

Abstract
Latent infection of mice with wild-type herpes simplex virus is established during an acute phase of ganglionic infection in which there is abundant viral replication and productive-cycle gene expression. Thymidine kinase-negative mutants establish latent infections but are severely impaired for acute ganglionic replication and productive-cycle gene expression. Indeed, by in situ hybridization assays, acute infection by these mutants resembles latency. To assess events during establishment of latency by wild-type and thymidine kinase-negative viruses, we quantified specific viral nucleic acid sequences in mouse trigeminal ganglia during acute ganglionic infection by using sensitive PCR-based assays. Through 32 h postinfection, viral DNA and transcripts representative of the three kinetic classes of productive-cycle genes accumulated to comparable levels in wild-type- and mutant-infected ganglia. At 48 and 72 h, although latency-associated transcripts accumulated to comparable levels in ganglia infected with wild-type or mutant virus, levels of DNA accumulating in wild-type-infected ganglia exceeded those in mutant-infected ganglia by 2 to 3 orders of magnitude. Coincident with this increase in DNA, wild-type-infected ganglia exhibited abundant expression of productive-cycle genes and high titers of infectious progeny. Nevertheless, the levels of productive-cycle RNAs expressed by mutant virus during acute infection greatly exceeded those expressed by wild-type virus during latency. The results thus distinguish acute infection of ganglia by a replication-compromised mutant from latent infection and may have implications for mechanisms of latency.
During infection of mammalian hosts, herpes simplex virus (HSV) replicates productively at peripheral sites and establishes a latent infection in sensory neurons that innervate those sites (7, 38). Productive replication is specified by a well-described cascade pattern of gene expression (14, 20) in which immediate-early (IE) genes are expressed first, followed by early (E) genes and finally by late (L) genes, resulting in viral amplification and cell death. In contrast, latent infection is characterized by the absence of infectious virus in tissues (50) containing viral DNA (vDNA) (11, 42), extremely limited productive-cycle gene expression (34, 51), and abundant expression of the latency-associated transcripts (LATs) (8, 43, 48, 51).
In animal models of HSV infection, establishment of latency by wild-type (wt) virus includes a period of viral replication in ganglia, typically for about 1 week (28). Viral mutants which cannot replicate or which are defective for replication in ganglia nevertheless can establish a latent infection (6, 10, 26, 35, 37, 45, 49, 52). Thymidine kinase-negative (TK−) mutants establish latency but exhibit a ≥105-fold reduction in infectious progeny virus during acute infection in ganglia and ordinarily do not reactivate (6, 10, 25). Acute infections with TK− virus permit studies of establishment of latency in the absence of acute replication. Because TK generates nucleotide precursors for DNA synthesis and because sensory neurons are nondividing cells that are presumably deficient in such precursors, it is thought that the block to productive infection of TK− mutants in ganglia is at the level of DNA replication, although evidence for this has been limited.
Previously, in situ hybridization analysis of representative genes of the three kinetic classes during acute ganglionic infection revealed abundant productive-cycle RNAs in wt-infected ganglia but little or none in TK− mutant-infected ganglia (29). Instead, these ganglia expressed abundant LATs, resembling latently infected ganglia. Given that IE and E expression in cell culture begins prior to and is not dependent upon vDNA synthesis, the failure to detect IE or E transcripts in TK− HSV-infected ganglia was unexpected. Kosz-Vnenchak et al. (29) suggested that wt and TK− viruses initiate transcription in neurons similarly but generate RNAs below the level of detection, and they hypothesized that expression of productive-cycle transcripts to levels detectable by in situ hybridization was dependent upon vDNA synthesis.
In this study we measured vDNA, LATs, and productive-cycle RNAs representative of the three kinetic classes by using quantitative PCR and quantitative reverse transcriptase PCR (QRPCR) assays in mouse trigeminal ganglia infected with wt or TK− virus during the establishment of latency. Our results indicate that although productive-cycle gene expression by TK− virus during establishment of latency is drastically reduced relative to that of wt virus, it greatly exceeds that observed during latency.
MATERIALS AND METHODS
Viruses and cells.
The wt HSV type 1 strain KOS and TK− deletion mutants dlsptk and dlsactk (6) were propagated and assayed on Vero cell monolayers as previously described (4).
Infection of mice.
Seven- to 8-week-old male CD-1 outbred mice were inoculated with 2 × 106 PFU of virus or mock infected with virus diluent via corneal scarification. At specified times postinoculation (p.i.), eye swabs and/or trigeminal ganglia were collected for determining titers of infectious virus as described previously (5).
Extraction of nucleic acids and reverse transcription.
Ganglia were collected and frozen, and nucleic acids were prepared as described previously (34). One-tenth of each ganglion homogenate was processed for vDNA and cellular DNA. The remainder was purified and reverse transcribed in the presence of mouse-specific (Act-2) and virus-specific (4-2, L-2, tk-2, gH-2, and gC-2) primers (Table 1) and other virus-specific oligonucleotides as described previously (34).
TABLE 1.
Primers and probes
| Gene | Oligonucleotidea | Sequence (5′ to 3′) | Primer pair and product size (nt) |
|---|---|---|---|
| tkb | tk-U | GTC CAC TTC GCA TAT TAA GG | U-2, 128 |
| gC | gC-1 | GGG TCC GTC CCC CCC AAT | 1-2, 109 |
| gC-2 | CGT TAG GTT GGG GGC GCT | ||
| gC-3 | TAG AGG AGG TCC TGA CGA ACA | ||
| gH | gH-U | ACG CAC GGG TGT TGG GTC | U-2, 81 |
| gH-1 | GTT CGG TCC CAG GGC TGG CA | 1-2, 42 | |
| gH-2 | GGT CTC GGT GGG GTA TCG | ||
| gH-3 | TAT CGA CAG AGT GCC AGC |
RNA standards.
Transcription plasmids for generating RNA quantification standards from the mouse β-actin (pSPMβA), ICP4 (pKS-5′ICP4), tk (pSVtk), and LAT (pKS-5′LAT) genes were previously described (34). Additionally, plasmid pBX1 (21), containing the EcoRI-PvuII fragment of the tk gene which includes the 5′ one-third of the gH gene (Fig. 1D), was kindly provided by Charles Hwang. Both pSVtk and pBX1 were used to generate transcripts for the two gH RNA standards (see Results), while only pSVtk1 was used to generate transcripts for tk RNA standards. Plasmid pKS+gCEB was constructed by cloning the EcoRI-BamHI 1.5-kb fragment of the gC gene (pEcoRI-BamHI-I-I) (12) into the EcoRI and BamHI sites of Bluescript II KS+ (Stratagene), placing it under the control of the bacterial T3 promoter (Fig. 1E). RNA was transcribed in the presence of labeled GTP and quantified as previously described (34), correcting for G+C contents (67% for gC [12] and 63% for the transcript containing tk and gH sequences [15, 23]). Known, equimolar amounts of synthetic RNAs from several viral genes were mixed, serially diluted, combined with a constant amount of uninfected mouse brain RNA, and reverse transcribed as previously described (34).
FIG. 1.
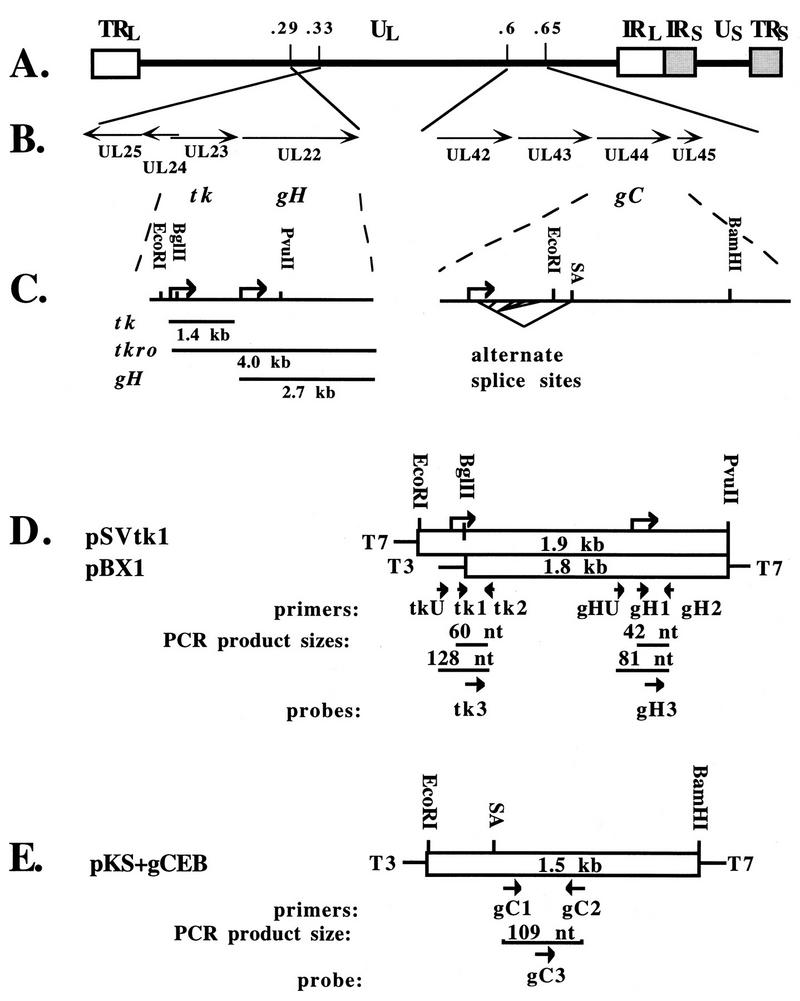
Regions of the HSV-1 genome assayed for L genes gH and gC. (A) Genome structure. The boxes represent the terminal- and internal-repeat (TR and IR) regions which bracket the unique (U) long and short (subscript L and S) sequences depicted in the prototype arrangement. Numbers above the line indicate map units and approximate locations. (B) Expanded regions showing the open reading frames and directions of transcription of the region from map unit 0.33 to 0.29 encompassing open reading frames UL22 to UL25 (left) and the region from map unit 0.60 to 0.65 encompassing open reading frames UL42 to UL45 (right). The open reading frames are indicated below the lines. (C) Features of tk and gH transcription (left) and of gC transcription (right). Restriction enzyme sites used for constructing transcription plasmids are indicated above the line. Bent arrows indicate the transcription start sites. Below the line on the left are depicted three size classes of RNAs originating from this region, a 1.4-kb transcript (tk), a 4.0-kb tk run-on transcript (tkro) 5′ coterminal with tk and 3′ coterminal with gH, and a 2.7-kb gH transcript. On the right the 3′-most splice acceptor (SA) site is indicated above the line, and alternate splice options of the 5′ terminus are indicated by bent lines below the line. (D and E) Open boxes represent the portions of each gene which were cloned into transcription plasmids, with the size of the insert indicated in each box. The restriction enzyme sites indicated above each box correspond to those shown in panel C. Transcription start sites are indicated as bent arrows above the boxes in panel D. The identities and directions of bacterial polymerase promoters are indicated at the sides of the boxes for T7 and T3 RNA polymerases. Below each box arrows indicate the directions and relative positions of PCR primers and probes, with the identities indicated underneath each. The size of each PCR product is indicated. (D) The tk transcription plasmid, pSVtk1, and the gH transcription plasmid, pBX1. (E) The gC transcription plasmid, pKS+gCEB, contains the 1.5-kb EcoRI-to-BamHI region cloned from gC(pEcoRI-BamHI-I-I) into Bluescript II KS+ (Stratagene). The SA site is shown relative to the primer locations.
PCR.
PCR was performed essentially as previously described (34), with a few modifications. Primers and probes for gC and gH and upstream primers for ICP4 and tk are listed in Table 1. Those for the mouse adipsin, mouse β-actin, ICP4, tk, and LAT genes are as previously reported (34). The gC and gH reaction mixtures contained 3.0 mM Mg2+ at an annealing temperature of 60°C; gC was amplified with 10% glycerol, and gH was amplified with Perfect Match Polymerase Enhancer (Stratagene). PCR components for the other assays were as previously described (34). For all, the hot-start method, which entailed heating reaction mixtures to 94°C for 10 min before adding Taq polymerase, was omitted. Instead, TaqStart antibody (Clontech), which reversibly inhibits Taq polymerase prior to the first denaturation step (27), was used, and the first denaturation time was held for 5 min; all subsequent temperature steps were held for 1 min. The reaction mixtures were prepared in batches, and Taq polymerase, TaqStart antibody, Perfect Match Polymerase Enhancer, and deoxynucleoside triphosphates were added just prior to use. In addition, Ficoll 400, at a final concentration of 0.5 to 1%, and the dye tartrazine, at a final concentration of 1 mM (54), were included in all reaction mixtures, precluding the need for a gel loading buffer and permitting direct application of the PCR products to acrylamide gels. Ficoll and tartrazine did not diminish the sensitivity of any assay (32). Positive and negative controls were included with each amplification. vDNA and cellular DNA were coamplified and analyzed as previously described (34) with the PCR modifications described above. QRPCR assays were performed separately for each gene by using a portion of the cDNA (0.5 μl for β-actin and 3.0 μl for each viral sequence). Product sizes are indicated in Fig. 1 and Table 1 and in reference 34. Product specificities of sequences amplified from infected tissue were verified by predicted size, hybridization with the internal oligonucleotide probe, and restriction endonuclease analysis (32). PCR products were quantified, vDNA was normalized to cellular DNA, vRNA was normalized to cellular RNA, and numbers of molecules were calculated on a per-ganglion basis, as previously described (34).
RESULTS
Previously, in situ hybridization analyses had detected abundant expression of LATs but little or no expression of productive-cycle genes in mouse ganglia acutely infected with TK− mutants during the establishment of latency, similar to what is observed in ganglia latently infected with wt or TK− HSV (29). We wished to determine whether gene expression during acute infection by TK− mutants was quantitatively akin to that of latent infection. Therefore, we compared the time course of appearance of infectious virus to that of HSV nucleic acids during acute infection of mouse trigeminal ganglia infected with wt or TK− virus.
Kinetics of appearance of infectious virus.
In previous studies, the TK− HSV mutants dlsptk and dlsactk replicated to wt titers at 24 h p.i. in the mouse eye, did not detectably replicate in ganglia, but nevertheless established latent infection defined by the presence of vDNA, LATs, and complementation activity in ganglia 30 days after corneal inoculation (6). In this study we observed similar eye swab titers of TK− and wt viruses (∼2 × 105 PFU/eye swab) at 24 and 48 h p.i. Progeny virus was not detected in wt-infected ganglia at 24 h p.i., but at 32 h p.i. 50% (8 of 16) of the ganglia infected with wt virus contained a few (5 to 50) PFU per ganglion (Table 2). Thus, infectious wt progeny virus could first be recovered between 24 and 32 h p.i. By 48 h all wt-infected ganglia contained relatively high titers similar to those previously observed (24). In contrast, no virus (<1 PFU/ganglion) was detected in all but one ganglia infected with the TK− virus dlsactk, similar to previous results (6, 25). The one exception was a single ganglion harvested at 48 h p.i. that contained 10 PFU. This titer, which represents ∼0.1% of the wt-infected-ganglion titer, may reflect very low-level replication in the ganglion or contamination from the eye during the dissection of the ganglion. In either event, these results reproduce previous reports of severely impaired replication of TK− mutants in ganglia.
TABLE 2.
Average ganglion titers of wt and TK− HSV
| Virus | Avg titera at:
|
||
|---|---|---|---|
| 24 h p.i. | 32 h p.i. | 48 h p.i. | |
| wt | <1 (0/6) | 25 (8/16) | 8,000 (4/4) |
| dlsactk | <1 (0/6) | <1 (0/16) | 10 (1/4) |
Values represent the numeric average of PFU per ganglion. Only values for positive samples were averaged. Values in parentheses represent number of ganglia which contained PFU/number assayed.
Kinetics of vDNA accumulation.
The accumulation of viral genomes in trigeminal ganglia following corneal inoculation was measured by quantitative PCR. Log10 means and standard deviations of numbers of viral genome copies per ganglion, plotted against time, are shown in Fig. 2. No vDNA signal was detected in corresponding ganglia from mock-infected mice, although cellular DNA levels were similar. Values comparing dlsptk and dlsactk were indistinguishable throughout; therefore, data from these two TK− mutants were pooled. The amounts of vDNA accumulated in wt- and TK− mutant-infected ganglia were indistinguishable through 32 h p.i. However, by 48 h p.i., the mean vDNA levels in wt-infected ganglia exceeded the levels in TK− mutant-infected ganglia by 2 orders of magnitude, and they continued to increase through 72 h p.i. to over 5 × 106 copies per ganglion. This amount is ∼200-fold higher than that observed in ganglia that are latently infected with wt virus (∼2 × 104 copies per ganglion) from 30 to 150 days p.i. (Fig. 2).
FIG. 2.

Time courses and extents of wt (▴) and TK− (○) HSV DNA accumulation and maintenance in trigeminal ganglia following corneal inoculation. Acute and latent vDNA is normalized to cellular DNA. Each point represents mean log10 vDNA per ganglion ± standard deviation, plotted against mean hours or days p.i. as indicated. The number of ganglia assayed for each point is provided in parentheses. vDNA values at 30 and 60 days p.i. (previously reported [34]) were part of an experimental group included in this study that extended to 150 days p.i. and are shown here for comparison.
In contrast, by 72 h p.i., the mean vDNA level in TK− mutant-infected ganglia was ∼3,000 copies per ganglion, a value 3 orders of magnitude lower than that seen in wt-infected ganglia. This value was indistinguishable from that observed at 30, 60, and 150 days p.i. (Fig. 2).
Kinetics of ICP4 and tk RNA accumulation.
To examine gene expression in wt- and TK− mutant-infected ganglia, we measured RNAs from genes representing the three major kinetic classes of transcripts in ganglia from mice infected with wt or TK− virus. In a pilot experiment, the IE ICP4 RNA was detected as early as 19 h p.i. in some ganglia in which vDNA was detected (32), indicating that IE transcription commenced approximately upon arrival of the viral genome in the ganglion. Assays for other transcripts were not performed with these samples. Subsequently, measurements were performed with a larger number of ganglia at 26, 32, 48, and 72 h p.i. Representative autoradiographs of QRPCR products derived from ICP4, tk, gC, and gH RNAs in ganglia at 32 h p.i. and from RNA standards are shown in Fig. 3. To address whether ICP4 and tk products were due to promoter-specific RNAs, primer pairs spanning the putative start sites of the ICP4 and tk genes, which have been shown to amplify synthetic RNA containing the upstream sequences (32, 34), were used to amplify cDNA from several samples. These assays did not generate a detectable signal, consistent with promoter specificity of the QRPCR products. ICP4 RNA accumulation in wt- and TK− mutant-infected ganglia is summarized in Fig. 4A. At 26 and 32 h p.i., ICP4 RNA was expressed to comparable levels in wt- and TK− mutant-infected ganglia. At these time points, accumulation of tk RNA in either wt- or TK− mutant-infected ganglia was, on average, only slightly less than that of ICP4 RNA, and the levels and ranges of tk RNA were comparable for the two viruses (Fig. 4B). However, at 48 and 72 h p.i., the levels of ICP4 and tk RNAs in wt-infected ganglia greatly exceeded (∼1,000-fold) those in TK− mutant-infected ganglia.
FIG. 3.
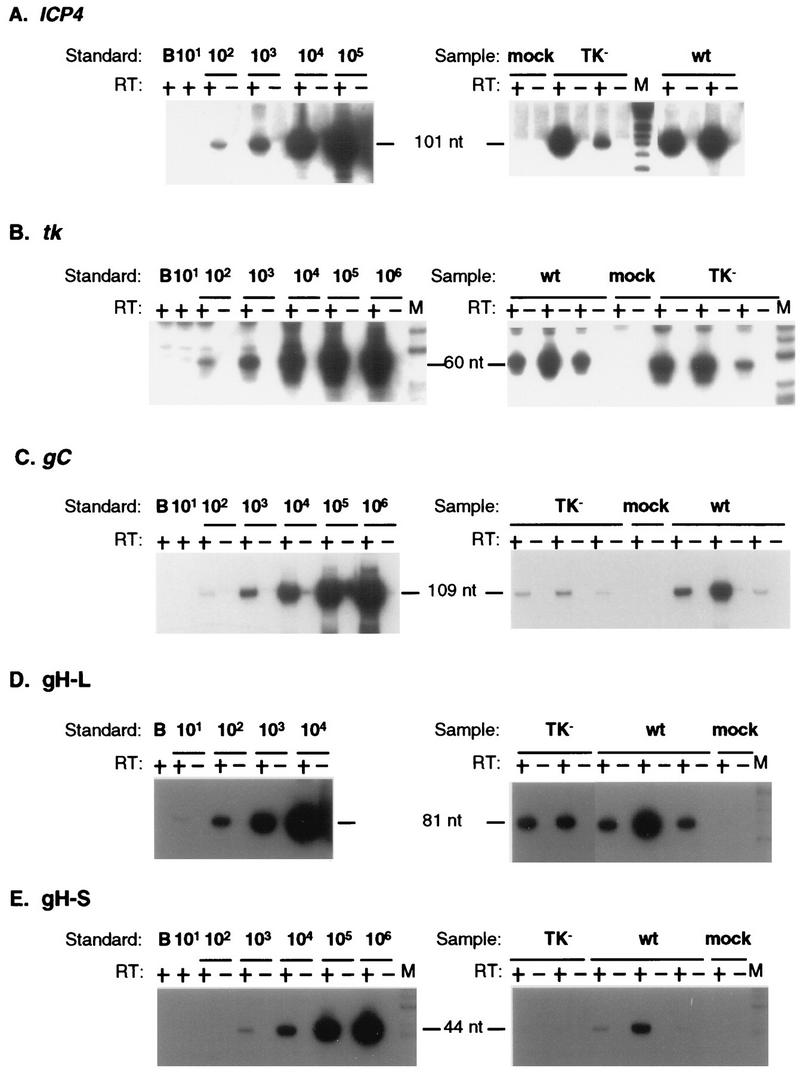
Quantitative measurement of productive-cycle transcripts at 33 h p.i. Autoradiographs of probed Southern blots of QRPCR products separated on polyacrylamide gels are shown. Synthetic transcript mixes (left) or individual ganglion RNAs (right) were reacted with (+) and without (−) reverse transcriptase (RT), and results are displayed in adjacent lanes. Standard, number of specific transcript molecules in serial dilution (indicated above the lines) or water blank (B) mixed with 5 μg of mouse brain RNA. Sample lanes: mock, individual ganglia from animals inoculated with medium not containing virus; TK−, two or three individual ganglia from animals inoculated with the tk deletion mutant dlsactk; wt, two or three individual ganglia from animals inoculated with wt HSV strain KOS; M, molecular weight marker (φX174 digested with HinfI and end labeled with 32P). (A) The ICP4 RNA 101-nt QRPCR product, separated on a 12% polyacrylamide gel, was detected by probing with oligonucleotide 4-3. (B) The tk RNA 60-nt QRPCR product, separated on a 10% polyacrylamide gel, was detected by probing with oligonucleotide tk-3. (C) The gC RNA 109-nt QRPCR product, separated on a 10% polyacrylamide gel, was detected by probing with oligonucleotide gC-3. (D) The gH-L 81-nt QRPCR product, separated on a 12% polyacrylamide gel, was detected by probing with oligonucleotide gH-3. (E) The gH-S 44-nt QRPCR product, separated on a 12% polyacrylamide gel, was detected by probing with oligonucleotide gH-3.
FIG. 4.

Productive-cycle transcript accumulation. Symbols are as in Fig. 2. Each point represents the mean log10 molecules of RNA per ganglion ± standard deviation, with the number of ganglia assayed for each point indicated in parentheses. Dashed lines delineate the lower detection limit for each assay; points which lie on the line represent maximum possible values of samples for which no product was detected. (A) ICP4 RNA. (B) tk RNA. (C) gC RNA. (D) gH RNA.
Kinetics of gC and gH RNA accumulation.
RNAs corresponding to two true L genes, gC and gH, were measured in wt- and TK− mutant-infected ganglia. The assay used to detect gC RNA is outlined in Fig. 1B and C (right), which show an expanded region around the gC locus from map unit 0.60 to 0.65 and an expanded view of the gC transcripts and known splice sites (12). PCR primers were located downstream of the 3′-most splice acceptor site (Fig. 1E). At 26 h p.i., gC RNA was not detected (Fig. 4C); however, the sensitivity of this assay was less than that of those for ICP4 and tk RNA. gC RNA was detectable in some ganglia by 32 h p.i. and was present and abundant in all ganglia infected with wt virus at 48 and 72 h p.i. (Fig. 3 and 4C). In ganglia infected with TK− virus, gC RNA was detectable from 32 to 72 h p.i., but the level remained low (Fig. 3 and 4C). At 32 h, in contrast to what was observed with ICP4 and tk RNAs, there was less gC RNA on average in mutant-infected ganglia than in wt-infected ganglia. By 48 h p.i., the average gC RNA level in wt-infected ganglia exceeded that in TK− mutant-infected ganglia by 700-fold. These results show an increase in gC RNA accumulation corresponding to the time when vDNA increased dramatically in wt-infected ganglia.
The low levels of gC RNA detected by our QRPCR assay at 32 h p.i. in wt-infected ganglia and at all times in TK− mutant-infected ganglia could represent authentic expression from the gC promoter or could represent transcription arising from another promoter, such as one upstream, like that of the E gene UL42 (Fig. 1B). (The kinetic class of the UL43 gene upstream of gC is not known; it could also be an E gene.) A PCR test to monitor the possible contribution of RNAs arising from other promoters was problematic due to the multiple splice options at the 5′ end of gC (Fig. 1C, right). Another L transcript, that of gH, is known to be colinear with a transcript arising from the E gene tk, located upstream of gH (17, 19, 46). To determine the level of promoter-specific RNA from gH, we asked if potential contributions to the QRPCR measurement from upstream run-on transcripts could be subtracted. The tk and gH genes and their transcripts and a tk run-on transcript (designated tkro) are depicted in Fig. 1B and C (left). To quantify promoter-specific gH RNA, we employed two PCR assays (Fig. 1D). In one assay, a primer pair (gH-1 and gH-2) downstream of the start site generates a 44-nucleotide (nt) product, gH-S (for short), and amplifies RNA that includes both promoter-specific gH RNA and tkro RNA. A second primer pair (gH-U and gH-2) bracketing the gH start site generates an 81-nt product, gH-L (for long), and amplifies only tkro. Each product, gH-S and gH-L, was quantified in separate assays (Fig. 3D and E), and the amount of gH RNA that did not arise from tkro was calculated by subtracting the amount of gH-L product from the amount of gH-S product. The gH-S assay was about 10-fold less sensitive than the gH-L assay, imposing a detection limit slightly higher than that for gC (Fig. 4C and D). By amplifying mixtures of gH-S and gH-L PCR products in stoichiometric ratios from 0.01 to 100, we found that promoter-specific gH RNA could be detected if the level of this RNA was at least half the level of tkro RNA (not shown).
Using these two assays, we measured the accumulation of promoter-specific gH RNA (Fig. 4D). In all ganglia at 26 h p.i. and in most ganglia at 32 h p.i., gH-S was equal to gH-L or not detectable; thus, there was no detectable gH RNA. In wt-infected ganglia there was detectable gH RNA at 32 h p.i. in two of six samples, and it was abundant in all samples at 48 and 72 h p.i. Interestingly, in the three wt-infected ganglia harvested at 48 h p.i. in which both L RNAs were measured, equivalent amounts of gC and gH RNA were observed (1.3 × 106 and 1.3 × 106, 6.3 × 105 and 7.9 × 105, and 2.5 × 105 and 2.5 × 105 molecules/ganglion). In TK− mutant-infected ganglia, gH RNA was below the limit of detection for all ganglia at 26, 32, and 48 h p.i. and for three of four ganglia at 72 h p.i. In the one exception at 72 h p.i., the amount of gH detected was ∼1,000-fold less than that in wt-infected ganglia at that time point.
Kinetics of accumulation of LATs.
LATs in acutely infected ganglia were assayed by QRPCR. Unlike for productive-cycle RNAs, the amounts of LATs were similar in wt- and TK− mutant-infected ganglia through 72 h p.i. (Fig. 5A). LATs were the most abundant viral nucleic acid measured at each time point. In TK− mutant-infected ganglia, the level of LATs normalized to vDNA (LAT/vDNA value) increased monotonically to the level achieved and maintained in latency (Fig. 5B). In wt-infected ganglia, the decrease in LAT/vDNA values at 72 h p.i. (Fig. 5B) were most plausibly due to the dramatic increase in vDNA. As previously reported (34), LAT/vDNA values were comparable in ganglia infected with wt and TK− viruses at 30 and 60 days p.i., although the amounts of both vDNA and LATs were 5- to 10-fold greater for wt virus than for TK− virus. Thus, the time course of expression of LATs, unlike those of productive-cycle genes, was very similar in ganglia infected with either wt or TK− virus.
FIG. 5.
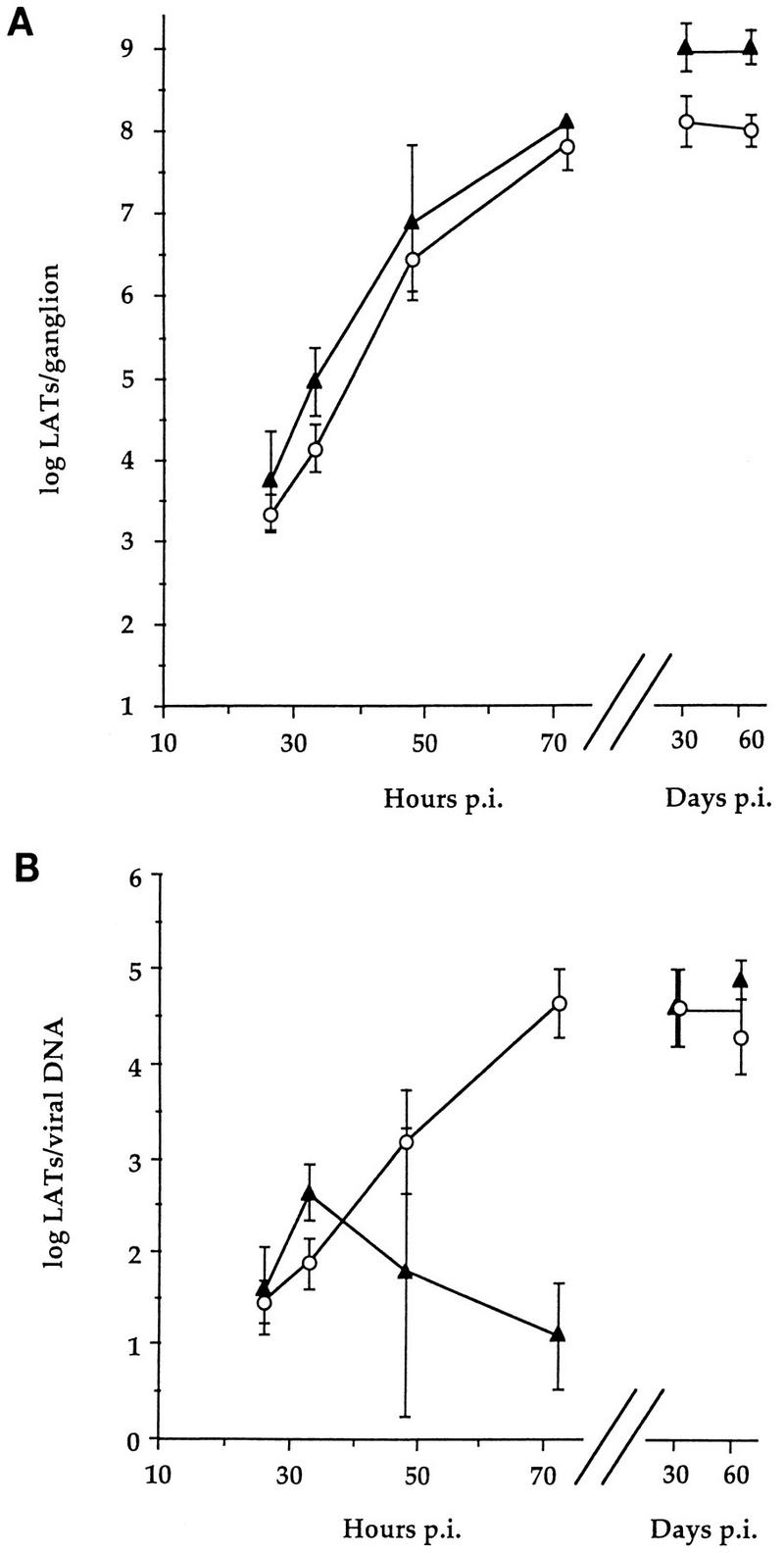
Time course of wt and TK− HSV LAT accumulation in trigeminal ganglia following corneal inoculation. Symbols are as in Fig. 2. Each point represents the mean log10 value ± standard deviation; n = 4 each. Data for 30 and 60 days p.i. were previously reported (34) and are included here for comparison. (A) Accumulation of LATs per ganglion. (B) LATs normalized to viral DNA.
Comparisons with levels of productive-cycle transcripts during the maintenance of latency.
We asked if levels of productive-cycle RNAs in ganglia latently infected with wt virus (30 days p.i.) were comparable to those in ganglia acutely infected with TK− virus (3 days p.i.), as they appeared to be by in situ hybridization (29). Levels of productive-cycle transcripts normalized to viral genomes measured in ganglia acutely infected with TK− virus greatly exceeded those in ganglia latently infected with wt virus (Table 3). (Productive-cycle transcripts in ganglia latently infected with TK− mutants were below the level of detection in the great majority of TK− mutant-infected ganglia, due at least in part to lower levels of vDNA in these ganglia [34].) Ganglia latently infected with wt virus contained nearly three times more vDNA than ganglia acutely infected with TK− virus but contained on average about 40 times less of each productive-cycle RNA detected, leading to a ∼100-fold difference in productive-cycle transcripts per genome. Thus, these results distinguish acute infection of ganglia by TK− virus from latent infection.
TABLE 3.
Productive-cycle RNAs present in ganglia in the absence of viral replication
| Virus | Days p.i. | Mean log10 molecules/ganglion ± SDa
|
||||
|---|---|---|---|---|---|---|
| vDNA | ICP4 RNA | tk RNA | gC RNA | gH RNA | ||
| wt | 30 | 4.1 ± 0.5 (30/30) | 3.1 ± 0.8 (29/30) | 3.0 ± 0.6 (19/22) | 3.2 ± 0.5 (3/10) | NDb (0/6) |
| TK− | 3 | 3.8 ± 0.6 (6/6) | 4.7 ± 0.9 (6/6) | 4.6 ± 0.7 (6/6) | 4.7 ± 1.5 (4/4) | 3.1 (1/4) |
Numbers in parentheses represent number of ganglia containing detectable signal/total number assayed in a given assay. Ganglia without a detectable level of a specific nucleic acid were assigned a number representing the lowest limit of detection for that assay for the purpose of calculating an average.
ND, none detected.
DISCUSSION
In this report we have characterized early molecular events of ganglionic infection by wt and TK− mutant HSV. During acute ganglionic infection, we found that a rapid increase in vDNA in wt-infected ganglia is associated with abundant accumulation of productive-cycle transcripts. This accumulation results in amounts detectable by in situ hybridization (29). In TK− mutant-infected ganglia, there was neither a rapid increase in vDNA nor abundant accumulation of productive-cycle transcripts, but accumulation of LATs was similar to that in wt-infected ganglia. Nevertheless, levels of productive-cycle transcripts in TK− mutant-infected ganglia were much greater than those observed during maintenance of latency by wt or TK− virus. We discuss these findings below.
Accumulation of vDNA.
We detected a few molecules of vDNA in ganglia from corneally infected mice as early as 19 h p.i. The rate of accumulation of vDNA in TK− mutant-infected ganglia remained low and constant at an average of about 10 copies per h through 72 h p.i. As TK− mutants were exceedingly impaired for replication in ganglia, we interpret this slow, constant increase in vDNA as the accumulation of input genomes from the cornea. This would be consistent with the notion that TK is required to supply nucleotide precursors for vDNA synthesis in neurons. The rate of accumulation of vDNA in wt-infected ganglia resembled that in TK− mutant-infected ganglia through 32 h p.i. but then increased to an average of about 300 copies per h from 32 to 48 h p.i. This correlates with the detection of a few PFU in about 50% of wt-infected ganglia at 32 h p.i. and the recovery of abundant infectious viral progeny at 48 h p.i. We interpret these results to mean that input wt genomes accumulate up to about 30 h, after which most vDNA results from new DNA synthesis. The remaining discussion will assume the interpretations in this paragraph, unless otherwise stated.
Accumulation of IE and E transcripts.
We observed similar levels of the IE transcript ICP4 and the E transcript tk during the initial stages of ganglionic infection by either wt or TK− virus (Fig. 4A and B). We saw no evidence during this period for any difference in the order in which genes were expressed relative to expression in cultured cells. ICP4 RNA could be detected in ganglia as early as vDNA (32). L RNA was detected only after detection of IE and E RNAs; however, this may have been due to the assays for L RNAs being less sensitive than those for IE and E RNAs. Indeed, it is worth noting that if the assay for tk transcripts had been more sensitive than that for ICP4 transcripts, it would have appeared that E genes were expressed earlier than IE genes.
The 100- to 1,000-fold increases in ICP4 and tk RNA levels between 32 and 48 h p.i. in wt-infected ganglia coincided with vDNA synthesis (Fig. 2 and 4). This is consistent with the proposal that IE- and E-gene expression is activated by vDNA replication in neurons (29, 30, 40). However, because vDNA reaches ganglia as early as 19 h p.i. and because infectious wt virus can be detected as early as 32 h p.i., we cannot exclude the possibility that the very large increases in IE- and E-gene expression were due to spread of wt virus to neighboring cells, resulting in infections that exhibit high levels of IE- and E-gene expression.
Accumulation of L transcripts.
As expected (although the sensitivity of the assay must be kept in mind), the true L-gene transcripts, those of gC and gH, which require vDNA synthesis for efficient expression, did not become detectable in wt-infected ganglia until the time of onset of vDNA synthesis and did not become abundant until vDNA synthesis was well under way (Fig. 4C and D). Similarly, promoter-specific gH transcripts were not detectable or were barely detectable in TK− mutant-infected ganglia (Fig. 4D) or in ganglia latently infected with wt or TK− virus (32, 33), consistent with the notion that TK is required for vDNA replication in ganglia. More difficult to understand is the presence of gC RNA in TK− mutant-infected ganglia, even as early as 32 h p.i. (Fig. 3 and 4C), and in latently infected ganglia (Table 3). This could represent RNA arising from transcription initiating from an upstream E promoter such as that of UL42 or possibly UL43 (Fig. 1B, right), authentic gC transcription that occurs in the absence of vDNA synthesis (basal expression), or a very low level of DNA replication by the TK− mutant. That the gC RNA detected could be arising from an upstream promoter is made more probable by the large amounts of RNA from an upstream promoter detected in the assays that measured gH RNA. Nevertheless, the one TK− mutant-infected ganglion that contained low but measurable promoter-specific gH RNA at 72 h p.i. is consistent with the other two possibilities. Evidence for basal expression of L RNA has been previously obtained (13, 16, 22, 33, 40, 52, 53, 55), so in the absence of evidence of vDNA synthesis in TK− mutant-infected ganglia, this appears to be the better supported of the two possibilities.
Accumulation of LATs.
We detected relatively abundant expression of LATs as early as 26 h p.i. Although we did not verify that the RNA detected arose from the LAT promoter, such early and abundant expression would be consistent with the neuronal specificity of the LAT promoter (1, 2, 56). In wt-infected ganglia, LATs exceeded ICP4 and tk RNAs by at least 10-fold at 26 and 32 h p.i. and exceeded gC and gH RNAs by a similar magnitude at 32 and 48 h p.i. While all four productive-cycle RNAs approached a plateau between 48 and 72 h p.i., LATs continued to increase. The pattern of LAT expression in TK− mutant-infected ganglia was essentially the same as that in wt-infected ganglia, consistent with LATs deriving mostly from input genomes, independent of and unperturbed by viral replication. This would be consistent, then, with observations of LAT expression in wt-infected ganglia occurring mainly in cells that do not abundantly express productive-cycle genes (37, 47).
TK− mutants initiate a productive infection which is then aborted.
Productive infection in cultured cells follows a well-characterized sequence of gene expression, commencing with IE- and E-gene expression, which is followed by vDNA synthesis and then activation of L-gene expression. Kosz-Vnenchak et al. (29) did not detect appreciable amounts of IE and E RNAs in TK− mutant-infected ganglia by using in situ hybridization. They proposed that in sensory neurons in vivo, limited IE- and E-gene expression occurs in the absence of vDNA synthesis (30). Nichol et al. (40) observed similar regulatory effects in cultured rat neurons. However, we found that TK− mutant-infected ganglia express representative IE and E RNAs as abundantly as wt-infected ganglia prior to vDNA synthesis. Subsequently, levels of productive-cycle RNAs in mutant-infected ganglia were about 1,000-fold lower than those in wt-infected ganglia. It is possible that this degree of expression accounts for the capacity of a TK-deficient virus to express a reporter gene downstream of an E promoter in a few neurons during the first several days following corneal inoculation (9, 18). Regardless, our results are consistent with TK− mutants initiating a productive infection that progresses up to the stage of vDNA replication and then aborts. These results support the suggestion by Kosz-Vnenchak et al. (30) that wt and TK− viruses initiate transcription similarly in ganglia.
Acute infection by TK− mutants entails levels of productive gene expression—both per viral genome and per ganglion—that are much greater than those during a latent infection by wt virus (Table 3) or TK− mutants (34). What, then, causes the drastic decrease in gene expression between days 3 and 30? There are two contrasting explanations for this decrease. One explanation begins with the idea that most productive-cycle gene expression at day 3 occurs within a few cells that are undergoing abortive productive infection, while most cells are expressing mainly LATs and the lower levels of productive-cycle transcripts characteristic of latently infected ganglia (32). By this explanation, the subpopulation of cells responsible for most productive-cycle gene expression is eliminated by day 30, accounting for the drastic decrease in productive-cycle gene expression. However, it should be emphasized that if such a subpopulation of cells exists, it does not express productive-cycle genes at wt levels at day 3, as such cells were not detected in in situ hybridization experiments (29).
An alternate explanation is that productive-cycle gene expression in TK− mutant-infected cells simply tapers off with time, as has been observed with reporter genes under the control of various promoters in TK+ and TK− backgrounds (9), perhaps due to nucleosomal repression of transcription (reference 36 and references therein). This explanation does not require the elimination of infected cells, even though there is much greater expression of productive-cycle genes during acute infection by TK− virus than there is in latently infected ganglia. Consistent with this explanation, in TK− mutant-infected ganglia the amounts of vDNA (in contrast with the case for wt-infected ganglia) (Fig. 2) and LATs (Fig. 5) at 3 days p.i. are indistinguishable (i.e., their standard deviations overlap) from those at ≥30 days p.i. Moreover, the numbers of LAT-positive cells in TK− mutant-infected ganglia measured by in situ hybridization also were similar at 3 and 30 days p.i. (31), and no evidence of degeneration or death of neurons or satellite cells was observed in histochemical analyses of TK− mutant-infected ganglia from 4 through 60 days p.i. (9). This explanation leads to the hypothesis that some neurons in latently infected ganglia could express levels of IE and E genes similar to those achieved during acute ganglion infection with TK− virus, which are high relative to those achieved in latently infected ganglia, without achieving full-blown reactivation or cell death. This might account for those latently infected ganglia that exhibit >1 ICP4 RNA and/or tk RNA per vDNA (3, 34). Such expression may be considered abortive reactivation; alternatively, it could represent a less repressed form of latency where the infected neuron has surmounted certain blocks to reactivation but not others that prevent, for example, vDNA replication. It should be possible to use methods such as in situ PCR and reverse transcriptase PCR (39, 41) or purification of neurons followed by QRPCR (44) to test specific predictions made from each of these contrasting explanations.
ACKNOWLEDGMENTS
We thank Cliff Cho and Kevin Torgerson for technical assistance.
This work was supported by NIH grants PO1 AI24010 and PO1 NS35138. M.F.K. was supported in part by a fellowship from the Albert J. Ryan Foundation.
REFERENCES
- 1.Batchelor A H, O’Hare P. Localization of cis-acting sequence requirements in the promoter of the latency-associated transcript of herpes simplex virus type 1 required for cell-type-specific activity. J Virol. 1992;66:3573–3582. doi: 10.1128/jvi.66.6.3573-3582.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batchelor A H, O’Hare P. Regulation and cell-type-specific activity of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. J Virol. 1990;64:3269–3279. doi: 10.1128/jvi.64.7.3269-3279.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S-H, Kramer M F, Schaffer P A, Coen D M. A viral function represses accumulation of transcripts from productive cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1997;71:5878–5884. doi: 10.1128/jvi.71.8.5878-5884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coen D M, Fleming J, Leslie H E, L K, Retondo M J. Sensitivity of arabinosyladenine-resistant mutants of herpes simplex virus to other antiviral drugs and mapping of drug hypersensitivity mutations to the DNA polymerase locus. J Virol. 1985;53:477–488. doi: 10.1128/jvi.53.2.477-488.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coen D M, Irmiere A F, Jacobson J G, Kerns K M. Low levels of herpes simplex virus thymidine- thymidylate kinase are not limiting for sensitivity to certain antiviral drugs or for latency in a mouse model. Virology. 1989;168:221–231. doi: 10.1016/0042-6822(89)90261-4. [DOI] [PubMed] [Google Scholar]
- 6.Coen D M, Kosz-Vnenchak M, Jacobson J G, Leib D A, Bogard C L, Schaffer P A, Tyler K L, Knipe D M. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci USA. 1989;86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook M L, Bastone V B, Stevens J G. Evidence that neurons harbor latent herpes simplex virus. Infect Immun. 1974;9:946–951. doi: 10.1128/iai.9.5.946-951.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croen K D, Ostrove J M, Dragovic L J, Smialek J E, Straus S E. Latent herpes simplex virus in human trigeminal ganglia: detection of an immediate early gene “anti-sense” transcript by in situ hybridization. N Engl J Med. 1987;317:1427–1431. doi: 10.1056/NEJM198712033172302. [DOI] [PubMed] [Google Scholar]
- 9.Davar G, Kramer M F, Garber D, Roca A L, Andersen J K, Bebrin W, Coen D M, Kosz-Vnenchak M, Knipe D M, Breakefield X O, Isacson O. Comparative efficacy of expression of genes delivered to mouse sensory neurons with herpes virus vectors. J Comp Neurol. 1994;393:3–11. doi: 10.1002/cne.903390103. [DOI] [PubMed] [Google Scholar]
- 10.Efstathiou S, Kemp S, Darby G, Minson A C. The role of herpes simplex virus type 1 thymidine kinase in pathogenesis. J Gen Virol. 1989;70:869–879. doi: 10.1099/0022-1317-70-4-869. [DOI] [PubMed] [Google Scholar]
- 11.Efstathiou S, Minson A C, Field H J, Anderson J R, Wildy P. Detection of herpes simplex virus-specific DNA sequences in latently infected mice and in humans. J Virol. 1986;57:446–455. doi: 10.1128/jvi.57.2.446-455.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frink R J, Eisenberg R, Cohen G, Wagner E K. Detailed analysis of the portion of the herpes simplex virus type 1 genome encoding glycoprotein C. J Virol. 1983;45:634–647. doi: 10.1128/jvi.45.2.634-647.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Godowski P J, Knipe D M. Identification of a herpes simplex virus function that represses late gene expression from parental genomes. J Virol. 1985;55:357–365. doi: 10.1128/jvi.55.2.357-365.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Godowski P J, Knipe D M. Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc Natl Acad Sci USA. 1986;83:256–260. doi: 10.1073/pnas.83.2.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gompels U, Minson A. The properties and sequence of glycoprotein H of herpes simplex virus type 1. Virology. 1986;153:230–247. doi: 10.1016/0042-6822(86)90026-7. [DOI] [PubMed] [Google Scholar]
- 16.Gu B, DeLuca N. Requirements for activation of the herpes simplex virus glycoprotein C promoter in vitro by the viral regulatory protein ICP4. J Virol. 1994;68:7953–7965. doi: 10.1128/jvi.68.12.7953-7965.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris-Hamilton E, Bachenheimer S L. Accumulation of herpes simplex virus type 1 RNAs of different kinetic classes in the cytoplasm of infected cells. J Virol. 1985;53:144–151. doi: 10.1128/jvi.53.1.144-151.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho D Y, Mocarski E S. β-Galactosidase as a marker in the peripheral and neural tissues of the herpes simplex virus-infected mouse. Virology. 1988;167:279–283. doi: 10.1016/0042-6822(88)90079-7. [DOI] [PubMed] [Google Scholar]
- 19.Holland L E, Sandri-Goldin R M, Goldin A, Glorioso J C, Levine M. Transcriptional and genetic analyses of the herpes simplex virus type 1 genome: coordinates 0.29 to 0.45. J Virol. 1984;49:947–959. doi: 10.1128/jvi.49.3.947-959.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Honess R W, Roizman B. Regulation of herpes virus macromolecular synthesis: sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc Natl Acad Sci USA. 1975;72:1276–1280. doi: 10.1073/pnas.72.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang C B C, Horsburgh B, Pelosi E, Roberts S, Digard P, Coen D M. A net +1 frameshift permits synthesis of thymidine kinase from a drug-resistant herpes simplex virus mutant. Proc Natl Acad Sci USA. 1994;91:5461–5465. doi: 10.1073/pnas.91.12.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imbalzano A N, Coen D M, DeLuca N A. Herpes simplex virus transactivator ICP4 operationally substitutes for the cellular transcription factor Sp1 for efficient expression of the viral thymidine kinase gene. J Virol. 1991;65:565–574. doi: 10.1128/jvi.65.2.565-574.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irmiere A F, Manos M M, Jacobson J G, Gibbs J S, Coen D M. Effect of an amber mutation in the herpes simplex virus thymidine kinase gene on polypeptide synthesis and stability. Virology. 1989;168:210–220. doi: 10.1016/0042-6822(89)90260-2. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson J G, Leib D A, Goldstein D J, Bogard C L, Schaffer P A, Weller S K, Coen D M. A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virology. 1989;173:276–283. doi: 10.1016/0042-6822(89)90244-4. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson J G, Ruffner K L, Kosz-Vnenchak M, Hwang C B C, Wobbe K K, Knipe D M, Coen D M. Herpes simplex virus thymidine kinase and specific stages of latency in murine trigeminal ganglia. J Virol. 1993;67:6903–6908. doi: 10.1128/jvi.67.11.6903-6908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katz J P, Bodin E T, Coen D M. Quantitative polymerase chain reaction analysis of herpes simplex virus DNA in ganglia of mice infected with replication-incompetent mutants. J Virol. 1990;64:4288–4295. doi: 10.1128/jvi.64.9.4288-4295.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kellogg D E, Rybalkin I, Chen S, Mukhamedora N, Vlasik T, Siebert P D, Chenchik A. TaqStart antibody: “hot start” PCR facilitated by a neutralizing monoclonal antibody directed against Taq DNA polymerase. BioTechniques. 1994;16:1134–1137. [PubMed] [Google Scholar]
- 28.Knotts F B, Cook M L, Stevens J G. Pathogenesis of herpetic encephalitis in mice after ophthalmic inoculation. J Infect Dis. 1974;130:16–27. doi: 10.1093/infdis/130.1.16. [DOI] [PubMed] [Google Scholar]
- 29.Kosz-Vnenchak M, Coen D M, Knipe D M. Restricted expression of herpes simplex virus lytic genes during establishment of latent infection by thymidine kinase-negative mutant viruses. J Virol. 1990;64:5396–5402. doi: 10.1128/jvi.64.11.5396-5402.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kosz-Vnenchak M, Jacobson J, Coen D M, Knipe D M. Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J Virol. 1993;67:5383–5393. doi: 10.1128/jvi.67.9.5383-5393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kosz-Vnenchak, M., and D. M. Knipe. Unpublished results.
- 32.Kramer M F. Ph.D. thesis. Cambridge, Mass: Harvard University; 1995. [Google Scholar]
- 33.Kramer, M. F., S.-H. Chen, and D. M. Coen. Unpublished results.
- 34.Kramer M F, Coen D M. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J Virol. 1995;69:1389–1399. doi: 10.1128/jvi.69.3.1389-1399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leib D A, Coen D M, Bogard C L, Hicks K A, Yager D R, Knipe D M, Tyler K L, Schaffer P A P A. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J Virol. 1989;63:759–768. doi: 10.1128/jvi.63.2.759-768.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lokensgard J R, Bloom D C, Dobson A T, Feldman L T. Long-term promoter activity during herpes simplex virus latency. J Virol. 1994;68:7148–7158. doi: 10.1128/jvi.68.11.7148-7158.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margolis T P, Sedarati F, Dobson A T, Feldman L T, Stevens J G. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology. 1992;189:150–160. doi: 10.1016/0042-6822(92)90690-q. [DOI] [PubMed] [Google Scholar]
- 38.McLennan J L, Darby G. Herpes simplex virus latency: the cellular location of virus in dorsal root ganglia and the fate of the infected cell following virus activation. J Gen Virol. 1980;51:233–243. doi: 10.1099/0022-1317-51-2-233. [DOI] [PubMed] [Google Scholar]
- 39.Mehta A, Maggioncalda J, Bagasra O, Thikkavarapu S, Saikumari P, Valyi-Nagy T, Fraser N W, Block T M. In situ DNA PCR and RNA hybridization detection of herpes simplex virus sequences in trigeminal ganglia of latently infected mice. Virology. 1995;206:633–640. doi: 10.1016/s0042-6822(95)80080-8. [DOI] [PubMed] [Google Scholar]
- 40.Nichol P F, Chang J Y, Johnson E M, Olivo P D. Herpes simplex virus gene expression in neurons: viral DNA synthesis is a critical regulatory event in the branch point between the lytic and latent pathways. J Virol. 1996;70:5476–5486. doi: 10.1128/jvi.70.8.5476-5486.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramakrishnan R, Levine M, Fink D. PCR-based analysis of herpes simplex virus type 1 latency in the rat trigeminal ganglion established with a ribonucleotide reductase-deficient mutant. J Virol. 1994;68:7083–7091. doi: 10.1128/jvi.68.11.7083-7091.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rock D L, Fraser N M. Detection of HSV-1 genome in the central nervous system of latently infected mice. Nature. 1983;302:523–525. doi: 10.1038/302523a0. [DOI] [PubMed] [Google Scholar]
- 43.Rock D L, Nesburn A B, Ghiasi H, Ong J, Lewis T L, Lokensgard J R, Wechsler S L. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits infected with herpes simplex virus type 1. J Virol. 1987;61:3820–3826. doi: 10.1128/jvi.61.12.3820-3826.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sawtell N M. Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol. 1997;71:5423–5431. doi: 10.1128/jvi.71.7.5423-5431.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sedarati F, Margolis T P, Stevens J G. Latent infection can be established with drastically restricted transcription and replication of the HSV-1 genome. Virology. 1993;192:687–691. doi: 10.1006/viro.1993.1089. [DOI] [PubMed] [Google Scholar]
- 46.Sharp J A, Wagner M J, Summers W C. Transcription of herpes simplex virus genes in vivo: overlap of a late promoter with the 3′ end of the early thymidine kinase gene. J Virol. 1983;45:10–17. doi: 10.1128/jvi.45.1.10-17.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Speck P G, Simmons A. Divergent molecular pathways of productive and latent infection with a virulent strain of herpes simplex virus type I. J Virol. 1991;65:4001–4005. doi: 10.1128/jvi.65.8.4001-4005.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spivack J G, Fraser N W. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J Virol. 1987;61:3841–3847. doi: 10.1128/jvi.61.12.3841-3847.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steiner I, Spivack J G, Deshmane S L, Ace C I, Preston C M, Fraser N W. A herpes simplex virus type 1 mutant containing a nontransducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia. J Virol. 1990;64:1630–1638. doi: 10.1128/jvi.64.4.1630-1638.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stevens J G, Cook M L. Latent herpes simplex virus in spinal ganglia of mice. Science. 1971;173:843–845. doi: 10.1126/science.173.3999.843. [DOI] [PubMed] [Google Scholar]
- 51.Stevens J G, Wagner E K, Devi-Rao G B, Cook M L. RNA complementary to a herpesvirus α gene mRNA is prominent in latently infected neurons. Science. 1987;235:1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- 52.Valyi-Nagy T, Deshmane S L, Spivack J G, Steiner I, Ace C I, Preston C M, Fraser N W. 1814, that does not replicate in mouse trigeminal ganglia. J. Gen. Virol. 72:641–649. 1991. Investigation of herpes simplex virus type 1 (HSV-1) gene expression and DNA synthesis during the establishment of latent infection by an HSV-1 mutant. [DOI] [PubMed] [Google Scholar]
- 53.Weir J P, Steffy K R, Sethna M. An insertion vector for the analysis of gene expression during herpes simplex virus infection. Gene. 1990;89:271–274. doi: 10.1016/0378-1119(90)90016-k. [DOI] [PubMed] [Google Scholar]
- 54.Wittwer C T, Garling D J. Rapid cycle DNA amplification: time and temperature optimization. BioTechniques. 1991;10:76–83. [PubMed] [Google Scholar]
- 55.Zhang Y-F, Wagner E K. The kinetics of expression of individual herpes simplex virus type 1 transcripts. Vol. 1. Boston, Mass: Martinus Nijhoff Publishing; 1987. [DOI] [PubMed] [Google Scholar]
- 56.Zwaagstra J C, Ghiasi H, Slanina S M, Nesburn A B, Wheatly S C, Lillycrop K, Wood J, Latchman D S, Patel K, Wechsler S L. Activity of herpes simplex virus type 1 latency-associated transcript (LAT) promoter in neuron-derived cells: evidence for neuron specificity and for a large LAT transcript. J Virol. 1990;64:5019–5028. doi: 10.1128/jvi.64.10.5019-5028.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]