

Abstract
Human T-cell lymphotropic virus type 1 (HTLV-1) is the etiologic agent for adult T-cell leukemia. HTLV-1 transforms lymphocytes, and there is increasing evidence that the virus-encoded protein, Tax, plays a primary role in viral transformation. We have shown that wild-type p53 in HTLV-1-transformed cells is stabilized. This study was initiated to directly analyze whether the p53 in HTLV-1-transformed cell lines was transcriptionally active and to identify the viral gene product responsible for stabilization and inactivation. Transfection experiments using a p53-responsive reporter plasmid and γ-irradiation studies demonstrate that the wild-type p53 in HTLV-1-transformed cell lines is not fully active. Further, we demonstrate that the HTLV-1-transforming protein, Tax, stabilizes and inactivates p53 function. Cotransfection of Tax with p53 results in a greater than 10-fold reduction in p53 transcription activity. Using Gal4-p53 fusion proteins, we demonstrate that Tax inhibition of p53 transactivation function is independent of sequence-specific DNA binding. Moreover, Tax inhibits p53 function by interfering with the activity of the N-terminal activation domain (amino acids 1 to 52). We conclude that Tax is involved in the inactivation of p53 function and stabilization of p53 in HTLV-1-infected cells. The functional interference of p53 function by Tax may be important for transformation and leukemogenesis.
Human T-cell leukemia virus type 1 (HTLV-1) is the etiologic agent of adult T-cell lymphoma/leukemia (44, 59) and the demyelinating syndrome tropical spastic paraparesis (21, 41). HTLV-1 transformation is not well understood. The HTLV genome does not encode an oncogene, nor is it integrated in the proximity of a cellular oncogene (48). The HTLV regulatory protein Tax, a potent transcriptional activator of the HTLV long terminal repeat (LTR) and several cellular genes, contributes to transformation of T cells (1, 19). Tax has been shown to immortalize T lymphocytes and, in conjunction with ras, transform rodent cells (22, 52). Transgenic mice that express Tax in mature T lymphocytes developed large granular lymphocytic leukemia, demonstrating that Tax is sufficient to induce leukemia (23).
The highly conserved p53 gene (51) encodes a tumor suppressor protein important for control of cellular growth. Inhibition of p53 transactivation function, through either mutation or interaction with viral transforming proteins, correlates strongly with the oncogenic potential of the protein. As a normal cellular response, DNA-damaging agents lead to stabilization of the p53 protein, resulting in the transactivation of several important cellular control genes, including those encoding MDM2 (2, 43, 57), Gadd45 (26, 32, 61), p21 (15, 16, 18), and Bax (34, 35, 60), which are essential to correct genomic alterations or induce apoptosis. For example, induction of p21waf1/cip1 results in arrest of the cell cycle at the G1/S border (15, 16), allowing the cell time to correct DNA damage before DNA replication. Recent studies further suggest that p21 may also regulate coordination of the S and M phases of the cell cycle (56). In contrast, induction of the Bax gene stimulates the cell to enter the programmed cell death apoptosis pathway (34, 35). p53 further stimulates apoptosis by repressing transcription of the bcl-2 gene (33, 34, 61).
In an effort to understand the leukemogenic mechanism in adult T-cell leukemia (ATL), p53 status in ATL patients and HTLV-1-infected cells has been analyzed by several groups (37, 38, 47, 58). These studies show that the p53 gene was mutated only in about 30% of the cases studied. Interestingly, the steady-state level of p53 protein expression was reported to be elevated in human T lymphocytes transformed by HTLV-1 (46). Further, it could be shown that the p53 gene was wild type and that expression was correlated with a significant increase in protein half-life. The present studies were initiated to analyze whether the stabilized wild-type p53 in HTLV-1-transformed cell lines was transcriptionally active. Data obtained from transfection experiments using a p53-responsive reporter plasmid and from γ-irradiation studies demonstrate that the wild-type p53 in these cell lines is not fully active. Further, we demonstrate that Tax alone in cotransfection studies is capable of inactivating p53 by inhibiting the N-terminal p53 activation domain.
MATERIALS AND METHODS
Cell lines, irradiation, and RNA isolation.
HTLV-1-transformed cell lines (C81, MT-2, MT-4, and HUT102) (24, 44, 49), T-lymphoblastoid cell line Jurkat (9, 50), and myeloid leukemia cell line ML-1 (27) were grown in RPMI 1640 medium supplemented with 10% fetal calf serum. Cell lines NIH 3T3 (3), OsA-Cl (39), and Saos-2 (5) were maintained in Dulbecco modified Eagle medium and 10% fetal calf serum. Exponentially growing cells were γ-irradiated with 20 Gy and incubated for 4 h. Cells were lysed in RNAzol B solution (Tel-Test, Inc.), and total cellular RNA was isolated according to the manufacturer’s instructions. Poly(A) mRNA was prepared, electrophoresed, and blotted to a Nytran membrane (Schleicher & Schuell). The blots were then sequentially probed with the following cDNA probes: MDM2, Gadd45, Waf1, Bax, and bcl-2. Probes were labeled by the random primer-labeling method (Amersham). Signals were quantitated by using a PhosphorImager and ImageQuant program (Molecular Dynamics). Northern hybridization against GAPDH was used as a control RNA.
Transfections, CAT, and Luc assays.
Transient transfection experiments with human T lymphocytes were performed by using the electroporation method as described previously (25) or Lipofectamine reagent (GIBCO-BRL) as described by the manufacturer. Adherent cells were transfected by using Lipofectamine reagent. The amount of DNA transfected was equalized by addition of a control vector. Cotransfection of a human growth hormone (hGH) reporter construct (Nichols Institute) was done to normalize for transfection efficiency. hGH expression, given as 125I counts per minute obtained in a GH ELISA (enzyme-linked immunosorbent assay), is presented in the legend to Fig. 1. Chloramphenicol acetyltransferase (CAT) activity was assayed 24 h posttransfection. Results were quantitated by using a PhosphorImager and the ImageQuant program. p53-responsive reporter plasmid p53G5BCAT contains six p53 consensus binding sites, 5′-CTAGAGGCATGTCT-3′ (17, 35), at the XbaI site positioned upstream of the E1B TATA sequence in plasmid pG5BCAT. Luciferase (Luc) activity was assayed 24 h posttransfection. Luc assays were performed with a Berthold LB9500C luminometer as described elsewhere (13). PG13pyLuc, which contains 13 p53 consensus binding sites, and its corresponding mutant MG13pyLuc were kindly provided by Bert Vogelstein (Johns Hopkins Oncology Center, Baltimore, Md.). Gal4(DBD [DNA binding domain])-p53 fusion plasmids were kindly provided by Thomas Shenk (Princeton University, Princeton, N.J.). The wild-type p53 expression plasmid used was either pCEP4-53 or pCMV-53. p53 or Tax protein was assayed directly from transfected lysates by Western blot analysis as described below.
FIG. 1.

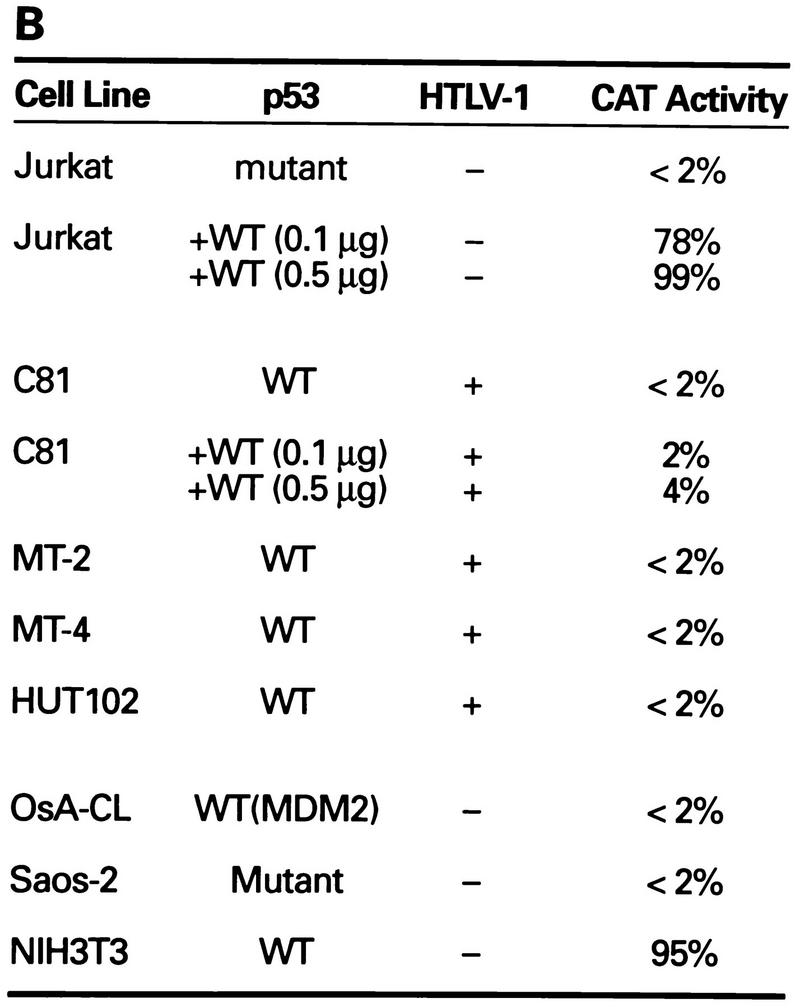

Transcriptional stimulation activity mediated by endogenous p53 protein in HTLV-1 transformed cell lines. (A) Diagrammatic representation of CAT reporter plasmids. The reporter plasmid pG5BCAT contains CAT coding sequences with five Gal4 DNA binding sites upstream of the E1B TATA box. Reporter p53G5BCAT contains six p53 consensus binding sites between the five Gal4 DNA binding sites and the E1B TATA sequence of pG5BCAT. (B) Transcriptional stimulation activity of p53 in different cell lines. pG5CAT or p53G5BCAT (8 μg) was transfected into different cell lines together with 8 μg of GH cDNA (pXGH5). The amounts of protein extract for CAT assays were adjusted to have the same GH activity. In Jurkat and C81 cells, the effect of exogenously introduced wild-type (WT) p53 was assayed by cotransfection of p53G5BCAT DNA (2 μg) with increasing amounts (0.1 and 0.5 μg) of the p53 expression vector. Numbers in the CAT activity column represent percent CAT conversion. (C) p53 transactivation in Jurkat and HTLV-1-transformed cells. p53G5BCAT (2 μg) was transfected into different cell lines together with 8 μg of GH cDNA (pXGH5). The effect of exogenously introduced wild-type p53 was assayed by cotransfection of p53G5BCAT DNA (2 μg) with increasing amounts (0.1 to 2.0 μg) of the p53 expression vector. All transfections were equalized for total DNA by addition of carrier DNA. CAT activity is presented as percent CAT conversion. Level of hGH expression, given as 125I counts per minute obtained in a GH ELISA, were as follows: lane 2, 630; lane 3, 540; lane 4, 423; lane 5, 500; lane 6, 657; lane 7, 558; lane 8, 705; and lane 9, 778. (D and E) Western blot analysis of HTLV-1 Tax and p53 following exposure of normal and HTLV-1-transformed cells to γ irradiation. Cells were lysed in RIPA buffer containing 1 mM PMSF, 1 μg of aprotinin per ml, 1 μg of leupeptin per ml, and 5 mM sodium fluoride at 0°C for 30 min. From each cell lysate, a total of 100 μg of protein was separated on an SDS–10% acrylamide denaturing gel, transferred to an Immobilon membrane (Millipore), and probed with antibodies against Tax (Tab 172) (D) and p53 (PAb421) (E).
Western blotting.
Cells were washed in phosphate-buffered saline and then lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) containing 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg of aprotinin per ml, 1 μg of leupeptin per ml, and 5 mM sodium fluoride at 0°C for 30 min. Lysates were cleared by centrifugation in a microcentrifuge at 14,000 rpm for 15 min. From each cell lysate, a total of 25 to 100 μg of protein, as determined by the Bio-Rad protein assay, was separated on an SDS-acrylamide denaturing gel, transferred to an Immobilon membrane (Millipore, Bedford, Mass.), and probed with antibodies against Tax and p53 (DO-1 and PAb421 [Oncogene]). Detection was performed with the Amersham Corp. (Arlington Heights, Ill.) enhanced chemiluminescence system.
RESULTS
p53 activity is suppressed in HTLV-1-transformed cell lines.
To investigate whether the p53 protein in HTLV-1-transformed cell lines is functional, we performed transfection analyses with the p53-responsive reporter plasmid p53G5BCAT, which contains multiple p53 consensus binding sites upstream of the TATA sequence (17) (Fig. 1A). We compared the activities of the p53 reporter plasmid in control Jurkat and HTLV-1-transformed lymphocyte cells (C81, MT-2, MT-4, and HUT102). When the p53 reporter construct was cotransfected with a p53-negative Jurkat T cells, low transcription activity (<2%) was observed (Fig. 1B and C). When the p53 reporter construct was cotransfected with a p53 expression vector, a 40- to 50-fold increase in CAT activity was observed in the Jurkat lymphocytes (Fig. 1B and C). The level of CAT activity in p53-positive HTLV-1-transformed cells lines was similar to the activity seen in the p53-negative Jurkat cells (<2%). Moreover, in contrast to the results obtained in the Jurkat cells, cotransfection of the p53 expression vector into HTLV-1-transformed C81 cells failed to induce transcription (Fig. 1B and C). Cotransfection of a plasmid expressing hGH was used to control for transfection efficiency of the different cell lines (Fig. 1C, see legend). The addition of this internal control eliminates the possibility that the differences in CAT activities are due to differences in transfection efficiency of the different cell lines.
Saos-2, OsA-Cl, and NIH 3T3 cells were included as controls. In NIH 3T3 cells, the endogenous p53 is wild type and functional (Fig. 1B). Saos-2 cells contain mutant p53, which is not transcriptionally active. OsA-Cl cells contain wild-type p53 which is not functional because of overexpression of the p53 suppressor protein, MDM2. Only basal levels of p53-dependent CAT activity (<2%) were observed in the Saos-2 and OsA-Cl cells (Fig. 1B). Thus, the p53 activity in the HTLV-1-transformed cell lines resembled that observed in p53-defective cell lines.
Transfection of the reporter construct HTLV-1 LTR CAT into the same cell lines gave a significantly different result. The transcriptional activity from this plasmid was 5- to 10-fold higher in the HTLV-1-transformed cells lines due to the presence of the endogenous Tax protein (data not shown). These results demonstrate that the inactivity of the p53-responsive plasmid was not due to inability to transfect the HTLV-1-transformed cells.
p53 function in HTLV-1 cell lines is not responsive to gamma-ray irradiation.
DNA damage induced by ionizing radiation or UV light has been shown to lead to accumulation of wild-type p53, resulting in transactivation of cellular genes such as those encoding MDM2, Gadd45, p21waf1/cip1, and Bax (6, 10, 26, 28, 29, 31). To further test whether the endogenous p53 in HTLV-1 cells is inactive, cellular responses to DNA damage were examined. Since the DNA damage pathway induced by γ-irradiation has been well studied in the wild-type p53-containing myeloid cell line ML-1 (27, 35, 56), these cells were used as controls to compare the levels of induction of MDM2, Gadd45, p21, and Bax RNAs. Exponentially growing cultures were exposed to 20 Gy of ionizing radiation and harvested 4 h later. Downstream target genes were analyzed by Northern blot analysis, and the level of induction of each transcript was related to the basal expression of these genes (Table 1). For comparative purposes, the constitutively expressed housekeeping gene GAPDH was analyzed. Consistent with earlier reports, in ML-1 cells harboring wild-type p53, a five- to eightfold induction of p53-responsive gene products MDM2, Gadd45, p21, and Bax was observed following irradiation (Table 1) (27, 35, 56). In contrast, in each of the HTLV-1-transformed cell lines, the level of cellular gene expression was not significantly altered (<2-fold) by irradiation, further suggesting that the p53 protein in HTLV-1-transformed cells is inactive.
TABLE 1.
Effect of γ irradiation on the expression of various DNA damage-inducible genes
| Cell line | HTLV-I | Fold increase in relative mRNAa
|
|||
|---|---|---|---|---|---|
| MDM2 | Gadd45 | p21waf1/cip1 | Bax | ||
| ML-1 | − | 4.8 | 5.2 | 7.7 | 6.7 |
| C81 | + | 1.2 | 1.5 | 1.9 | 1.7 |
| Hut102 | + | 1.1 | 1.0 | 1.0 | 0.4 |
| MT-2 | + | 2.1 | 1.7 | 2.2 | 1.0 |
Relative signals for the Northern blot analyses were quantitated by using a PhosphorImager and the ImageQuant program. Values represent the ratio of the signal in the γ-irradiated samples compared with untreated controls. Briefly, exponentially growing cells were γ-irradiated with 20 Gy and incubated for 4 h, at which time poly(A) mRNA was prepared, electrophoresed, and blotted to a Nytran membrane (Schleicher & Schuell). The blots were then sequentially probed with the following cDNA probes: MDM-FL4, a full-length MDM2 clone (39); pHulB2, a human Gadd45 clone (42); pZL-Waf1, a full-length Waf1 cDNA clone (18); and pBAX N7, a full-length human Bax cDNA clone (8). The MDM2 and p21 probes were kindly provided by Bert Vogelstein (Johns Hopkins Oncology Center, Baltimore, Md.). The Gadd45 and Bax probes were a kind gift from Albert J. Fornace, Jr. (National Institutes of Health, Bethesda, Md.).
We compared the levels of p53 expression in ML-1 and HTLV-1-transformed cells following exposure to γ-irradiation. Consistent with previous observations, Western blot analysis of the HTLV-1-transformed cells demonstrated that the constitutive level of p53 protein in C81, HUT102, and MT-2 cells is elevated (Fig. 1E), similar to that seen in ML-1 cells in response to γ-irradiation. The level of p53 or Tax (Fig. 1D) in HTLV-1-transformed cells is not significantly increased following irradiation.
The HTLV-1 Tax protein stabilizes p53 and inhibits p53 transactivation function.
It was of interest to identify the virus-encoded protein which was important for p53 stabilization (46) and transcriptional inactivation. We first studied the ability of Tax to stabilize p53. As seen in Fig. 2, cotransfection of pCMV-53 and pHTLV-Tax into the p53-negative human Jurkat T lymphocytes resulted in a significant accumulation of p53 protein (lanes 11 and 12). In the absence of pHTLV-Tax, no p53 protein was detected by Western blotting, consistent with the short half-life of the wild-type protein (Fig. 2, lane 10). Control studies in which a cytomegalovirus (CMV)-CAT reporter construct was cotransfected with the HTLV-I Tax protein demonstrated that the increase in p53 expression was not due to transactivation of the CMV promoter located upstream of the p53 coding sequences (data not shown). Further, Tax expression was equivalent in the presence or absence of p53 protein (Fig. 2, lanes 3 and 6). These results demonstrate that Tax expression is sufficient for p53 stabilization.
FIG. 2.

Stabilization of p53 by Tax. An expression vector for wild-type p53 was cotransfected into Jurkat cells with the expression vector for Tax or the control expression vector. Cells were lysed in RIPA buffer containing 1 mM PMSF, 1 μg of aprotinin per ml, 1 μg of leupeptin per ml, and 5 mM sodium fluoride at 0°C for 30 min. From each cell lysate, a total of 100 μg of protein was separated on an SDS–10% acrylamide denaturing gel, transferred to an Immobilon membrane (Millipore), and probed with antibodies against Tax (Tab 172) and p53 (PAb421).
To determine if Tax inhibited p53 transcription function, transient transfection assays in Jurkat T cells were performed. Reporter construct PG13pyLuc (Fig. 3A), which contains 13 copies of the p53 binding site upstream of the polyomavirus promoter, is stimulated by cotransfection of a plasmid expressing p53 (Fig. 3B, bar 4). In contrast, the control promoter MG13pyLuc, which contains a mutated p53 binding site, was not activated (Fig. 3B, bar 6). Cotransfection of a plasmid encoding the Tax protein inhibited the ability of p53 to activate expression from the promoter in a dose-dependent fashion (Fig. 3B, bars 1 to 3). A 33-fold reduction in p53-dependent Luc activity was observed with 6 μg of Tax expression vector. Transfection of a control plasmid without the Tax insert had no effect on activation by p53 (Fig. 3B, bar 4), ruling out the possibility that the inhibition was due to promoter competition.
FIG. 3.

Dose-dependent repression of p53 transactivation. (A) Diagrammatic representation of Luc reporter constructs. PG13pyLuc contains 13 copies of a p53 consensus binding site upstream of the polyomavirus promoter. MG13pyLuc contains 13 mutated p53 binding sites upstream of the polyomavirus promoter. (B) Repression of p53 activation. By using Lipofectamine reagent, Jurkat cells were cotransfected with 3 μg of reporter plasmid and 3 μg of wild-type p53 in the pCEP4 vector (kindly provided by Jennifer Pietenpol, Vanderbilt Cancer Center, Nashville, Tenn.) along with increasing amounts of pcTax. DNA concentrations were adjusted with vector control so that equivalent amounts of DNA were used for all transfections. Cells were harvested 24 h after transfection and assayed for Luc activity by using a Berthold LB9500C luminometer. (C) Effect of Tax mutations on p53 activity. p53 transcriptional activity was measured by cotransfection (as described above) of wild-type p53 (3 μg) and PG13pyLuc (3 μg) in the presence of wild-type (6 μg), M22 (6 μg), and M32 (6 μg) Tax plasmids.
To convincingly demonstrate that Tax inhibited p53, it was important to identify Tax mutants which failed to inhibit p53 transactivation. Following an initial screen of multiple Tax mutants, we obtained data for two Tax mutants, M32, which contains amino acid substitutions at positions 196 and 197, and M22, which contains amino acid substitutions at positions 130 and 131 (49). Importantly, these mutants have been shown to have no effect on nuclear localization of the Tax protein and were expressed to similar levels as wild-type Tax protein (data not shown). Consistent with the data presented above, wild-type Tax inhibited p53 transactivation (Fig. 3C). In contrast, Tax mutants M22 and M32 failed to inhibit p53 function. These studies provide conclusive evidence that Tax inhibits the p53 transactivation function. An exhaustive analysis of Tax mutants is under way to define the domains of Tax involved in p53 inactivation.
Tax inhibits p53 transactivation function independent of sequence-specific DNA binding.
To determine if Tax inhibited p53 function by blocking the sequence-specific interaction of p53 with the DNA or interfering with its transactivation function, we used Gal4(DBD)-p53 fusion proteins and the GAL-TK (thymidine kinase)-Luc reporter construct. The DBD of Gal4 was fused either to the full-length wild-type p53 (Gal53) or to the first 52 amino acids, or activation domain, of p53 (GalN53). As shown in Fig. 4B, the GAL-TK-Luc promoter was activated in the presence of either Gal53 or GalN53 (bars 2 and 5). Cotransfection of the GalN53 plasmid with the reporter construct resulted in a level of induction equivalent to the full-length Gal53 protein. When Gal4(DBD)-p53 fusion constructs were transfected with Tax, transactivation was repressed (Fig. 4B, bars 3 and 6). These results demonstrate that Tax inhibition of p53 activation is independent of site-specific DNA binding and delimits the target of Tax inhibition to the N-terminal 52 amino acids of p53. Consistent with this observation, Tax does not inhibit p53 DNA binding in band shift or biotinylated DNA binding assays (data not shown).
FIG. 4.

Inhibition of p53 transcriptional activation domain. (A) Diagrammatic representation of the Gal4-Luc reporter plasmid. GAL-TK-Luc has five Gal4 DNA binding sites positioned upstream of the TK promoter. (B) Tax-dependent repression of p53 function independent of DNA binding. Jurkat cells were cotransfected with 3 μg of reporter construct and 3 μg of either pGal53 or pGalN53 in the presence or absence of Tax (6 μg).
DISCUSSION
Our studies provide the first experimental evidence that the HTLV-1-transforming protein, Tax, stabilizes and inactivates the transactivation function of p53. Moreover, Tax inhibits p53 activity by interfering with the N-terminal activation domain of p53, independent of p53 DNA binding activity. These important experiments confirm and extend earlier observations from this and other laboratories on p53 stabilization and lack of p53 transcription activity in HTLV-1-transformed cells. Gartenhaus and Wang (20) reported that wild-type p53 was functionally inactive in HTLV-1-transformed cell lines. Subsequently, Cereseto et al. (4) demonstrated that p53 regulation of cellular genes such as p21 and Gadd45 are inactivated in HTLV-1-transformed cell lines. Our present studies further demonstrate that p53 transactivation of the MDM2 and Bax promoters is inhibited in HTLV-1-transformed cells.
Several viral proteins interfere with the transcriptional function of the p53 protein (7, 11, 12, 14, 30, 36, 40, 53, 54, 62). Interestingly, the mode of interference of p53 function is accomplished by targeting different domains of p53. The adenovirus E1B 55-kDa (45) and cellular MDM2 (7, 36, 40) proteins target the N terminus of p53, interfering with the interaction of the p53 transcriptional activation domain with TAFII31 and TATA binding protein. Simian virus 40 T antigen interacts with p53 through the sequence-specific DBD, inhibiting the interaction of p53 with DNA (12, 36). The human CMV IE2 (54), adenovirus E4orf6 (14), and Epstein-Barr virus BZLF1 (62) proteins interact with the carboxy terminus of p53. Of interest, the interaction of the E4orf6 protein with p53 at the carboxy terminus inhibits the interaction of TAFII31 with the N terminus of p53 (14). Adenovirus E1A inhibits the transactivation function in an indirect fashion, apparently by increasing homo- or hetero-oligomerization of p53 (27).
Our studies with the Gal4-p53 fusion proteins demonstrate that Tax is able to interfere with the transactivation domain of p53 located within amino acids 1 to 52. The ability of Tax to block the N-terminal p53 transactivation domain is novel, in that it is unlikely that Tax inhibits transactivation through a direct physical interaction with p53. Studies by our group, as well as others (4, 20), fail to find an in vivo association between Tax and p53. Our most recent results suggest that Tax may inactivate p53 function through a novel pathway involving posttranslational modification of p53.
The functional inactivation of p53 by Tax could play an important role in the development of ATL. It has been postulated by several groups that following viral infection and immortalization, a “second hit” is responsible for transformation and development of ATL. Certainly, if the p53 protection pathway is inactivated, the development of chromosomal abnormalities or mutations during the chronic viral infection is increased. In fact, Tax may combine two pathways, transcriptional repression (55) and protein inactivation, to fully inactivate p53 function. Our results further suggest that p53’s regulatory function of apoptotic genes is impaired in HTLV-1-transformed cells. The functional interference of p53 by Tax may contribute to the resistance of ATL cells to radio- and chemotherapeutic agents.
REFERENCES
- 1.Ballard D W, Bohnlein E, Lowenthal J W, Wano Y, Franza B R, Greene W C. HTLV-I tax induces cellular proteins that activate the kappa B element in the IL-2 receptor alpha gene. Science. 1988;241:1652–1655. doi: 10.1126/science.241.4873.1652. [DOI] [PubMed] [Google Scholar]
- 2.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Butz K, Shahabeddin L, Geisen C, Spitkovsky D, Ullmann A, Hoppe-Seyler F. Functional p53 protein in human papillomavirus-positive cancer cells. Oncogene. 1995;10:927–936. [PubMed] [Google Scholar]
- 4.Cereseto A, Diella F, Mulloy J C, Cara A, Michieli P, Grassmann R, Franchini G, Klotman M E. p53 functional impairment and high p21waf1/cip1 expression in human T-cell lymphotropic/leukemia virus type I-transformed T cells. Blood. 1996;88:1551–1560. [PubMed] [Google Scholar]
- 5.Chandar N, Billig B, McMaster J, Novak J. Inactivation of p53 gene in human and murine osteosarcoma cells. Br J Cancer. 1992;65:208–214. doi: 10.1038/bjc.1992.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C Y, Oliner J D, Zhan Q, Fornace A J, Jr, Vogelstein B, Kastan M B. Interactions between p53 and MDM2 in a mammalian cell cycle checkpoint pathway. Proc Natl Acad Sci USA. 1994;91:2684–2688. doi: 10.1073/pnas.91.7.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Marechal V, Levine A J. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–4114. doi: 10.1128/mcb.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen T H, Brody J R, Romantsev F E, Yu J G, Kayler A E, Voneiff E, Kuhajda F P, Pasternack G R. Structure of pp32, an acidic nuclear protein which inhibits oncogene-induced formation of transformed foci. Mol Biol Cell. 1996;7:2045–2056. doi: 10.1091/mbc.7.12.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng J, Haas M. Frequent mutations in the p53 tumor suppressor gene in human leukemia T-cell lines. Mol Cell Biol. 1990;10:5502–5509. doi: 10.1128/mcb.10.10.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke A R, Purdie C A, Harrison D J, Morris R G, Bird C C, Hooper M L, Wyllie A H. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 11.Demers G W, Halbert C L, Galloway D A. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology. 1994;198:169–174. doi: 10.1006/viro.1994.1019. [DOI] [PubMed] [Google Scholar]
- 12.Deppert W, Haug M, Steinmayer T. Modulation of p53 protein expression during cellular transformation with simian virus 40. Mol Cell Biol. 1987;7:4453–4463. doi: 10.1128/mcb.7.12.4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Wet J R, Wood K V, DeLuca M, Helinski D R, Subramani S. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol. 1987;7:725–737. doi: 10.1128/mcb.7.2.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobner T, Horikoshi N, Rubenwolf S, Shenk T. Blockage by adenovirus E4orf6 of transcriptional activation by the p53 tumor suppressor. Science. 1996;272:1470–1473. doi: 10.1126/science.272.5267.1470. [DOI] [PubMed] [Google Scholar]
- 15.Dulic V, Kaufmann W K, Wilson S J, Tlsty T D, Lees E, Harper J W, Elledge S J, Reed S I. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 16.El-Deiry W S, Harper J W, O’Connor P M, Velculescu V E, Canman C E, Jackman J, Pietenpol J A, Burrell M, Hill D E, Wang Y. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54:1169–1174. [PubMed] [Google Scholar]
- 17.El-Deiry W S, Kern S E, Pietenpol J A, Kinzler K W, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 18.El-Deiry W S, Tokino T, Velculescu V E, Levy D B, Parsons R, Trent J M, Lin D, Mercer W E, Kinzler K W, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 19.Fujii M, Sassone-Corsi P, Verma I M. c-fos promoter trans-activation by the tax1 protein of human T-cell leukemia virus type I. Proc Natl Acad Sci USA. 1988;85:8526–8530. doi: 10.1073/pnas.85.22.8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gartenhaus R B, Wang P. Functional inactivation of wild-type p53 protein correlates with loss of IL-2 dependence in HTLV-I transformed human T lymphocytes. Leukemia. 1995;9:2082–2086. [PubMed] [Google Scholar]
- 21.Gessain A, Barin F, Vernant J C, Gout O, Maurs L, Calender A, de The G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet. 1985;ii:407–410. doi: 10.1016/s0140-6736(85)92734-5. [DOI] [PubMed] [Google Scholar]
- 22.Grassmann R, Dengler C, Muller-Fleckenstein I, Fleckenstein B, McGuire K, Dokhelar M C, Sodroski J G, Haseltine W A. Transformation to continuous growth of primary human T lymphocytes by human T-cell leukemia virus type I X-region genes transduced by a herpesvirus saimiri vector. Proc Natl Acad Sci USA. 1989;86:3351–3355. doi: 10.1073/pnas.86.9.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grossman W J, Kimata J T, Wong F H, Zutter M, Ley T J, Ratner L. Development of leukemia in mice transgenic for the tax gene of human T-cell leukemia virus type I. Proc Natl Acad Sci USA. 1995;92:1057–1061. doi: 10.1073/pnas.92.4.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada S, Koyanagi Y, Yamamoto N. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science. 1985;229:563–566. doi: 10.1126/science.2992081. [DOI] [PubMed] [Google Scholar]
- 25.Kashanchi F, Duvall J F, Brady J N. Electroporation of viral transactivator proteins into lymphocyte suspension cells. Nucleic Acids Res. 1992;20:4673–4674. doi: 10.1093/nar/20.17.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kastan M B, Zhan Q, El-Deiry W S, Carrier F, Jacks T, Walsh W V, Plunkett B S, Vogelstein B, Fornace A J., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 27.Kastan M B, Onyekwere O, Sidransky D, Vogelstein B, Craig R W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 28.Ko L J, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 29.Kuerbitz S J, Plunkett B S, Walsh W V, Kastan M B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowe S W, Ruley H E. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev. 1993;7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- 31.Lowe S W, Schmitt E M, Smith S W, Osborne B A, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 32.Lu X, Lane D P. Differential induction of transcriptionally active p53 following UV or ionizing radiation: defects in chromosome instability syndromes? Cell. 1993;75:765–778. doi: 10.1016/0092-8674(93)90496-d. [DOI] [PubMed] [Google Scholar]
- 33.Miyashita T, Harigai M, Hanada M, Reed J C. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res. 1994;54:3131–3135. [PubMed] [Google Scholar]
- 34.Miyashita T, Krajewski S, Krajewska M, Wang H G, Lin H K, Liebermann D A, Hoffman B, Reed J C. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 35.Miyashita T, Reed J C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 36.Momand J, Zambetti G P, Olson D C, George D, Levine A J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 37.Nagai H, Kinoshita T, Imamura J, Murakami Y, Hayashi K, Mukai K, Ikeda S, Tobinai K, Saito H, Shimoyama M. Genetic alteration of p53 in some patients with adult T-cell leukemia. Jpn J Cancer Res. 1991;82:1421–1427. doi: 10.1111/j.1349-7006.1991.tb01815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newcomb E W. P53 gene mutations in lymphoid diseases and their possible relevance to drug resistance. Leuk Lymphoma. 1995;17:211–221. doi: 10.3109/10428199509056825. [DOI] [PubMed] [Google Scholar]
- 39.Oliner J D, Kinzler K W, Meltzer P S, George D L, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 40.Oliner J D, Pietenpol J A, Thiagalingam S, Gyuris J, Kinzler K W, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 41.Osame M, Usuku K, Izumo S, Ijichi N, Amitani H, Igata A, Matsumoto M, Tara M. HTLV-I associated myelopathy, a new clinical entity. Lancet. 1986;ii:1031–1032. doi: 10.1016/s0140-6736(86)91298-5. . (Letter.) [DOI] [PubMed] [Google Scholar]
- 42.Papathanasiou M A, Kerr N C, Robbins J H, McBride O W, Alamo I, Jr, Barrett S F, Hickson I D, Fornace A J., Jr Induction by ionizing radiation of the gadd45 gene in cultured human cells: lack of mediation by protein kinase C. Mol Cell Biol. 1991;11:1009–1016. doi: 10.1128/mcb.11.2.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perry M E, Piette J, Zawadzki J A, Harvey D, Levine A J. The mdm-2 gene is induced in response to UV light in a p53-dependent manner. Proc Natl Acad Sci USA. 1993;90:11623–11627. doi: 10.1073/pnas.90.24.11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poiesz B J, Ruscetti F W, Gazdar A F, Bunn P A, Minna J D, Gallo R C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA. 1980;77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prives C, Manfredi J J. The p53 tumor suppressor protein: meeting review. Genes Dev. 1993;7:529–534. doi: 10.1101/gad.7.4.529. [DOI] [PubMed] [Google Scholar]
- 46.Reid R L, Lindholm P F, Mireskandari A, Dittmer J, Brady J N. Stabilization of wild-type p53 in human T-lymphocytes transformed by HTLV-I. Oncogene. 1993;8:3029–3036. [PubMed] [Google Scholar]
- 47.Sakashita A, Hattori T, Miller C W, Suzushima H, Asou N, Takatsuki K, Koeffler H P. Mutations of the p53 gene in adult T-cell leukemia. Blood. 1992;79:477–480. [PubMed] [Google Scholar]
- 48.Seiki M, Eddy R, Shows T B, Yoshida M. Nonspecific integration of the HTLV provirus genome into adult T-cell leukaemia cells. Nature. 1984;309:640–642. doi: 10.1038/309640a0. [DOI] [PubMed] [Google Scholar]
- 49.Smith M R, Greene W C. Type I human T cell leukemia virus tax protein transforms rat fibroblasts through the cyclic adenosine monophosphate response element binding protein/activating transcription factor pathway. J Clin Invest. 1991;88:1038–1042. doi: 10.1172/JCI115364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snow K, Judd W. Heterogeneity of a human T-lymphoblastoid cell line. Exp Cell Res. 1987;171:389–403. doi: 10.1016/0014-4827(87)90171-6. [DOI] [PubMed] [Google Scholar]
- 51.Soussi T, Caron de Fromentel C, May P. Structural aspects of the p53 protein in relation to gene evolution. Oncogene. 1990;5:945–952. [PubMed] [Google Scholar]
- 52.Tanaka A, Takahashi C, Yamaoka S, Nosaka T, Maki M, Hatanaka M. Oncogenic transformation by the tax gene of human T-cell leukemia virus type I in vitro. Proc Natl Acad Sci USA. 1990;87:1071–1075. doi: 10.1073/pnas.87.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tiemann F, Zerrahn J, Deppert W. Cooperation of simian virus 40 large and small T antigens in metabolic stabilization of tumor suppressor p53 during cellular transformation. J Virol. 1995;69:6115–6121. doi: 10.1128/jvi.69.10.6115-6121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsai H L, Kou G H, Chen S C, Wu C W, Lin Y S. Human cytomegalovirus immediate-early protein IE2 tethers a transcriptional repression domain to p53. J Biol Chem. 1996;271:3534–3540. [PubMed] [Google Scholar]
- 55.Uittenbogaard M N, Giebler H A, Reisman D, Nyborg J K. Transcriptional repression of p53 by human T-cell leukemia virus type I Tax protein. J Biol Chem. 1995;270:28503–28506. doi: 10.1074/jbc.270.48.28503. [DOI] [PubMed] [Google Scholar]
- 56.Waldman T, Lengauer C, Kinzler K W, Vogelstein B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature. 1996;381:713–716. doi: 10.1038/381713a0. [DOI] [PubMed] [Google Scholar]
- 57.Wu X, Bayle J H, Olson D, Levine A J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 58.Yamato K, Oka T, Hiroi M, Iwahara Y, Sugito S, Tsuchida N, Miyoshi I. Aberrant expression of the p53 tumor suppressor gene in adult T-cell leukemia and HTLV-I-infected cells. Jpn J Cancer Res. 1993;84:4–8. doi: 10.1111/j.1349-7006.1993.tb02775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshida M, Seiki M, Yamaguchi K, Takatsuki K. Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proc Natl Acad Sci USA. 1984;81:2534–2537. doi: 10.1073/pnas.81.8.2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhan Q, Fan S, Bae I, Guillouf C, Liebermann D A, O’Connor P M, Fornace A J., Jr Induction of bax by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene. 1994;9:3743–3751. . (Erratum, 10:1259, 1995.) [PubMed] [Google Scholar]
- 61.Zhan Q, Bae I, Kastan M B, Fornace A J., Jr The p53-dependent gamma-ray response of GADD45. Cancer Res. 1994;54:2755–2760. [PubMed] [Google Scholar]
- 62.Zhang Q, Gutsch D, Kenney S. Functional and physical interaction between p53 and BZLF1: implications for Epstein-Barr virus latency. Mol Cell Biol. 1994;14:1929–1938. doi: 10.1128/mcb.14.3.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]