suspended in 5 ml of RPMI medium–10% FBS. Cell-free supernatants were collected for viral assays at regular intervals, and fresh medium with or without interferon (5,000 U per ml) was added every 2 days beginning 5 days after infection.
Quantitation of HIV p24 levels.
Cell-free culture supernatants (10 to 20 μl of 1:10 to 1:100 dilutions) were harvested to determine p24 protein levels with an HIV p24 antigen capture kit (Coulter). Briefly, cell-free culture supernatants and lyse buffer were added to wells of microtiter strip plates coated with a monoclonal antibody to p24 and allowed to incubate at room temperature for 1 h. The wells were washed three to four times, biotinylated anti-HIV-1 immunoglobulin G was added, and the wells were incubated at 37°C for 1 h. Following another wash, streptavidin-horseradish peroxidase was added, and the wells were incubated for 30 min at 37°C. After washing, tetramethylbenzidine was added at room temperature, and development of color at 650 nm was detected as a function of time for a 15-min period. Amounts of p24 protein, which do not always correlate with TCID50 levels, were determined in comparison to a calibrated p24 antigen standard in a Coulter microplate reader.
PCR amplification of RNase L cDNA from HIV proviral DNA.
Total cellular DNA was used for PCR with Taq polymerase (Perkin-Elmer Cetus). Primer pairs (5′-GCA GCC GGA TCC TTA GC-3′ and 5′-GAT ATA CCA TGG ATC-3′) homologous to a sequence 320 bp 5′ and 1,508 bp 3′ of RNase L cDNA in the recombinant HIV proviral DNA were used. DNA (1 μg) was added to 100 μl of reaction mixture containing 0.2 mM each deoxynucleoside triphosphate (dNTP), 500 nM each oligonucleotide primer, 4.0 mM MgCl2, 50 mM KCl, 10 mM Tris-HCl (pH 8.3), 0.01% gelatin (wt/vol), and 2.5 U of DNA Taq polymerase (Perkin-Elmer Cetus). PCR was carried out for 30 cycles with denaturation at 94°C for 1.5 min, annealing at 50°C for 1.5 min, and extension at 72°C for 2 min.
Detection of RNase L in Western blots.
RNase L levels were determined with monoclonal antibody against RNase L in Western blots as described elsewhere (11, 41). Cells were lysed with RIPA buffer containing 10 mM Tris (pH 7.4), 1 mM EDTA, 0.15 M NaCl, 0.1% sodium dodecyl sulfate (SDS), 1% Triton X-100, 1% sodium deoxycholate, and 1 mM phenylmethylsulfonyl fluoride.
Northern blots for detection of HIV RNA and 18S rRNA.
Total cellular RNAs from Jurkat cells were harvested at 48, 72, and 96 h posttransfection with RNAzol reagent (Cinna/Biotex). RNAs were separated in 1.2% agarose–formamide gels. RNA was transferred onto Nylon transfer membranes (Micron Separations) with 10× SSC (1× SSC is 0.15 M NaCl, 0.015 M sodium citrate) for 18 to 20 h. The membranes were placed in prehybridization solution (GIBCO-BRL) containing 6× SSC, 5× Denhardt’s solution, 100 μg of sheared, denatured salmon sperm DNA per ml, 20 mM sodium phosphate buffer (pH 6.5), and 50% formamide for 4 h at 65°C. PCR-amplified HIV LTR labeled with [α-32P]dCTP with a random priming kit (Boehringer Mannheim) was used as a probe. About 107 cpm of probe was added to the prehybridization buffer, and the blots were incubated for 16 h at 42°C. The membranes were washed with solutions of SSC-SDS of increasing stringency, and the RNA levels were measured with a PhosphorImager (Molecular Dynamics). The blots were stripped with 0.1× SSC for 10 to 15 min at 100°C and probed with a randomly primed, 32P-labeled DNA probe of 18S rRNA (American Type Culture Collection).
DNA fragmentation assay.
DNA fragmentation in situ was measured with the Trevigen Apoptotic Cell System by using terminal deoxynucleotide transferase (15 U) in 0.05 M Tris (pH 7.5)–5 mM MgCl2–0.6 mM β-mercaptoethanesulfonic acid–50 μg of bovine serum albumin–0.25 mM biotinylated nucleotides (dNTPs) at 37°C for 60 min. Reactions were terminated with 0.1 M EDTA (pH 8.0). Streptavidin-horseradish peroxidase conjugate was applied at room temperature for 10 min. Fragmented DNA was stained blue, and labeled cells were visualized through a microscope and counted.
RESULTS
Expression of RNase L cDNA from a recombinant HIV suppresses virus replication.
To determine the effects of regulating RNase L levels on HIV-1 replication, we subcloned human RNase L cDNA into a modified HIV-1 proviral DNA (5). RNase L cDNA was inserted in either the forward (pNL4-3/sRL) or reverse (pNL4-3/aRL) orientation in place of a 272-bp segment of the nef gene in pNL4-3Δnef DNA (Fig. 1). Virus production after transfection of Jurkat cells or PBL with provirus was determined by measuring levels of the viral gag or core protein, p24, in the culture supernatants. Transfections with an HIV-1 provirus (pNL4-3, pNL4-3Δnef, or pNL4-3/aRL) produced similar viral growth curves in Jurkat cells (Fig. 2A). In contrast, virus replication was very significantly repressed after transfection of Jurkat cells with the sense RNase L construct, pNL4-3/sRL (Fig. 2A). For instance, at 16 days posttransfection of Jurkat cells with pNL4-3/sRL, the viral yield was about 1,000-fold lower than that in pNL4-3-transfected cells. However, by 28 days posttransfection, viral yields from pNL4-3/sRL, pNL4-3, pNL4-3/Δnef, and pNL4-3/aRL were similar. Transfections of PBL produced similar results, except that the level of virus produced by pNL4-3/sRL was closer to 30- to 40-fold less than that with pNL4-3 or pNL4-3/Δnef from 8 to 12 days and recovery of viral growth was observed earlier, between 12 and 16 days posttransfection (Fig. 2B). Perhaps the smaller antiviral effect of RNase L in the PBL was because the HIV replicated more rapidly and to much higher titers in PBL than in Jurkat cells. There was a modest but consistent increase (as much as three- to sixfold) in viral yield obtained after transfections with the antisense vector, pNL4-3/aRL, in the Jurkat cells and PBL. Therefore, forward orientation RNase L cDNA greatly but transiently suppressed virus production, while the reverse orientation constructs modestly enhanced virus growth.
FIG. 2.

Expression of RNase L cDNA from a recombinant HIV provirus suppresses HIV replication in transfected Jurkat cells (A) and PBL (B). The cells were transfected with pNL4-3 (○), pNL4-3/Δnef (◊), pNL4-3/aRL (▴), or pNL4-3/sRL (•). p24 antigen in the cell-free culture supernatant was collected at regular intervals and measured. d, days.
RNase L expression was terminated by a deletion during long-term cultures of transfected cells.
Viral yields were correlated with levels of RNase L determined by probing Western blots with monoclonal antibody against RNase L (Fig. 3A and B, top panel). Levels of RNase L were similar in cells transfected with pNL4-3 or pNL4-3Δnef. However, transfection with pNL4-3/aRL led to a transient decrease in RNase L levels by 4 days, followed by a gradual increase to control levels by 21 days. In contrast, cells transfected with pNL4-3/sRL produced four- to fivefold higher levels of RNase L by 4 to 9 days. Subsequently, there was a gradual decline in RNase L levels in the pNL4-3/sRL-transfected cells to basal levels by 24 days. These findings suggested that expression of high levels of RNase L from the recombinant HIV provirus, pNL4-3/sRL, suppressed virus production at early times posttransfection. Later, however, a decline in expression levels which correlated with increased virus production occurred.
FIG. 3.
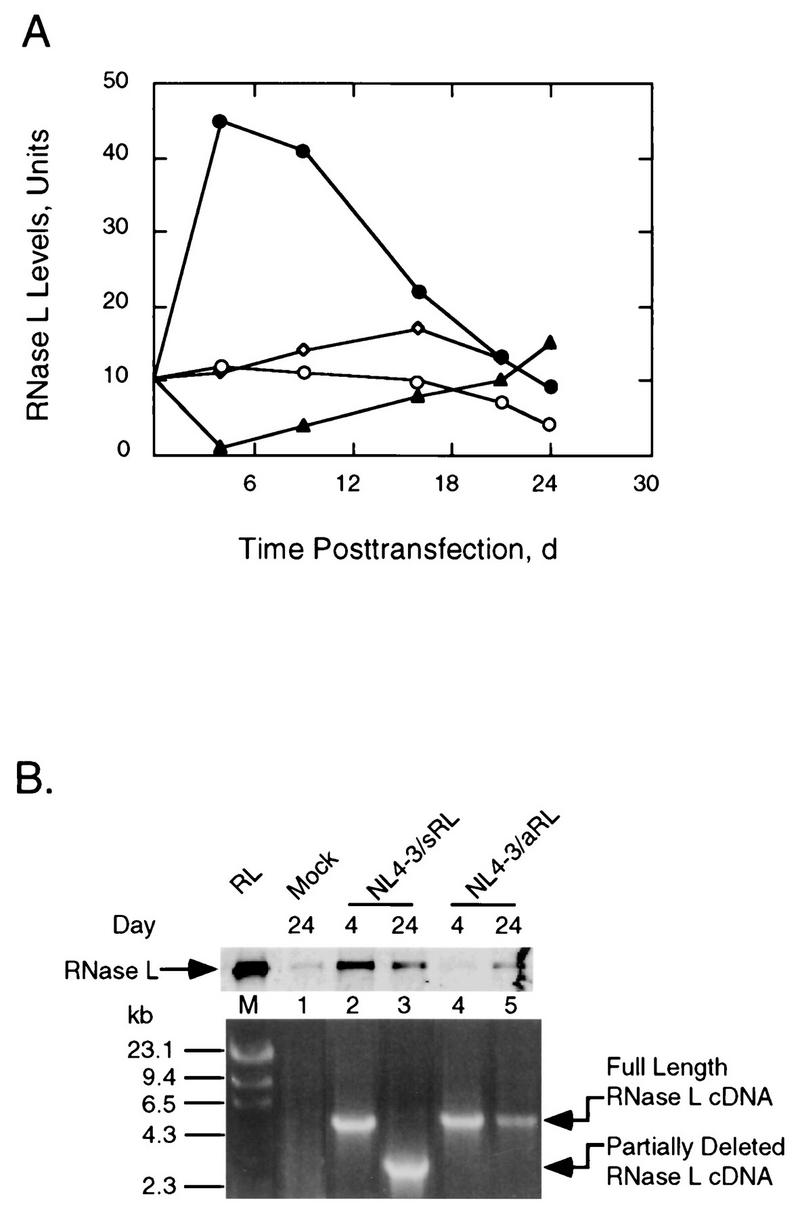
RNase L levels increase and then decline after transfection with NL4-3/sRL due to a deletion of RNase L coding sequence. (A) RNase L levels from Jurkat cells transfected with pNL4-3 (○), pNL4-3/Δnef (◊), pNL4-3/aRL (▴), or pNL4-3/sRL (•). The basal level of RNase L in untreated cells was an average of values obtained at 4, 16, and 24 days (d) of cell culture. (B) Upper panel, Western blot analysis of RNase L from transfected Jurkat cells probed with monoclonal antibody against human RNase L; lower panel, PCR amplification of the integrated RNase L cDNA. RL, 3 ng of RNase L; M, λ/HindIII molecular size markers (numbers to left are in kilobases). Plasmid names and times posttransfection in days are indicated.
To determine the cause of the declining levels of RNase L at late times posttransfection with pNL4-3/sRL, the stability of the integrated RNase L cDNA was determined. At 4 days posttransfection, a DNA fragment of the expected size (4.4 kb) was obtained with PCR primers to sequences 320 nucleotides upstream and 1,508 nucleotides downstream of the integrated RNase L cDNA (Fig. 3B, lower panel, lane 2). However, at 24 days posttransfection, the amplified DNA fragment was reduced in size by about 2 kb (lane 3). In contrast, the RNase L cDNA was stable in the antisense construct, pNL4-3/aRL, at 24 days posttransfection (lane 5). These findings suggest that the emergence of virus at late times posttransfection with pNL4-3/sRL was due to a partial deletion of the RNase L coding sequence. It was apparent from these experiments that the presence of the sense orientation RNase L cDNA in the viral genome imposed negative selective pressure on the recombinant virus.
Transfection with recombinant HIV provirus expressing RNase L leads to decreased levels of HIV RNA and increased rates of cell death.
The mechanism of the anti-HIV effect of overexpressing RNase L was investigated. Total levels of the HIV RNA were detected and measured in Northern blots at 48, 72, and 96 h posttransfection of Jurkat cells. During this time, amounts of HIV RNA decreased to undetectable levels in cells transfected with pNL4-3/sRL, while viral RNA levels increased by >10-fold in cells transfected with pNL4-3/Δnef and pNL4-3/aRL (Fig. 4A and B). These results were consistent with RNase L activity against HIV RNA in the pNL4-3/sRL-transfected cells. However, we have been unable to clearly demonstrate cleavage products of either HIV RNA or rRNA in the pNL4-3/sRL-transfected cells (Fig. 4A). Perhaps HIV RNA fragments escaped detection due to their rapid degradation relative to the time scale of these experiments. The reason for the lack of rRNA cleavage products was unexpected but suggests that HIV RNA may be more susceptible to cleavage by RNase L than rRNA.
FIG. 4.
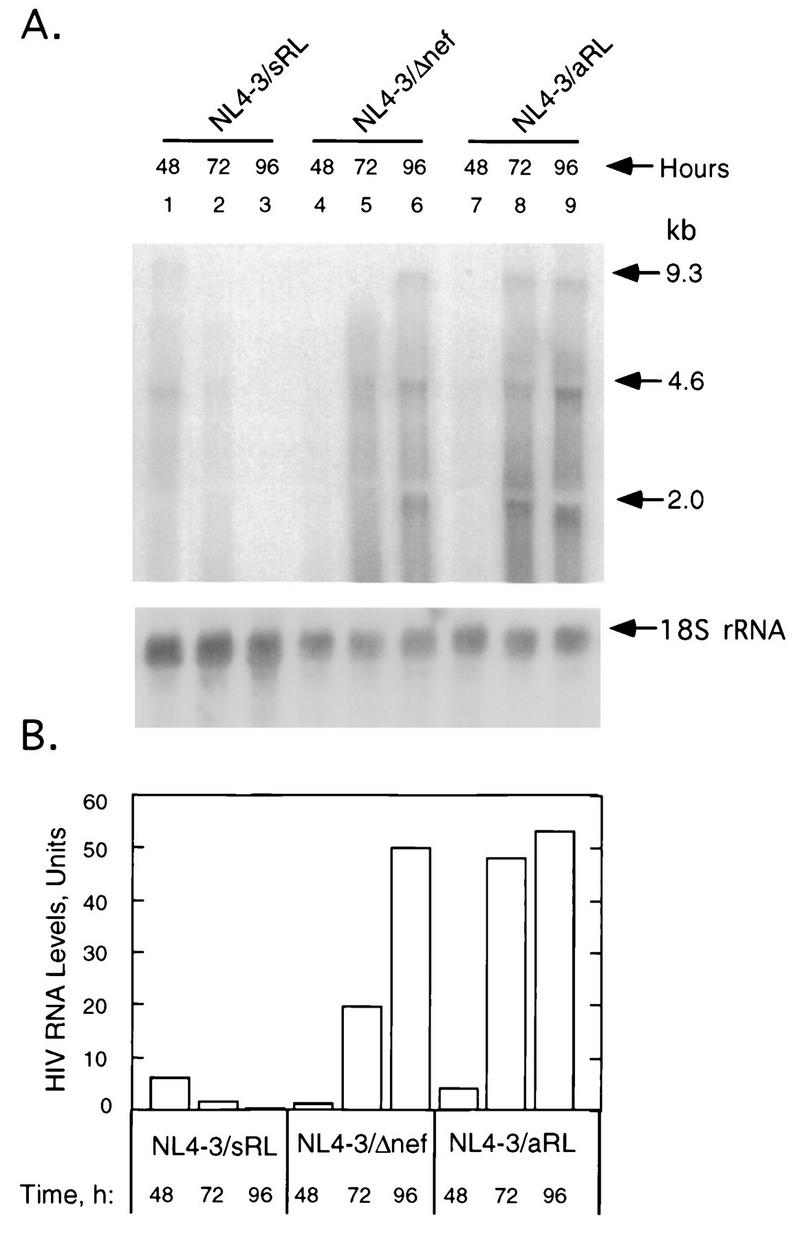
Reduced levels of HIV mRNAs in Jurkat cells transfected with NL4-3/sRL. (A) Upper panel, Northern blot of HIV RNA from Jurkat cells at 48, 72, and 96 h posttransfection with pNL4-3/sRL, pNL4-3/Δnef, or pNL4-3/aRL as indicated; lower panel, Northern blot of 18S rRNA. (B) Quantitation of total HIV mRNA with a PhosphorImager.
The effect of RNase L overexpression from pNL4-3/sRL on cell death was determined by an in situ DNA fragmentation assay. Transfection with the control, salmon sperm DNA, did not cause cell death, whereas transfection of Jurkat cells with all of the HIV proviruses led to varying levels of cell death (Fig. 5). However, transfection with pNL4-3/sRL caused cell death earlier than transfection with pNL4-3, pNL4-3Δnef, or pNL4-3/aRL. The peak of cell death, with 35 and 40% of the cells dying, occurred by 6 days with NL4-3/sRL and by 12 days with the other three proviruses. Therefore, while HIV itself causes cell death, the overexpression of RNase L accelerates this process.
FIG. 5.

Overexpression of RNase L from NL4-3 recombinant proviral DNA (NL4-3/sRL) accelerates HIV-induced cell death. Jurkat cells were transfected with pNL4-3 (○), pNL4-3/Δnef (◊), pNL4-3/aRL (▴), or pNL4-3/sRL (•) or were mock transfected with salmon sperm DNA (□), and the percentage of apoptotic cells as a function of time was determined by in situ detection of DNA fragmentation. d, days.
Reducing expression of RNase L increases the rate of HIV replication and transiently reduces the anti-HIV effect of interferon.
The effect of decreasing RNase L levels on viral growth was further explored in infections with NL4-3/aRL virus, which encodes RNase L in the antisense orientation. Infections rather than transfections were performed to enable the viral genomes to enter a higher proportion of the cells. Amounts of RNase L were measured at different times postinfection of Jurkat cells with NL4-3 and NL4-3/aRL (Fig. 6). RNase L concentrations decreased to almost undetectable levels at 5 days postinfection with NL4-3/aRL, an apparent antisense effect (Fig. 6, lane 6). Subsequently, there was a gradual increase in RNase L levels. Interferon treatment induced RNase L in cells infected with either NL4-3 or NL4-3/aRL. However, at 9 days postinfection, interferon-induced levels of RNase L were approximately fourfold higher in NL4-3 infected cells than those in NL4-3/aRL-infected cells (Fig. 6; compare lanes 3 and 7).
FIG. 6.

Levels of RNase L in NL4-3 and NL4-3/aRL virus-infected Jurkat cells without or with interferon pretreatment (5,000 U per ml). Upper panel, quantitation of RNase L; lower panel, Western blot used to prepare graph shown in upper panel. d, days.
NL4-3/aRL virus replicated more rapidly and to higher titers than did either NL4-3 (Fig. 7) or NL4-3Δnef viruses (data not shown). There was as much as 5- to 12-fold more virus from Jurkat cells and PBL infected with NL4-3/aRL than from those infected with NL4-3. These results suggest that the decrease in RNase L levels led to an increase in viral yields. Similar results were seen in transfection experiments (Fig. 2). To determine if the 2-5A system was involved in the anti-HIV effect of interferon, infections were performed in cells pretreated with 5,000 Units of recombinant, human alpha interferon per ml for 18 to 24 h with the same concentrations of interferon added again every 2 days postinfection beginning on day 5 (Fig. 7). In Jurkat cells, NL4-3 virus replication was completely blocked by the interferon treatments, whereas interferon treatment reduced production of NL4-3 virus by 24-fold in PBL (Fig. 7A and B, respectively). The inhibitory effects of interferon on NL4-3 and NL4-3Δnef replication were similar (data not shown). In contrast, NL4-3/aRL replication was reduced but not blocked in Jurkat cells during the first 10 days of interferon treatment (Fig. 7A). Subsequently, interferon prevented further increases in NL4-3/aRL titers in the Jurkat cells. In PBL, there was no observable effect of interferon on NL4-3/aRL replication during the first 9 days of infection (Fig. 7B). However, at later times postinfection, NL4-3/aRL replication was suppressed by interferon treatment. Therefore, the antiviral effect of interferon was transiently reduced for NL4-3/aRL compared to the wild-type virus, NL4-3. The increase in the activity of interferon against NL4-3/aRL at later times postinfection could be due to increasing levels of RNase L (Fig. 6). These findings suggest a role for the 2-5A system in the antiviral mechanism of action of interferons against HIV.
FIG. 7.

Replication of NL4-3 virus (○ and •) and pNL4-3/aRL virus (▵ and ▴) in the presence (• and ▴) or absence (○ and ▵) of 5,000 U of alpha interferon per ml in Jurkat cells (A) and PBL (B).
Expression of RNase L from a separate plasmid suppresses HIV replication.
Because RNase L cDNA was unstable in recombinant HIV provirus (Fig. 3B), the RNase L cDNA was expressed under the control of a cytomegalovirus (CMV) promoter in a plasmid vector (Fig. 8A). In cotransfections with the wild type, pNL4-3, the RNase L expression plasmid, pcDNAneo/sRL, suppressed HIV-1 yields by 8.4-fold in comparison to control transfections in which pNL4-3 was introduced together with the empty vector or with salmon sperm DNA (Fig. 8B). In contrast, cotransfection of pNL4-3 with plasmid containing reverse orientation RNase L cDNA, pcDNAneo/aRL, modestly enhanced virus production by about twofold. These findings confirm in a two-plasmid system the ability of RNase L to control of HIV replication in infected human cells. Presumably, therefore, stable or inducible expression of RNase L could lead to a sustained inhibition of HIV replication.
DISCUSSION
RNase L, an interferon-inducible antiviral protein, suppresses HIV replication when overexpressed in human cells.
Here, we show that RNase L can function as a potent suppressor of HIV replication when it is overexpressed in transfected or infected human cells. This report thus extends several studies in which antiviral activity was achieved by elevating levels of individual, interferon-inducible proteins from plasmids or recombinant viruses. These strategies effectively bypass the requirement for interferon treatment to obtain inhibition of virus growth. For instance, expression of 2-5A synthetase cDNA under the control of strong constitutive promoters inhibited replication of mengovirus, EMCV, and vaccinia virus while not affecting herpes simplex virus type 2 and VSV replication (6, 7, 10, 27). Recently, Diaz-Guerra et al. reported that expression of cDNAs encoding RNase L and 2-5A synthetase from recombinant vaccinia virus resulted in characteristic cleavage of rRNA and a potent suppression of vaccinia virus protein synthesis and growth (10). These investigators found that expression of RNase L produced a much greater antiviral effect than did expression of 2-5A synthetase. Similarly, expression of a 2-5A synthetase cDNA from an HIV-1 LTR reduced HIV replication in HeLa T4+ cells by 10- to 20-fold as measured by HIV reverse transcriptase activity (29). Here, we have obtained up to 1,000-fold suppression of HIV-1 replication in Jurkat cells by overexpressing RNase L (Fig. 2).
Expression of proteins from alternative, interferon-regulated pathways also provides differing levels of protection from viral infections. For example, in cotransfection experiments with wild-type HIV provirus, a recombinant HIV provirus encoding the dsRNA-dependent protein kinase PKR potently suppressed virus production (3, 20a). The TAR binding protein TRBP blocked the anti-HIV effect of PKR through a direct protein-protein interaction (3). Similarly, TAT/72 protein is able to directly bind with and inhibit PKR function (21). Overexpression of PKR also suppressed replication of EMCV but not of vesicular stomatitis virus (VSV) (22). In addition, expression of the interferon-inducible MxA protein in cells and/or transgenic mice inhibited replication of several RNA viruses, including influenza A virus, measles virus, Thogoto virus, VSV, and human parainfluenza virus type 3 (24, 28, 44). Clearly, direct expression of several interferon-regulated proteins can have profound antiviral effects. In addition, expression of interferon cDNAs has been shown to repress viral replication (37, 39).
The anti-HIV effects of the 2-5A system are probably due to the stimulation of 2-5A synthetase activity by highly structured regions of HIV RNA. In particular, the TAR region of HIV RNA is sufficiently double stranded to stimulate both PKR and 2-5A synthetase activities (12, 20, 32). Presumably, by overexpressing RNase L, even low levels of 2-5A would be capable of producing enough RNA cleavage to result in a substantial antiviral effect. Therefore, when the RNase L cDNA was expressed from a recombinant HIV provirus, a severalfold increase in RNase L levels translated into a surprising 1,000-fold suppression of HIV growth in Jurkat cells at 2 weeks posttransfection (Fig. 2 and 3A). Levels of RNase L, therefore, represent a critical, rate-limiting component to the 2-5A system. Perhaps the potency of this approach was partially due to insertion of the RNase L cDNA in the HIV nef gene, thus coordinating RNase L production with virus gene expression. The negative selective pressure on the virus imposed by the presence of sense orientation RNase L cDNA led to its eventual deletion (Fig. 3B). Following the partial deletion of the RNase L cDNA, virus production increased markedly (Fig. 2). In contrast, the RNase L cDNA was stable in the antisense construct, pNL4-3/aRL, presumably because of a lack of selective pressure.
The presence of RNase L cDNA in recombinant provirus-accelerated, virus-induced cell death in transfected cells.
The depletion of CD4+ T cells in patients with AIDS occurs as a result of apoptosis, mostly in uninfected bystander cells (2, 13). HIV-1 Tat and gp120 proteins have been suggested to cause apoptosis in T cells by induction of Fas ligand expression, resulting in sensitization to Fas-mediated apoptosis (2, 4, 19, 42). Recently, we have shown that mice lacking RNase L have a major apoptotic defect resulting in enlarged thymuses containing thymocytes which are resistant to anti-Fas-mediated apoptosis (46). Perhaps, therefore, the overexpression of RNase L sensitizes cells to Fas-Fas ligand-induced killing. The finding that overexpression of RNase L from NL4-3/sRL accelerates cell death accompanied by DNA fragmentation is consistent with the involvement of RNase L in virus-mediated apoptosis (Fig. 5).
The 2-5A system restricts HIV replication in interferon-treated and untreated cells.
HIV replication was enhanced when RNase L levels were depressed by expression of antisense orientation RNase L cDNA (Fig. 7). NL4-3/aRL replicated to higher titers than the control viruses even in the absence of interferon treatment, suggesting that basal levels of RNase L and 2-5A synthetase were sufficient to dampen HIV replication rates. Because the 2-5A system limits the extent of HIV replication, it could contribute to establishment of chronic infections. In cells treated with alpha interferon, NL4-3/aRL virus also replicated to higher titers than did the control viruses NL4-3 and NL4-3/Δnef. The antiviral effect of interferon was transiently reduced for NL4-3/aRL compared to NL4-3 or NL4-3/Δnef (Fig. 7 and data not shown). Long-term treatments with interferon led to an increased inhibition of replication of NL4-3/aRL, perhaps due to induction of RNase L by interferon (Fig. 6). These findings suggest a role for the 2-5A system in the anti-HIV activity of interferon. Previously, it was reported that steady-state levels of HIV RNAs and the integrity of rRNA were unaffected by alpha interferon treatment of chronically infected U937 and acutely infected CEM T cells, arguing against a role for the 2-5A system in the anti-HIV effect of interferon (8, 33). However, we used a 10-fold higher concentration of alpha interferon (5,000 versus 500 U per ml), and treatment was extended for several days, compared with 2 days or fewer in the other studies, which are experimental parameters that may be necessary to observe the effect of the 2-5A system on HIV replication. In addition, different cell types were analyzed in the different studies. Relatively high levels of interferon (5,000 U per ml) were used here because concentrations of less than 1,000 U per ml were ineffective (data not shown). The relative insensitivity of HIV to interferons could be the result of inhibition of the antiviral pathways by HIV proteins (21, 30).
Destabilizing RNA as an anti-HIV strategy.
By inducing overexpression of RNase L, HIV replication becomes severely impaired, presumably as the result of RNA decay. Accordingly, levels of HIV RNA declined after 48 h of posttransfection with NL4-3/sRL (Fig. 4). Perhaps, the anti-HIV effect could be further improved by either controlling RNase L expression from an inducible promoter or by selectively targeting HIV RNA for decay (38). Destabilizing RNA with the RNases onconase (a frog RNase) and bovine seminal RNase A also causes potent inhibition of HIV replication in H9 cells (43). The advantage of RNase L over other RNases is that it can exist in either a silent or an active form. Previously, we showed that there is sufficient secondary structure in HIV TAR RNA to stimulate 2-5A synthetase activity (20). Our results here demonstrate that antiviral approaches involving the 2-5A system have the potential to provide potent suppression of HIV infections in vivo.
ACKNOWLEDGMENTS
We thank Amiya Banerjee (Cleveland) for comments made during preparation of the manuscript, Aimin Zhou (Cleveland) for pcDNAneo/aRL and pcDNAneo/sRL, and H. Kestler (Cleveland) for pNL4-3/Δnef.
This investigation was supported by U.S. Public Health Service grant CA 44059, which was awarded to R.H.S. by the Department of Health and Human Services, National Cancer Institute.
REFERENCES
- 1.Adachi A, Gendelman H E, Koenig S, Folks T, Willey R, Robon A, Martin M A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and non-human cells transfected with an infectious molecular clone. J Virol. 1986;59:284–289. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badley A D, Mcelhinny J A, Leibson P J, Lynch D H, Alderson M R, Paya C V. Upregulation of Fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T Lymphocytes. J Virol. 1996;70:199–206. doi: 10.1128/jvi.70.1.199-206.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benkirane M, Neuveut C, Chun R F, Smith S M, Samuel C E, Gatignol A, Jeang K T. Oncogenic potential of TAR RNA binding protein TRBP and its regulatory interaction with RNA-dependent protein kinase PKR. EMBO J. 1997;16:611–624. doi: 10.1093/emboj/16.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campioni D, Corallini A, Zauli G, Possati L, Altavilla G, Barbanti-Brodano G. HIV type 1 extracellular Tat protein stimulates growth and protects cells of BK virus/tat transgenic mice from apoptosis. AIDS Res Hum Retroviruses. 1995;11:1039–1048. doi: 10.1089/aid.1995.11.1039. [DOI] [PubMed] [Google Scholar]
- 5.Chakrabarty B K, Maitra R K, Ma X-Z, Kestler H W. A candidate live inactivatable attenuated vaccine for AIDS. Proc Natl Acad Sci USA. 1996;93:9810–9815. doi: 10.1073/pnas.93.18.9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chebath J, Benech P, Revel M, Vigneron M. Constitutive expression of (2′-5′)oligo A synthetase confers resistance to picornavirus infection. Nature. 1987;330:587–588. doi: 10.1038/330587a0. [DOI] [PubMed] [Google Scholar]
- 7.Coccia E M, Romeo G, Nissim A, Marziali G, Albertini R, Affabris E, Battistini A, Fiorucci G, Orsatti R, Rossi G B, Chebath J. A full-length murine 2-5A synthetase cDNA transfected in NIH-3T3 cells impairs EMCV but not VSV replication. Virology. 1990;179:228–233. doi: 10.1016/0042-6822(90)90292-y. [DOI] [PubMed] [Google Scholar]
- 8.Coccia E M, Krust B, Hovenessian A G. Specific inhibition of viral protein synthesis in HIV-infected cells in response to interferon treatment. J Biol Chem. 1994;269:23087–23094. [PubMed] [Google Scholar]
- 9.Cole J L, Carroll S S, Kuo L C. Stoichiometry of 2′,5′-oligoadenylate-induced dimerization of ribonuclease L. J Biol Chem. 1996;271:3979–3981. doi: 10.1074/jbc.271.8.3979. [DOI] [PubMed] [Google Scholar]
- 10.Diaz-Guerra M, Rivas C, Esteban M. Inducible expression of the 2-5A synthetase/RNase L system results in inhibition of vaccinia virus replication. Virology. 1997;227:220–228. doi: 10.1006/viro.1996.8294. [DOI] [PubMed] [Google Scholar]
- 11.Dong B, Silverman R H. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J Biol Chem. 1995;270:4133–4137. doi: 10.1074/jbc.270.8.4133. [DOI] [PubMed] [Google Scholar]
- 12.Edery I, Petryshyn R, Sonenberg N. Activation of double-stranded RNA-dependent kinase (dsI) by the TAR region of HIV-1 mRNA: a novel translational control mechanism. Cell. 1989;56:303–312. doi: 10.1016/0092-8674(89)90904-5. [DOI] [PubMed] [Google Scholar]
- 13.Finkel T H, Tudor-Williams G, Banda N K, Cotton M F, Curiel T, Monks C, Baba T W, Ruprecht R M, Kupfer A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nature Med. 1995;1:129–134. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 14.Friedman R M, Pitha P M. The effect of interferon on membrane associated virus. In: Friedman R M, editor. Interferon 3: mechanism of production and action. Amsterdam, The Netherlands: Elsevier; 1984. pp. 319–336. [Google Scholar]
- 15.Gendelman H E, Baca L M, Turpin J, Kalter D C, Hansen A, Orenstein C W, Diffenbach C W, Friedman R M, Meltzer M S. Regulation of HIV replication in infected monocytes by interferon α: mechanisms for viral restriction. J Immunol. 1990;145:2669–2676. [PubMed] [Google Scholar]
- 16.Hansen B D, Nara P L, Maheshwari R K, Sidhu G S, Bernbaum J G, Hoekzema D, Meltzer M S, Gendelman H E. Loss of infectivity by progeny virus from alpha interferon-treated human immunodeficiency virus type 1-infected T cells is associated with defective assembly of envelope gp120. J Virol. 1992;66:7543–7548. doi: 10.1128/jvi.66.12.7543-7548.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson V A, Byington R E. Quantitative assays for virus infectivity. In: Aldovini A, Walker B D, editors. Techniques in HIV research. New York, N.Y: Stockton; 1990. pp. 71–76. [Google Scholar]
- 18.Kerr I M, Brown R E. pppA2′p5′A2′p5′A2′p5′A: an inhibitor of protein synthesis synthesized with an enzyme fraction from interferon-treated cells. Proc Natl Acad Sci USA. 1978;75:256–260. doi: 10.1073/pnas.75.1.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laurent-Crawford A G, Krust B, Riviere Y, Desgranges C, Muller S, Kieny M P, Dauguet C, Hovanessian A G. Membrane expression of HIV envelope glycoproteins triggers apoptosis in CD4 cells. AIDS Res Hum Retroviruses. 1993;9:761–773. doi: 10.1089/aid.1993.9.761. [DOI] [PubMed] [Google Scholar]
- 20.Maitra R K, McMillan N A J, Desai S, McSwiggen J, Hovanessian A, Sen G, Williams B R G, Silverman R H. HIV-1 TAR RNA has an intrinsic ability to activate interferon-inducible enzymes. Virology. 1994;204:823–827. doi: 10.1006/viro.1994.1601. [DOI] [PubMed] [Google Scholar]
- 20a.Maitra, R. K., and B. R. G. Williams. Unpublished data.
- 21.McMillan N A J, Chun R F, Siderovski D P, Galabru J, Toone W M, Samuel C E, Mak T W, Hovanessian A G, Jeang K-T, Williams B R G. HIV-1 Tat directly interacts with interferon-induced, double-stranded RNA-dependent kinase, PKR. Virology. 1995;213:413–424. doi: 10.1006/viro.1995.0014. [DOI] [PubMed] [Google Scholar]
- 22.Meurs E F, Watanabe Y, Kadereit S, Barber G N, Katze M G, Chong K, Williams B R G, Hovanessian A G. Constitutive expression of human double-stranded RNA-activated p68 kinase in murine cells mediates phosphorylation of eukaryotic initiation factor 2 and partial resistance to encephalomyocarditis virus growth. J Virol. 1992;66:5805–5814. doi: 10.1128/jvi.66.10.5805-5814.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meylan P R, Guatelli J C, Munis J R, Richman D D, Kornbluth R S. Mechanisms for the inhibition of HIV replication by interferon-α, -β, and -γ in primary human macrophages. Virology. 1993;193:138–148. doi: 10.1006/viro.1993.1110. [DOI] [PubMed] [Google Scholar]
- 24.Pavlovic J, Arzet H A, Hefti H P, Frese M, Rost D, Ernst B, Kolb E, Staeheli P, Haller O. Enhanced virus resistance of transgenic mice expressing the human MxA protein. J Virol. 1995;69:4506–4510. doi: 10.1128/jvi.69.7.4506-4510.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pitha P. Multiple effects of interferon on HIV-1 replication. J Interferon Res. 1991;11:313–318. doi: 10.1089/jir.1991.11.313. [DOI] [PubMed] [Google Scholar]
- 26.Poli G, Orenstein J M, Kinter A, Folks T M, Fauci A S. Interferon alpha but not AZT suppresses HIV expression in chronically infected cell lines. Science. 1989;244:575–577. doi: 10.1126/science.2470148. [DOI] [PubMed] [Google Scholar]
- 27.Rysiecki G, Gewert D R, Williams B R G. Constitutive expression of a 2′-5′-oligoadenylate synthetase cDNA results in increased antiviral activity and growth suppression. J Interferon Res. 1989;9:649–657. doi: 10.1089/jir.1989.9.649. [DOI] [PubMed] [Google Scholar]
- 28.Schneider-Schaulies S, Schneider-Schaulies J, Schuster A, Bayer M, Pavlovic J, Meulen V T. Cell-type-specific MxA-mediated inhibition of measles virus transcription in human brain cells. J Virol. 1994;68:6910–6917. doi: 10.1128/jvi.68.11.6910-6917.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroder H C, Ugarkovic D, Merz H, Kuchino Y, Okamoto T, Muller W E G. Protection of HeLa-T4 cells against human immunodeficiency virus (HIV) infection after stable transfection with HIV LTR-2′-5′-oligoadenylate synthetase hybrid gene. FASEB J. 1990;4:3124–3130. doi: 10.1096/fasebj.4.13.1698680. [DOI] [PubMed] [Google Scholar]
- 30.Schroder H C, Ugarkovic D, Wenger R, Reuter P, Okamoto T, Muller W E G. Binding of Tat protein to TAR region of human immunodeficiency virus type-1 blocks TAR-mediated activation of (2′-5′) oligoadenylate synthetase. AIDS Res Hum Retrov. 1990;6:659–672. doi: 10.1089/aid.1990.6.659. [DOI] [PubMed] [Google Scholar]
- 31.Schroder H C, Wenger R, Kuchino Y, Muller W E G. Modulation of nuclear matrix-associated 2′,5′-oligoadenylate metabolism and ribonuclease L activity in H9 cells by human immunodeficiency virus. J Biol Chem. 1989;264:5669–5673. [PubMed] [Google Scholar]
- 32.Sengupta D N, Silverman R H. Activation of interferon regulated dsRNA dependent enzymes by human immunodeficiency virus 1 leader RNA. Nucleic Acids Res. 1989;17:969–978. doi: 10.1093/nar/17.3.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shirazi Y, Pitha P M. Alpha interferon inhibits early stages of human immunodeficiency virus type 1 replication cycle. J Virol. 1992;66:1321–1328. doi: 10.1128/jvi.66.3.1321-1328.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silverman R H, Cirino N M. RNA decay by the interferon-regulated 2-5A system as a host defense against viruses. In: Hartford J B, Morris D R, editors. mRNA metabolism and posttranscriptional gene regulation. New York, N.Y: Wiley-Liss, Inc.; 1997. pp. 295–309. [Google Scholar]
- 35.Sobol R W, Fisher W L, Reichenbach N L, Kumar A, Beard W A, Wilson S H, Charubala R, Pfleiderer W, Suhadolnik R J. HIV-1 reverse transcriptase: inhibition by 2′,5′-oligoadenylates. Biochemistry. 1993;32:12112–12118. doi: 10.1021/bi00096a023. [DOI] [PubMed] [Google Scholar]
- 36.Sobol R W, Henderson E E, Kon N, Shao J, Hitzges P, Mordechai E, Reichenbach N L, Charubala R, Schirmeister H, Pfleiderer W, Suhadolnik R J. Inhibition of HIV replication and activation of RNase L by phosphorothioate/phosphodiester 2′,5′-oligoadenylate derivatives. J Biol Chem. 1995;270:5963–5978. doi: 10.1074/jbc.270.11.5963. [DOI] [PubMed] [Google Scholar]
- 37.Su Y, Popic W, Pitha P M. Inhibition of human immunodeficiency virus type-1 replication by a Tat-activated transduced interferon gene: targeted expression to human immunodeficiency virus type 1-infected cells. J Virol. 1995;69:110–121. doi: 10.1128/jvi.69.1.110-121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torrence P F, Maitra R K, Lesiak K, Khamnei S, Zhou A, Silverman R H. Targeting RNA for degradation with a (2′-5′)oligoadenylate-antisense chimera. Proc Natl Acad Sci USA. 1993;90:1300–1304. doi: 10.1073/pnas.90.4.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vieillard V, Lauret E, Rousseau V, Maeter E D. Blocking of retroviral infection at a step prior to reverse transcription in cells transformed to constitutively express interferon β. Proc Natl Acad Sci USA. 1994;91:2689–2693. doi: 10.1073/pnas.91.7.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vilcek J, Sen G C. Interferon and other cytokines. In: Fields B N, Knipe D N, Howley P M, editors. Fields virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 375–399. [Google Scholar]
- 41.Wang L, Zhou A, Vasavada S, Dong B, Nie H, Church J M, Williams B R G, Banerjee S, Silverman R H. Elevated levels of 2′,5′-linked oligoadenylate-dependent ribonuclease L occur as an early event in colorectal tumorigenesis. Clin Cancer Res. 1995;1:1421–1428. [PubMed] [Google Scholar]
- 42.Westendorp M O, Rainer F, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin K-M, Krammer P H. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- 43.Youle R J, Wu Y N, Mikulski S M, Shogen K, Hamilton R S, Newton D, D’Alessio G, Gravell M. RNase inhibition of human immunodeficiency virus infection of H9 cells. Proc Natl Acad Sci USA. 1994;91:6012–6016. doi: 10.1073/pnas.91.13.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao H, De B B, Das T, Banerjee A K. Inhibition of human parainfluenza virus-3 replication by interferon and human MxA. Virology. 1996;220:330–338. doi: 10.1006/viro.1996.0321. [DOI] [PubMed] [Google Scholar]
- 45.Zhou A, Hassel B A, Silverman R H. Expression cloning of 2-5A-dependent RNase—a uniquely regulated mediator of interferon action. Cell. 1993;72:743–765. doi: 10.1016/0092-8674(93)90403-d. [DOI] [PubMed] [Google Scholar]
- 46.Zhou A, Paranjape J, Brown T L, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman R H. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou, A., and R. H. Silverman. Unpublished data.