

Abstract
R13-1 is an intertypic recombinant virus in which the left-hand 18% of the herpes simplex virus type 1 (HSV-1) genome is replaced by homologous sequences from HSV-2. R13-1 is nonneurovirulent and defective in DNA replication in neurons. The defect was localized to the UL5 open reading frame by using marker rescue analysis (D. C. Bloom and J. G. Stevens, J. Virol. 68:3761–3772, 1994). To provide conclusive evidence that UL5 is the only HSV-2 gene involved in the restricted replication phenotype of R13-1, we have characterized the phenotype of a recombinant virus (IB1) in which only the UL5 gene of HSV-1 was replaced by HSV-2 UL5. Data from 50% lethal dose determinations and the in vivo yields of virus suggested that IB1 has the same phenotypic characteristics as R13-1. UL5 is the helicase component of a complex with helicase and primase activities. All three subunits of this complex (UL5, UL8, and UL52) are required for viral DNA replication in all cell types. The intertypic complex HSV-2 UL5–HSV-1 UL8–HSV-1 UL52 was purified and biochemically characterized. The primase activity of the intertypic complex was 10-fold lower than that of HSV-1 UL5–HSV-1 UL8–HSV-1 UL52. The ATPase activity was comparable to that of the HSV-1 enzyme complex, and although the helicase activity was threefold lower, this did not interfere with the synthesis of leading strands by the HSV polymerase. One explanation for these findings is that the interactions between the subunits of the helicase-primase intertypic complex that are important for the full function of each subunit are inappropriate or weak.
Infection of humans by herpes simplex virus (HSV) is typically characterized by a primary infection of mucosal epithelial cells, followed by secondary infection of sensory neurons near the site of primary infection. In most cases, the virus travels up the axons of the infected neurons to the ganglion, where it becomes latent. In a small fraction of cases, however, the virus invades the central nervous system, leading to encephalitis and severe neurological damage or death. Thus, a key element of HSV pathogenesis is the interaction of the virus with the nervous system. One well-studied model for investigating this interaction is the experimental infection of mice via intracranial (i.c.) inoculation. Many wild-type strains of both HSV type 1 (HSV-1) and HSV-2 are highly virulent when tested with this model, but a number of mutant strains of HSV with defects in virulence have been identified and used to attempt to gain an understanding of the genetic and molecular basis for HSV pathogenesis (reviewed in references 2, 37, and 38). One such virus is R13-1, an HSV-1–HSV-2 intertypic recombinant virus in which the left-hand 18% of the genome (around 30 kb) is derived from the HG52 strain of HSV-2 and the remainder is derived from HSV-1 strain 17syn+ (3, 23). As far as is known, the DNA sequences of both the HSV-1 and HSV-2 segments of R13-1 are identical to those of their respective parental genomes. Nevertheless, studies on the phenotype of R13-1 show that it is 10,000-fold less neurovirulent in mice than either the wild-type HSV-1 (strain 17syn+) or HSV-2 (strain HG52) parental virus (23). Moreover, R13-1 progress into the nervous system following footpad inoculation is primarily restricted at the level of spinal ganglia, and viral antigen is detected in supporting cells but in few neurons. In cell culture experiments, R13-1 replicates to near-wild-type levels in Vero cells, primary mouse glial cells, and Rat-2 fibroblast cells but is restricted in primary neurons and in rat pheochromocytoma PC12 cells. The inability of R13-1 to replicate in neuronal cells can be accounted for by a specific defect in viral DNA synthesis (3). In neuronal cells, immediate-early gene expression and early gene expression occur at wild-type levels, but DNA replication and late gene expression are greatly reduced. In contrast, in nonneuronal cells, both DNA replication and late gene expression occur at the same levels as those observed with the wild-type parental virus.
To establish which region(s) of the R13-1 genome is responsible for the nonneurovirulent phenotype, marker rescue experiments were carried out. In these experiments, plasmid DNAs containing selected HSV-1 sequences were cotransfected with full-length R13-1 DNA into rabbit skin cells in culture, and the resulting virus was used to infect mice. The data showed that a region of wild-type HSV-1 DNA containing the UL5 open reading frame (ORF) was sufficient to restore the wild-type neurovirulence phenotype (3). Thus, in the intertypic recombinant R13-1, replacement of UL5 from HSV-1 with its homolog from HSV-2 apparently attenuates the virus for neurovirulence, although other, more complex interactions between HSV-1 and HSV-2 genes are still a formal possibility, since both R13-1 and the neurovirulent rescued virus contain other HSV-2 genes.
The HSV UL5 gene is one of seven viral genes that are directly involved in and essential for viral DNA replication (for recent reviews, see references 5 and 45), a fact that is at least consistent with the idea that the primary defect of R13-1 in neuronal cells is a defect in DNA replication. The product of the HSV UL5 gene is a 95-kDa polypeptide which comprises one subunit of a three-subunit (UL5, UL8, and UL52) enzyme that has single-stranded DNA (ssDNA)-dependent ATPase, ssDNA-dependent GTPase, 5′-to-3′ DNA helicase, and DNA primase activities (4, 7–10). The function of the three subunits of the helicase-primase complex has been investigated by both biochemical and molecular genetic experiments. The results show that UL5 contains the helicase active site (15, 16, 17, 18, 22, 47), UL52 contains the primase active site (12, 24), and UL8 stimulates both activities (16a, 36, 40, 41). The full function of the complex, however, clearly depends on interactions between the three subunits, since none of the three polypeptides has appreciable biochemical activity when expressed singly (4, 11, 36).
In this report, we present both additional genetic evidence and biochemical evidence that the nonneurovirulent phenotype of R13-1 is due to expression by this virus of an intertypic helicase-primase complex containing subunits of both HSV-1 and HSV-2 origin. An intertypic recombinant virus was constructed by replacement of only the HSV-1 UL5 gene with the wild-type HSV-2 UL5 gene. Characterization of this recombinant virus showed that it had essentially the same attenuated phenotype as that of R13-1. We also investigated the biochemical activities of the intertypic HSV-2 UL5–HSV-1 UL8–HSV-1 UL52 (IT 5/8/52) helicase-primase complex and compared them to the activities of the HSV-1 homotypic complex. The DNA-dependent ATPase activity of the intertypic complex was identical to that of the HSV-1 homotypic enzyme. The helicase activity of the IT 5/8/52 complex was slightly reduced, as measured by a strand displacement assay, but was identical to that of the HSV-1 homotypic complex when assayed for the ability to support leading-strand DNA synthesis in combination with the other virus replication fork proteins in vitro. In contrast, the primase activity of the intertypic complex was greatly decreased compared to that of the HSV-1 homotypic complex. One explanation for these findings is that the interactions between the subunits of the helicase-primase intertypic complex are inappropriate or weak, leading to a defect in the overall function of the enzyme. Our findings are consistent with the idea that the primary defect of R13-1 in neurons is at the level of DNA replication. The mechanism by which a partial defect in an enzyme necessary for DNA replication leads to a cell-type-specific defect in DNA synthesis remains to be established.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney cells (Vero cells; American Type Culture Collection, Rockville, Md.) and L2-5 cells (Vero cell line derivative expressing HSV-1 UL5 protein; provided by S. Weller [48]) were grown as previously described (46). Rabbit skin cells were prepared and passaged as described previously (3). Spodoptera frugiperda (Sf9) cells were grown on TMNFH (GIBCO) medium (24). hr99 (48) and its wild-type parent, HSV-1 strain KOS, HSV-1 strain 17syn+, HSV-2 strain HG52, R13-1 (23), and IB1 (this report) were passaged on Vero or L2-5 cells, as appropriate (6). Recombinant baculoviruses were propagated as described previously (36).
R13-1 UL5 DNA cloning and sequencing.
The HindIII-C fragment containing the UL5 ORF was obtained from purified R13-1 DNA and cloned into pUC19 by standard molecular biology techniques (1). Sequencing of the R13-1 UL5 gene (construct pUCRUL5) and the HSV-2 UL5 (strain HG52) (construct pATHindC; provided by D. McGeoch) was done by using the PRISM Ready Reaction Dyedeoxy terminator cycle sequencing kit (Perkin-Elmer) and the automated 373A DNA sequencer (Applied Biosystems) as specified by the manufacturers but with the modification of incubating the linearized template in the presence of 5% dimethyl sulfoxide due to the high GC content of the template.
Construction of IB1 recombinant HSV virus.
HSV-1 strain hr99 is an insertion mutant virus containing the lacZ gene within the UL5 sequence (48). A permissive cell line, L2-5, which contains the wild-type UL5 gene, is capable of supporting hr99 growth (48). Recombinant virus IB1, whose HSV-1 UL5 gene was replaced with its HSV-2 counterpart, was obtained by standard Ca2PO4 cotransfection (1) of hr99 DNA and pATHindC plasmid containing the sequence spanning the HSV-2 UL5 gene on L2-5 cells. Recombinant viruses were identified by infecting Vero cells and screening for loss of β-galactosidase activity (white plaques in the presence of X-Gal [5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside]). Several recombinant viruses were picked, and three rounds of purification were carried out with each recombinant virus. Characterization of the recombination sites and consequent loss of the lacZ cassette was carried out by Southern blot analysis with the probes described previously (48). To ensure that recombination took place outside the coding region of the UL5 gene, the HSV-2 UL5 flanking sequences were amplified by PCR. A 212-bp PCR product from the 5′-end flanking sequence of the gene was obtained by using pN1b primer (HSV-2 sequence from positions 6070 to 6101) (27a, 28) and pN2b (HSV-2 sequence from positions 6282 to 6258). The 237-bp PCR product for the 3′-end flanking sequence was obtained by using pC1b primer (HSV-2 sequence from positions 3357 to 3388) and pC2a (HSV-2 sequence from positions 3595 to 3573). PCRs were carried out by use of the GeneAmp kit (Perkin-Elmer) as indicated by the manufacturer but with 5% dimethyl sulfoxide added when pN1b and pN2b were used.
Determination of LD50s following i.c. inoculation.
Four-week-old female CD1 mice were anesthetized with xylazine-acepromazine-ketamine and inoculated with 10-fold serial dilutions of each virus (0.02 ml per mouse) into the left cerebral hemisphere with a 27-gauge needle. Five mice per dilution per virus were scored for lethal endpoints during an observation period of 21 days. Fifty percent lethal dose (LD50) values were calculated by the method of Reed and Muench (35).
In vivo yields of virus in the central nervous system following i.c. inoculation.
Four-week-old female CD1 mice were anesthetized with xylazine-acepromazine-ketamine and inoculated with 5,000 PFU of each virus (in 0.02 ml per mouse) into the left cerebral hemisphere with a 27-gauge needle. Three days (72 h) postinoculation, four mice for each virus were sacrificed, and the brains were removed and snap-frozen in liquid nitrogen. Tissue was homogenized at 10% (wt/vol) solutions in Dulbecco’s modified Eagle’s medium (GIBCO) and clarified by centrifugation at 3,000 × g for 5 min at 4°C. The titer of the supernatant for infectious virus was determined on rabbit skin cells.
Construction of recombinant baculovirus.
R13-1 UL5 and HSV-2 UL5 genes were cloned into the pBluebac III baculovirus vector (Invitrogene) and then recombined as indicated by the suppliers into the Autographa californica nuclear polyhedrosis virus genome by using BaculoGold linearized baculovirus DNA (Pharmingen) as the target DNA. Plaques of recombinant viruses were identified by X-Gal screening, and one round of plaque purification was performed for each recombinant virus. Lysates of infected Sf9 cells were screened for the expression of UL5 protein by Western immunoblot analysis of sodium dodecyl sulfate (SDS) protein gels by using rabbit antisera against HSV-1 UL5, kindly provided by S. Weller.
Protein purification.
The IT 5/8/52 complex was purified as described before (24), except that the last purification step, Biosil SEC 250 size-exclusion chromatography, was omitted. The homotypic HSV-1 UL5–HSV-1 UL8–HSV-1 UL52 (HSV-1 5/8/52) and the HSV-1 UL5–HSV-1 UL52 (HSV-1 5/52) primase-helicase complex preparations were obtained from D. Klinedinst. ICP8, UL8, and UL30-UL42 preparations were a gift from J. Gottlieb. It is important to note that all recombinant baculoviruses expressing HSV-1 genes were derived from HSV-1 (KOS) (33).
Enzymatic assays. (i) ATPase assay.
DNA-dependent ATPase activity was characterized as described previously (36).
(ii) Helicase assay.
Single-primed helicase substrates were prepared by annealing 2 pmol of M13mp18 ssDNA with 10 pmol of a 32P-radiolabeled 5′-end 45-mer (22 bases annealed, 23 bases 3′ tail) (8), 53-mer (30 bases annealed, 23 bases 3′ tail), or 83-mer (60 bases annealed, 23 bases 3′ tail) oligonucleotide separately (gift from J. Gottlieb) in the presence of 10 mM Tris-HCl (pH 7.5)–100 mM NaCl–0.1 mM EDTA. The reaction mixtures were incubated at 90°C for 5 min and allowed to cool at room temperature. Helicase substrates were separated from unannealed oligonucleotide by Bio-Gel A-15M (Bio-Rad) chromatography in 10 mM Tris-HCl (pH 8)–1 mM EDTA–50 mM NaCl. Helicase assay mixtures (20 μl) containing 1 pmol of either IT 5/8/52, HSV-1 5/8/52, or HSV-1 5/52 with or without a threefold molar excess of HSV-1 UL8, 25 fmol of helicase substrate, 25 mM Tris-HCl (pH 7.5), 25 mM NaCl, 5 mM MgCl2, 2 mM rATP, and 1 mM dithiothreitol were incubated at 37°C for 1 h. Reactions were stopped by adding 20 mM EDTA and 0.1% SDS. The reaction products were run on a 10% acrylamide nondenaturing gel in 100 mM Tris-HCl (pH 8.3)–100 mM boric acid–2 mM EDTA buffer at 150 to 200 V at 4°C until the gel dye had migrated approximately half way down the gel and analyzed with a Phosphorimager (ImageQuant; Molecular Dynamics).
(iii) Primase assay.
RNA primer synthesis reaction mixtures (25 μl) contained 25 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 25 mM NaCl, 5 μg of acetylated bovine serum albumin, 1 mM dithiothreitol, 1 mM GTP, 2 mM ATP, 0.1 mM UTP, 10 μCi of [α-32P]CTP (3,000 Ci/mmol), 1 pmol of a 50-mer DNA oligonucleotide template containing the primase initiation site from pBluescript plasmid DNA (3′-GTCCTTCCG-5′, with primer synthesis starting at the underlined T residue [16a]), and 0.75 pmol of either IT 5/8/52, HSV-1 5/8/52, or HSV-1 5/52 with or without a threefold molar excess of HSV-1 UL8. The reaction mixtures were incubated for 1.5 h at 30°C. Control reactions for absence of substrate degradation were also carried out: after 1.5 h of incubation with the intertypic helicase-primase complex containing the R13-1 UL5 subunit, an equal amount of homotypic HSV-1 helicase-primase complex was added to the reaction mix and the mixture was incubated for an additional 1.5 h. Reactions were stopped by the addition of 1 μl of 250 mM EDTA, the mixtures were heated for 5 min at 65°C and vacuum dried, and the reaction products were resuspended in 15 μl of 50% formamide. Five microliters of the reaction mixture was denatured for 2 min at 90°C before being loaded onto an 18% Hydrolink Long Ranger (AT Biochem, Malvern, Pa.)–7 M urea sequencing gel. The RNA primers were analyzed with a Phosphorimager.
(iv) Leading-strand synthesis assay.
Leading-strand replication assay mixtures were prepared as described previously (24). However, the preformed fork substrate was preincubated with polymerase-UL42 in the presence of ICP8.
RESULTS
R13-1 cell restrictive phenotype is not due to a mutation.
The R13-1 intertypic virus was originally isolated by selection of replication-competent virus following infection of cells with a temperature-sensitive HSV-1 mutant and cotransfection with fragmented HSV-2 genomes (39). One possible explanation for the defective phenotype of R13-1 is the introduction of a mutation(s) into the virus during the original isolation. Since cloned fragments of wild-type HSV-1 DNA containing only the UL5 gene can rescue the neurovirulence phenotype of R13-1, if such a mutation exists in R13-1, it must be located within the R13-1 UL5 gene. To rule out this possibility, a DNA fragment from R13-1 virion DNA containing the UL5 ORF was cloned into a pUC19 vector and sequenced (data not shown). The R13-1 sequence was found to be identical to the reported sequence of the UL5 gene from HSV-2 strain HG52 (27a, 28). Thus, mutations are not responsible for the attenuated phenotype of R13-1.
IB1 exhibits the same phenotype as that of R13-1.
R13-1 virus has 18% of the left end of the HSV-1 genome (from the end to approximately the UL13 gene) replaced by the homologous sequence of HSV-2 (3). Thus, R13-1 contains two subunits of helicase-primase derived from HSV-2 (UL5 and UL8), and the virulent rescued virus still contains HSV-2 UL8 as well as a number of other HSV-2 genes. To provide conclusive evidence that the HSV-2 UL5 gene alone is responsible for the biological properties of R13-1, we constructed a recombinant virus (IB1) in which only the UL5 gene of HSV-1 was replaced by the corresponding sequences from HSV-2 and compared its phenotype to those of wild-type HSV-1 and R13-1. IB1 was constructed by using the defective virus HSV-1 (KOS) hr99 (48) as the parent. This virus contains an insertion of the lacZ gene (under control of the HSV-1 ICP6 promoter) in the UL5 coding sequence; the UL5 defect can be complemented by using cells that express the UL5 gene. A plasmid containing the HSV-2 (HG52) UL5 gene and flanking sequences was transfected into cells infected with hr99, and replication-competent recombinants were selected by plaquing the resulting virus progeny on Vero cells that did not express UL5. Individual plaque isolates were screened by Southern blot analysis for the loss of the lacZ gene and by PCR for the presence of HSV-2 sequences flanking both sides of the UL5 gene (see Materials and Methods). Finally, both ends of the UL5 gene contained within IB1 were sequenced (data not shown). The results showed that IB1 contains the complete HSV-2 UL5 gene. The kinetics of virus replication in Vero cells was analyzed by use of a one-step growth curve (Fig. 1). The results showed that like R13-1, IB1 is indistinguishable from its wild-type parent in both the rate of production and final yield of progeny virus.
FIG. 1.

Kinetics of replication of HSV-1 (KOS) and IB1 viruses in Vero cells. Cultures were infected at a multiplicity of infection of 0.1 PFU per cell with HSV-1 (◘) or IB1 (•). At the indicated times, the cells were harvested and the virus was titrated by plaque assay on Vero cells. Mean values of two duplicate infections are shown.
Neurovirulence was measured by i.c. inoculation of mice because this route is not influenced by viral neuroinvasiveness (42). Table 1 shows the LD50 values after i.c. inoculation as well as the viral yields in the brain of HSV-1 strains KOS and 17syn+ and IB1 and R13-1 viruses. When compared to its parental strain, KOS, IB1 was attenuated. The slightly higher attenuation observed in the case of R13-1 virus in comparison to that of IB1 may be related to the difference in parental strain virulence, since the 17syn+ strain is more virulent than the KOS strain (LD50s of 2.4 versus 31 PFU) (42).
TABLE 1.
LD50s and in vivo yields of virus in the central nervous system following i.c. inoculationa
| Virus | LD50 (PFU) | In vivo yield (PFU/g) (104) |
|---|---|---|
| KOS | 31 | 2.3 |
| IB1 | 3.1 × 103 | 2.4 |
| 17syn+ | 2.4 | 5.6b |
| R13-1 | 3.7 × 103 | 5.8 |
Determined as described in Materials and Methods.
Only three mice survived to 72 h.
A novel feature of the R13-1 phenotype is the observation that despite the fact that the virus is defective for replication in neurons, the yield of virus following i.c. inoculation is similar to that observed with wild-type parental virus. Previous studies have shown that this is due to virus replication in supporting cells. To directly assay the ability of each of the four viruses to replicate within the central nervous system tissue, the viral yields obtained in the brain after i.c. inoculation were compared. The intertypic viruses both replicated at the same level that their parental strains did (Table 1). Thus, IB1 and R13-1 have closely similar phenotypes when compared to those of their respective parental strains: they both are highly attenuated for neurovirulence but nevertheless replicate well in the central nervous system. These data rule out a substantive contribution to the phenotype of R13-1 by UL8 or any HSV-2 gene apart from UL5. On the basis of the data presented here and those published previously, we therefore conclude that replacement of HSV-1 UL5 by HSV-2 UL5 causes a specific defect in the efficiency of viral DNA replication in neuronal cells and a consequent defect in neurovirulence.
Purification of intertypic helicase-primase.
The most straightforward explanation of the genetic data regarding R13-1 and IB1 is that the intertypic helicase-primase complex expressed by these viruses has an altered biochemical function. To test this general idea, a recombinant baculovirus expressing HSV-2 UL5 was constructed, and Sf9 cells were triply infected with recombinant baculoviruses expressing HSV-2 UL5, HSV-1 UL8, and HSV-1 UL52. The IT 5/8/52 complex was purified from the infected cells by a protocol similar to that used for the purification of the HSV-1 homotypic enzyme. The IT 5/8/52 complex remained associated as a trimer, as shown by gel filtration chromatography on Superose 12 (Fig. 2). ATPase activity was detected and coeluted with the peak of protein (data not shown).
FIG. 2.

Superose 12 gel filtration chromatography of the IT 5/8/52 protein complex. One hundred micrograms of IT 5/8/52 preparation, purified as described in Materials and Methods, was loaded onto a Superose 12 column (3.2 by 300 mm, 2.4 ml; Pharmacia). The indicated fractions were analyzed by SDS-polyacrylamide gel electrophoresis. Proteins in the gel were visualized by silver staining. Lane L, fraction loaded onto column.
Comparison of the biochemical activities of intertypic and homotypic helicase-primase.
To investigate the molecular basis of the phenotypic defect of IB1, we compared the different biochemical activities of the purified intertypic and HSV-1 homotypic helicase-primase enzymes in vitro. Both biochemical activities that are independent of the other HSV-1 replication proteins, i.e., ATPase, helicase, and primase activities, and the activity of the enzyme in conjunction with other HSV replication proteins were characterized. The specific ATPase activities of the complexes were determined by use of a colorimetric assay in which each complex was incubated in the presence of ATP and denatured calf thymus DNA. The activities of the complexes were equivalent (Table 2).
TABLE 2.
Biochemical activities of HSV-1 5/8/52, IT 5/8/52, and HSV-1 5/52 in the absence or presence of UL8
| Complex | Biochemical activity
|
|||
|---|---|---|---|---|
| ATPasea | Helicaseb | Primaseb | Leading strandb | |
| HSV-1 5/8/52 | 1.35 | 100 | 100 | 100 |
| IT 5/8/52 | ||||
| −UL8 | 1.40 | 26 | 7 | 195 |
| +UL8 | NDc | 31 | 6.6 | ND |
| HSV-1 5/52 | ||||
| −UL8 | 1.45 | 4.8 | 9 | 0 |
| +UL8 | ND | 32.6 | 53.2 | ND |
Expressed as nanomoles of Pi released per picomole of enzyme per hour.
Expressed as percentage of HSV-1 5/8/52 activity.
ND, not done.
The helicase-primase complexes were tested for their abilities to displace oligonucleotides of different lengths annealed to a circular ssDNA molecule. Duplex regions ranged in length from 22 to 60 bp, and all substrates had a 23-base 3′ tail (8). ATPase activity measurements were carried out in parallel with the strand displacement assays as a control for the functionality of the protein preparations. Figure 3 and Table 2 show the data obtained when the 83-mer oligonucleotide was used. There was a threefold reduction in the helicase activity of the IT 5/8/52 complex (Fig. 3, lane e) compared to that of the homotypic HSV-1 5/8/52 complex (lane c). The reduction of the helicase activity of the IT 5/8/52 complex was also about two- to threefold when the substrates consisted of a circular ssDNA molecule annealed to shorter oligonucleotides (data not shown).
FIG. 3.
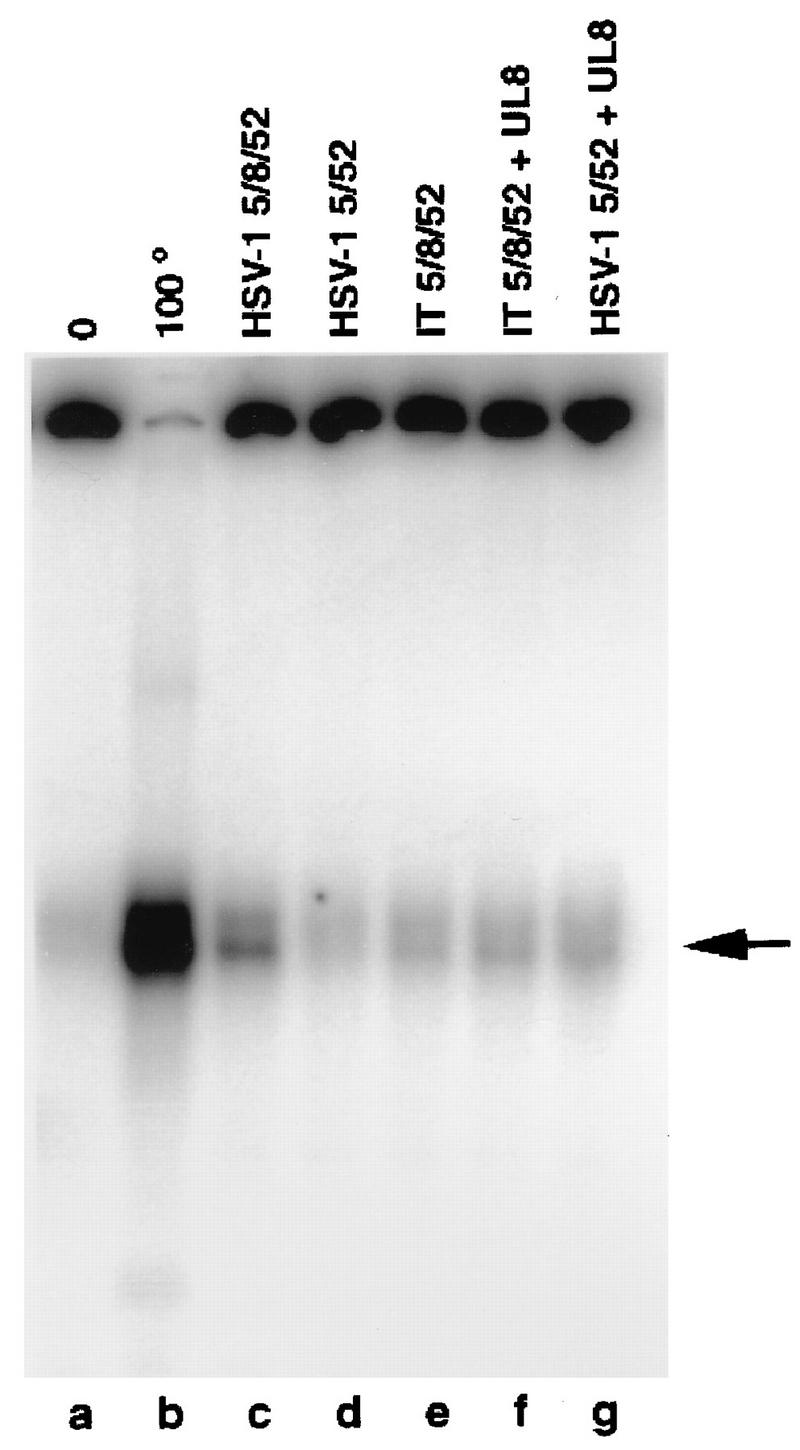
Comparison of the helicase activities of HSV-1 5/8/52, HSV-1 5/52, and IT 5/8/52 by strand displacement assay. One-picomole amounts of helicase-primase complexes were incubated at 37°C for 1 h with a circular ssDNA template previously annealed to a 32P-radiolabeled oligonucleotide. The reaction products were resolved by using nondenaturing polyacrylamide electrophoresis and visualized and quantitated with the Phosphorimager. Lanes: a, no protein; b, heat-denatured substrate. The arrow indicates the position of the displaced oligonucleotide. The reaction mixtures in lanes f and g were incubated in the presence of a threefold molar excess of HSV-1 UL8.
UL8 is known to stimulate both the helicase activity (16a) and the primase activity (36, 40, 41) of the two-subunit complex containing HSV-1 UL5 and UL52 (HSV-1 5/52). Although, as shown above, the three subunits of the intertypic complex interact well enough to remain associated during gel filtration chromatography, we considered the possibility that the interaction of UL52 and UL5 with UL8 in the intertypic enzyme was not strong enough to survive the relatively low protein concentrations that exist in the in vitro assays and that this weak interaction accounted for the reduced helicase activity of the intertypic enzyme. To test this possibility, all of the biochemical assays were also carried out in the presence of high concentrations of purified HSV-1 UL8. As shown in Fig. 3, lane f, added UL8 had no effect on the helicase activity of the intertypic enzyme, although it did stimulate the activity of purified HSV-1 5/52 by nearly eightfold (see Fig. 5; compare lanes d and g).
FIG. 5.
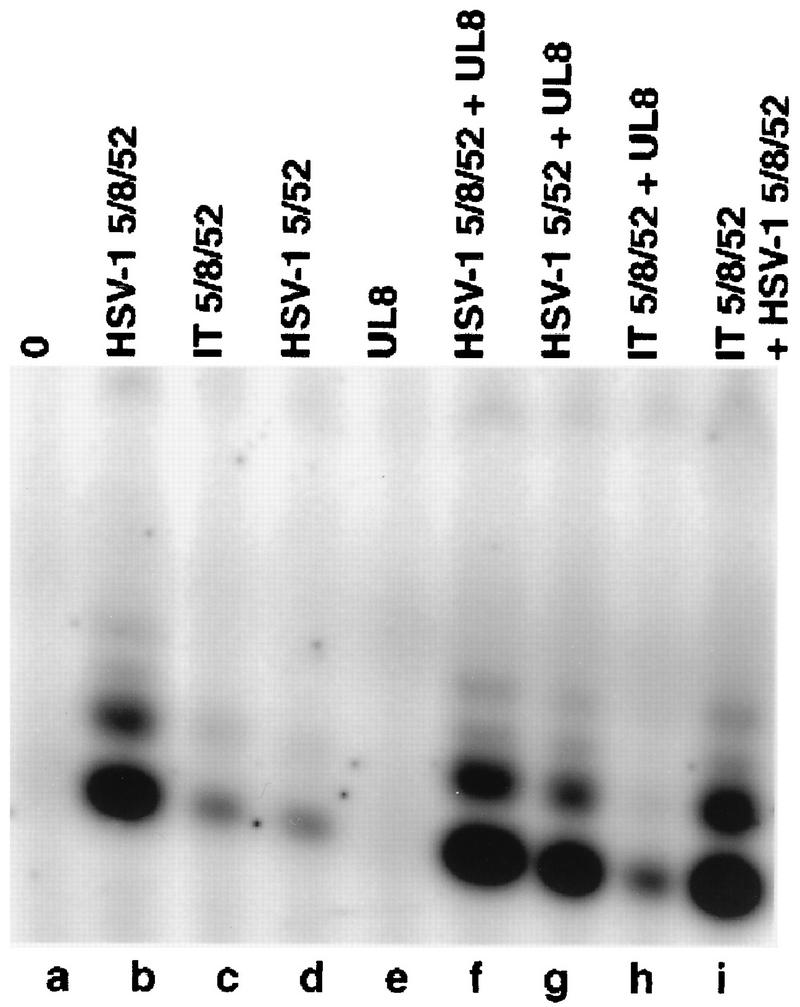
Comparison of the abilities of HSV-1 5/8/52, HSV-1 5/52, and IT 5/8/52 to synthesize RNA primers. Helicase-primase complexes were incubated in 0.75-pmol amounts with a 50-mer oligonucleotide containing the primase initiation site as the DNA template and GTP, ATP, UTP, and [α-32P]CTP in the absence (lanes b to d) or presence (lanes e to h) of exogenous UL8. The products were resolved by using denaturing polyacrylamide gel electrophoresis and visualized and quantitated with the Phosphorimager. Lane a contains no added protein.
Since the defect in the helicase activity of the intertypic enzyme by itself was quite modest, we thought it possible that the enzyme would show a greater defect in an assay in which the helicase function of this enzyme was coupled to the activity of other HSV-1 replication proteins. One such test is the leading-strand synthesis assay (24) (Fig. 4). Briefly, a prefork substrate (consisting of pBS(+) ssDNA molecules primed with a 62-mer oligonucleotide (32-base nonhomologous 5′ tail) was incubated with the UL30-UL42 polymerase complex, ICP8, ribonucleotides, and deoxynucleotides. The polymerase extended the oligonucleotide 3′-OH, but it stopped when it reached the duplex region, leading to a radioactively labeled open duplex circular plasmid with a free 5′ tail product of about 3.2 kb (Fig. 4). The addition of HSV-1 5/8/52 allows unwinding of the duplex sequences and further elongation of the leading strand (Fig. 4, lanes a to d; Table 2). Absence of UL8 in the homotypic complex abolishes leading-strand synthesis (Fig. 4, lanes i to l; Table 2) (16a). The IT 5/8/52 complex was tested for its ability to support leading-strand synthesis (Fig. 4, lanes e to h; Table 2). Although, as shown above, a threefold reduction in the helicase activity was observed for the intertypic complex in a simple strand displacement assay, this reduction did not seem to interfere in the ability of the IT 5/8/52 complex to assist in the synthesis of leading strands by the polymerase. In fact, in the experiment shown, the level of leading-strand synthesis was approximately 20% greater than that seen with the homotypic complex; this difference was not reproducible and was considered to be insignificant. Thus, the IT 5/8/52 complex was able to interact with HSV-1 polymerase-UL42 and/or HSV-1 ICP8 strongly enough to promote leading-strand elongation as well as the homotypic enzyme.
FIG. 4.
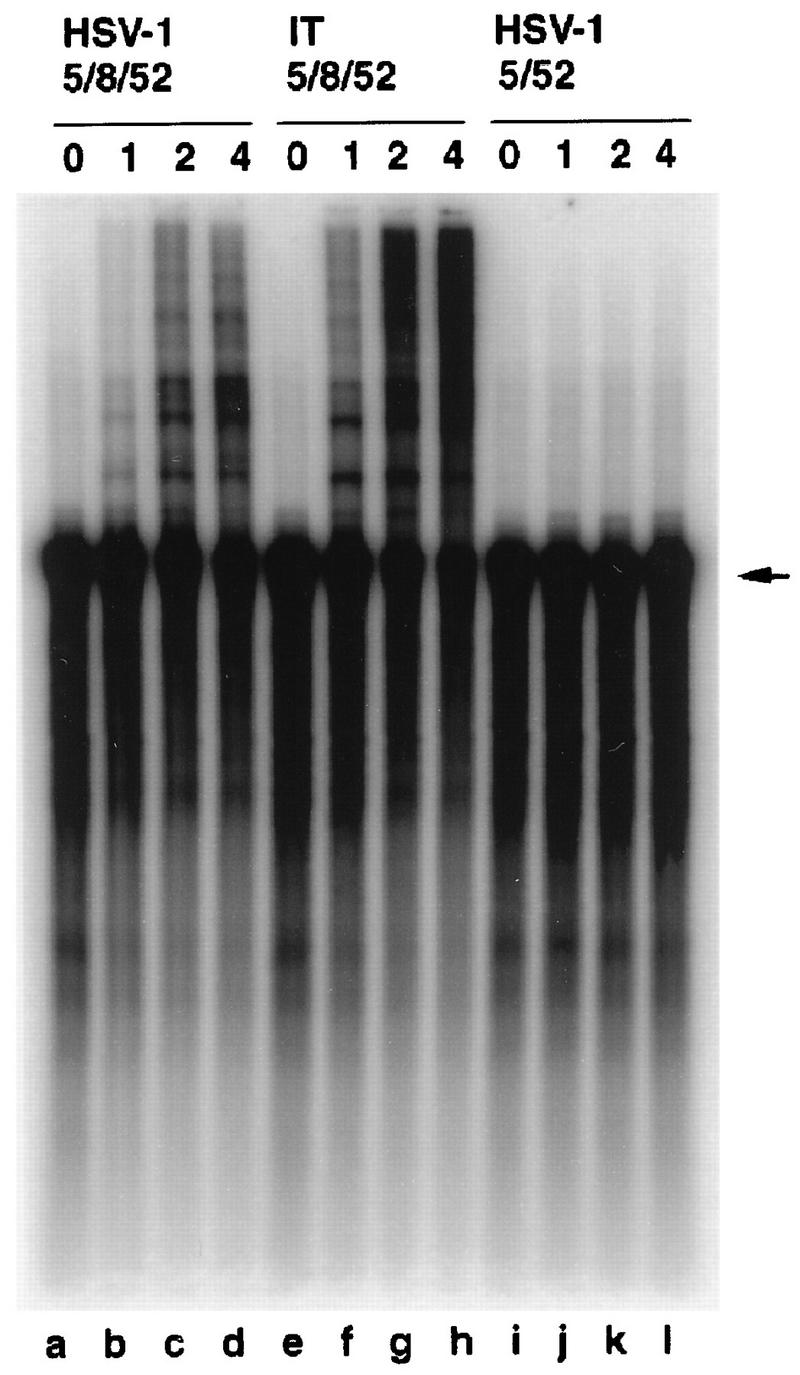
Comparison of the abilities of HSV-1 5/8/52, IT 5/8/52, and HSV-1 5/52 to support leading-strand synthesis. Enzyme in 0-, 1-, 2-, and 4-pmol amounts (as indicated at the tops of the lanes) was incubated with a preformed fork substrate [single-stranded pBS(+) single primed with an oligonucleotide and extended with polymerase-UL42 to yield double-stranded pBS with a free tail] in the presence of polymerase-UL42, ICP8, [α-32P]dCTP, the other three deoxynucleoside triphosphates, and the four ribonucleoside triphosphates for 1.5 h at 30°C. Following alkaline gel electrophoresis, products were visualized and quantitated with the Phosphorimager. The arrow indicates the position of the preformed fork substrate.
The ability of the intertypic complex to synthesize RNA primers was compared to that of the homotypic enzyme by using a 50-mer oligonucleotide containing a primase initiation site in the presence of GTP, ATP, UTP, and [α-32P]CTP. ATPase assays were carried out in parallel to the priming reaction as a control for the functionality of the protein preparations. As shown in Fig. 5 and Table 2, the IT 5/8/52 complex (Fig. 5, lane c) shows an approximate 10-fold decrease in specific primase activity as compared to that of the homotypic HSV-1 5/8/52 complex (lane b). When circular M13mp18 ssDNA substrate containing multiple primase initiation sites was used as the template, a similar 10-fold deficiency in the specific activity of the intertypic complex was observed, but the distribution of products was qualitatively identical to that observed with the homotypic enzyme (data not shown). As in the case of helicase assays, the addition of HSV-1 UL8 had no effect on the primase activity of the intertypic complex (Fig. 5, lane h), suggesting that the deficiency in activity was not due to a defect in the interactions of UL8 within the IT 5/8/52 complex. The deficiency of the IT 5/8/52 complex in synthesizing RNA primers was not due to an inhibitory contaminant in the intertypic complex preparation, because there was no effect on the activity of the homotypic enzyme when the two enzyme preparations were mixed (Fig. 5, lane i). Thus, the presence in the intertypic complex of the HSV-2 subunit thought to contain the helicase active site causes a significant decrease in the primase activity of the enzyme.
DISCUSSION
Previous studies on the intertypic recombinant virus R13-1 suggested that introduction of the HSV-2 UL5 gene into a predominately HSV-1 genetic background causes a defect in the ability of this virus to carry out DNA replication in neuronal cells (3). In the present study, we have extended these findings in two ways. First, we have refined the genetic analysis to rule out more complicated interactions between HSV-2 and HSV-1 genes: an HSV-1 recombinant virus (named IB1) in which only the UL5 gene was replaced by its HSV-2 homolog had the same nonneurovirulent phenotype that R13-1 did, in which the left 18% of the genome (approximately 27 kbp) is of HSV-2 origin. Second, we have documented biochemical defects in a key DNA replication enzyme that must be produced in cells infected by IB1, an intertypic helicase-primase complex containing a UL5 polypeptide of HSV-2 origin and UL52 and UL8 polypeptides of HSV-1 origin. This intertypic enzyme complex has threefold-lower helicase activity and 10-fold lower primase activity than the homotypic enzyme, in which all three subunits are of HSV-1 origin.
DNA sequence analysis of HSV-1 and HSV-2 has shown that the genomes of these two viruses are highly homologous, averaging about 90% identity in predicted amino acid sequence. Virus replication, however, requires a large number of specific interactions between viral polypeptides, and therefore it is perhaps not surprising that many intertypic recombinants containing a mixture of HSV-1 and HSV-2 sequences have modest growth defects (and attenuated virulence) that could come about as the result of nonoptimal interactions between HSV-1 and HSV-2 polypeptides (20, 21, 31, 34, 43). In fact, most intertypic recombinants have historically been produced by selection of replication-competent recombinant viruses following coinfection of cells with replication-defective HSV-1 and HSV-2 parents (reviewed in reference 19). It is therefore possible that many different combinations of HSV-1 and HSV-2 genes might lead to a replication defect if produced in the absence of selection. Not all combinations of HSV-1 and HSV-2 polypeptides, however, are deleterious, even in cases where the polypeptides clearly interact. For example, the helicase primase produced by R13-1 contains not only the UL5 subunit but also the UL8 subunit derived from HSV-2. Marker rescue of R13-1 with the HSV-1 UL5 gene produced a neurovirulent rescuant that still retains the HSV-2 UL8 gene (3). Moreover, UL8 is known to interact not only with the other two subunits of the helicase-primase but also with the origin-binding protein UL9 (29), the polymerase accessory protein, UL42 (27), and the ssDNA-binding protein, ICP8 (14). Nevertheless, even though the HSV-2 and HSV-1 UL8 proteins have a smaller degree of sequence identity than that of the corresponding UL5 polypeptides (80 versus 89% [27a]), the presence of a HSV-2 UL8 gene in an otherwise HSV-1 genetic background is tolerated with no known phenotypic consequences. It should be noted in this context that the structures of IB1 and R13-1 are such that the HSV-2 UL5 gene is expressed from an HSV-2, rather than an HSV-1, promoter. It is possible, therefore, that the phenotypes of these viruses are due to altered expression of the UL5 polypeptide. To rule out this possibility, the steady-state levels of UL5 in IB1- and KOS-infected cells were compared at several times postinfection. No differences were observed (25a). It therefore seems likely that the phenotype of IB1 is due to differences in the activity of the heterotypic helicase-primase rather than to differences in the level of the enzyme in infected cells.
The finding that the intertypic helicase-primase complex has reduced helicase and primase activities is consistent with previous data regarding the functional interdependence of the UL5 and UL52 polypeptides. The UL5 subunit, which contains the sequences most likely to be involved in helicase catalysis (15, 16, 22, 47), is completely inactive in the absence of UL52 (4, 11, 36). Conversely, the UL52 subunit, which is likely to be the site of primase catalysis (12, 24), is insoluble in the absence of UL5 (4, 10, 36). In addition, not only do mutations within the UL5 helicase motifs affect ATPase and helicase activity (18), but also many of these mutations show increased primase activity (17). There are also examples of helicase-primase complexes in other systems that require the presence of both subunits for optimal biochemical function. The bacteriophage T4 gene 41 helicase and gene 61 primase stimulate each other’s activities in vitro (44). In the case of bacteriophage T7, the products of the g4 ORF can be found as a long product (63 kDa) with helicase-primase activities and as a short product (56 kDa) with only the helicase activity. When both products are associated, there is a 100-fold increase of the primase activity when compared to the activity of the 63-kDa product (30). In Escherichia coli, DnaB helicase and DnaG primase also appear to stimulate each other (26). All of these data lend support to the view that the reduced biochemical function of the intertypic enzyme containing HSV-1 UL5 and HSV-1 UL52 is due to defective or inappropriate interactions between the two polypeptides, leading to structural distortions and consequent loss of activity.
Our data show that the intertypic helicase-primase complex has reduced helicase and primase activities. We have no data, however, that bear on the question of which of these two defects, or both, accounts for the specific defect in DNA replication in neuronal cells that is the most striking feature of the nonneurovirulent phenotype of R13-1. As discussed previously (3), a number of HSV mutants with defects in nucleotide metabolism are restricted for virus growth in nervous system tissues. These include mutants with alterations in the thymidine kinase, dUTPase, and ribonucleotide reductase genes (reviewed in references 2, 37, and 38). These mutants differ from R13-1, however, because they fail to grow in all nondividing cells, including central nervous system cells and serum-starved cells in culture, while R13-1 replicates to high titers in nonneuronal cells in the central nervous system and under conditions of serum starvation in tissue culture (3). The neuronal restriction of R13-1 appears to be more similar to that of certain drug-resistant DNA polymerase mutants which are impaired for replication in the central nervous system but not in the periphery (13). On the basis of the data presented in this report and these other examples of neuronal restriction, we propose two models to explain the specific DNA replication defect in neuronal cells exhibited by R13-1 and IB-1. In the first model, the principal defect of R13-1 is the substantial defect in primase activity of the intertypic helicase-primase complex. According to this model, this lack of adequate primase can be complemented in nonneuronal cells by the cellular polymerase α-primase but not in neuronal cells. The second model depends in part on the different patterns of gene expression that occur in neuronal and nonneuronal cells: in contrast to the classic pattern of gene expression observed for most cell types, maximum levels of both immediate-early and early gene expression are dependent on the initial rounds of DNA synthesis in neuronal cells (25, 32). Thus, according to this model, the defects in the intertypic helicase-primase complex cause a small decrease in the initial rate of DNA replication in all cell types. In nonneuronal cells, this small decrease in rate has little or no effect on virus replication because DNA replication is not a limiting step in the virus multiplication cycle. In neuronal cells, on the other hand, a small defect in the initial rate of DNA replication leads to a large defect in virus replication overall because the small decrease in rate of DNA replication is amplified by consequent differences in early gene expression. Additional data are required to prove which of these two models, if either, is correct.
In summary, we have shown that an intertypic helicase-primase complex produced by an HSV-1–HSV-2 intertypic recombinant virus has quantifiable defects in biochemical function. Further investigation of the mechanism by which these defects lead to a loss of neurovirulence should lead to a greater understanding of the interaction of HSV with the nervous system of infected individuals.
ACKNOWLEDGMENTS
We are grateful to John Gottlieb and Donna Klinedinst for purified proteins and assistance with the biochemical assays and to William Nelson and Sandra Weller for helpful comments on the manuscript.
This work was supported by the Schweizerische Nationalfonds and the National Institutes of Health.
REFERENCES
- 1.Ausubel F M, Brent R, Kingstone R E, Moore D D, Smiths J A, Struhl K. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1987. [Google Scholar]
- 2.Bennett J L, Gilden D. The molecular genetics of herpes simplex virus latency and pathogenesis: a puzzle with many pieces still missing. J Neurovirol. 1996;2:225–229. doi: 10.3109/13550289609146884. [DOI] [PubMed] [Google Scholar]
- 3.Bloom D C, Stevens J G. Neuron-specific restriction of a herpes simplex virus recombinant maps to the UL5 gene. J Virol. 1994;68:3761–3772. doi: 10.1128/jvi.68.6.3761-3772.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calder J M, Stow N D. Herpes simplex virus helicase-primase: the UL8 protein is not required for DNA-dependent ATPase and DNA helicase activities. Nucleic Acids Res. 1990;25:3573–3578. doi: 10.1093/nar/18.12.3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Challberg M D. Herpesvirus DNA replication. In: DePamphilis M L, editor. DNA replication in eukaryotic cells. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 721–749. [Google Scholar]
- 6.Challberg M D. A method for identifying the viral genes required for herpesvirus DNA replication. Proc Natl Acad Sci USA. 1986;83:9094–9098. doi: 10.1073/pnas.83.23.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crute J J, Lehman I R. Herpes simplex virus-1 helicase-primase. Physical and catalytic properties. J Biol Chem. 1991;266:4484–4488. [PubMed] [Google Scholar]
- 8.Crute J J, Mocarski E S, Lehman I R. A DNA helicase induced by herpes simplex virus type 1. Nucleic Acids Res. 1988;16:6585–6596. doi: 10.1093/nar/16.14.6585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crute J J, Tsurumi T, Zhu L, Weller S K, Olivo P D, Challberg M D, Mocarski E S, Lehman I R. Herpes simplex virus 1 helicase-primase: a complex of three herpes-encoded gene products. Proc Natl Acad Sci USA. 1989;86:2186–2189. doi: 10.1073/pnas.86.7.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodson M S, Crute J J, Bruckner R C, Lehman I R. Overexpression and assembly of the herpes simplex virus type-1 helicase-primase in insect cells. J Biol Chem. 1989;264:20835–20838. [PubMed] [Google Scholar]
- 11.Dodson M S, Lehman I R. Association of DNA helicase and primase activities with a subassembly of the herpes simplex virus 1 helicase-primase composed of the UL5 and UL52 gene products. Proc Natl Acad Sci USA. 1991;88:1105–1109. doi: 10.1073/pnas.88.4.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dracheva S, Koonin E, Crute J J. Identification of the primase active site of the herpes simplex virus type 1 helicase-primase. J Biol Chem. 1995;270:14148–14153. doi: 10.1074/jbc.270.23.14148. [DOI] [PubMed] [Google Scholar]
- 13.Field H J, Coen D M. Pathogenicity of herpes simplex virus mutants containing drug-resistant mutations in the viral DNA polymerase gene. J Virol. 1986;60:286–289. doi: 10.1128/jvi.60.1.286-289.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gac N T L, Villani G, Hoffmann J S, Boehmer P E. The UL8 subunit of the herpes simplex virus type-1 DNA helicase-primase optimizes utilization of DNA templates covered by the homologous single-strand DNA-binding protein ICP8. J Biol Chem. 1996;271:21645–21651. doi: 10.1074/jbc.271.35.21645. [DOI] [PubMed] [Google Scholar]
- 15.Gorbalenya A E, E. V. K, Donchenko A P, Blinov V M. Two related superfamilies of putative helicases involved in replication, repair and expression of DNA and RNA genomes. Nucleic Acids Res. 1989;17:4713–4730. doi: 10.1093/nar/17.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorbalenya A E, Koonin E B, Donchenko A P, Blinov V M. A conserved NTP-motif in putative helicases. Nature. 1988;333:22. doi: 10.1038/333022a0. [DOI] [PubMed] [Google Scholar]
- 16a.Gottlieb, J., and M. Challberg. Unpublished data.
- 17.Graves-Woodward K L, Gottlieb J, Challberg M D, Weller S K. Biochemical analyses of mutations in the HSV-1 helicase-primase that alter ATP hydrolysis, DNA unwinding, and coupling between hydrolysis and unwinding. J Biol Chem. 1997;272:4623–4630. doi: 10.1074/jbc.272.7.4623. [DOI] [PubMed] [Google Scholar]
- 18.Graves-Woodward K L, Weller S K. Replacement of Gly 815 in helicase motif V alters the single-stranded DNA-dependent ATPase activity of the herpes simplex virus type I helicase-primase. J Biol Chem. 1996;271:13629–13635. doi: 10.1074/jbc.271.23.13629. [DOI] [PubMed] [Google Scholar]
- 19.Halliburton I W. Intertypic recombinants of herpes simplex viruses. J Gen Virol. 1980;48:1–23. doi: 10.1099/0022-1317-48-1-1. [DOI] [PubMed] [Google Scholar]
- 20.Halliburton I W, Honess R W, Killington R A. Virulence is not conserved in recombinants between herpes simplex virus types 1 and 2. J Gen Virol. 1987;68:1435–1440. doi: 10.1099/0022-1317-68-5-1435. [DOI] [PubMed] [Google Scholar]
- 21.Halliburton I W, Randall R E, Killington R A, Watson D H. Some properties of recombinants between type 1 and type 2 herpes simplex viruses. J Gen Virol. 1977;36:471–478. doi: 10.1099/0022-1317-36-3-471. [DOI] [PubMed] [Google Scholar]
- 22.Hodgman T C. A new superfamily of replicative proteins. Nature. 1988;333:22–23. doi: 10.1038/333022b0. [DOI] [PubMed] [Google Scholar]
- 23.Javier R T, Izumi K M, Stevens J G. Localization of a herpes simplex virus neurovirulence gene dissociated from a high-titer virus replication in the brain. J Virol. 1988;62:1381–1387. doi: 10.1128/jvi.62.4.1381-1387.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klinedinst D K, Challberg M D. Helicase-primase complex of herpes simplex virus type 1: a mutation in the UL52 subunit abolishes primase activity. J Virol. 1994;68:3693–3701. doi: 10.1128/jvi.68.6.3693-3701.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosz-Vnenchak M, Jacobson J, Coen D M, Knipe D M. Evidence for a novel regulatory pathway for herpes simplex virus gene expression in trigeminal ganglion neurons. J Virol. 1993;67:5383–5393. doi: 10.1128/jvi.67.9.5383-5393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25a.Lantz, T., and M. Challberg. Unpublished results.
- 26.LeBowitz J H, McMacken R. The Escherichia coli dnaB replication protein is a DNA helicase. J Biol Chem. 1986;261:4738–4748. [PubMed] [Google Scholar]
- 27.Marsden H S, McLean G W, Barnard E, Francis G J, MacEachran K, Murphy M, McVey G, Abbotts A, Cross A, Stow N D. 21st Herpesvirus Workshop, DeKalb, Ill. 1996. The catalytic subunit of the DNA polymerase of HSV-1 interacts specifically with the C-terminus of the UL8 replication protein, abstr. 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27a.McGeoch, D. Personal communication.
- 28.McGeoch D J, Cunningham C, McIntyre G, Dolan A. Comparative sequence analysis of the long repeat regions and adjoining parts of the long unique regions in the genomes of herpes simplex viruses types 1 and 2. J Gen Virol. 1991;72:3057–3075. doi: 10.1099/0022-1317-72-12-3057. [DOI] [PubMed] [Google Scholar]
- 29.McLean G W, Abbotts A P, Parry M E, Marsden H S, Stow N D. The herpes simplex virus type 1 origin-binding protein interacts specifically with the viral UL8 protein. J Gen Virol. 1994;75:2699–2706. doi: 10.1099/0022-1317-75-10-2699. [DOI] [PubMed] [Google Scholar]
- 30.Mendelman L V, Richardson C C. Requirements for primer synthesis by bacteriophage T7 63-kDa gene 4 protein. J Biol Chem. 1991;266:23240–23250. [PubMed] [Google Scholar]
- 31.Morse L S, Buchman T G, Roizman B, Schaffer P A. Anatomy of herpes simplex virus DNA. IX. Apparent exclusion of some parental DNA arrangements in the generation of intertypic (HSV-1 × HSV-2) recombinants. J Virol. 1977;24:231–248. doi: 10.1128/jvi.24.1.231-248.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nichol P F, Chang J Y, Johnson E M, Olivo P D. Herpes simplex virus gene expression in neurons: viral DNA synthesis is a critical regulatory event in the branch point between the lytic and latent pathways. J Virol. 1996;70:5476–5486. doi: 10.1128/jvi.70.8.5476-5486.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olivo P D, Nelson N J, Challberg M D. Herpes simplex virus type 1 gene products required for DNA replication: identification and overexpression. J Virol. 1989;63:196–204. doi: 10.1128/jvi.63.1.196-204.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Preston V G, Davison A J, Marsden H S, Timbury M C, Subak-Sharpe J H, Wilkie N M. Recombinants between herpes simplex virus types 1 and 2: analyses of genome structures and expression of immediate early polypeptides. J Virol. 1978;28:499–517. doi: 10.1128/jvi.28.2.499-517.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed L J, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 36.Sherman G, Gottlieb J, Challberg M D. The UL8 subunit of the herpes simplex virus helicase-primase complex is required for efficient primer utilization. J Virol. 1992;66:4884–4892. doi: 10.1128/jvi.66.8.4884-4892.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens J G. Herpes simplex virus: neuroinvasiveness, neurovirulence and latency. Semin Neurosci. 1991;3:141–147. [Google Scholar]
- 38.Stevens J G. HSV-1 neuroinvasiveness. Intervirology. 1993;35:152–163. doi: 10.1159/000150306. [DOI] [PubMed] [Google Scholar]
- 39.Stow N D, Wilkie N M. Physical mapping of temperature-sensitive mutations of herpes simplex virus type 1 by intertypic marker rescue. Virology. 1978;90:1–11. doi: 10.1016/0042-6822(78)90327-6. [DOI] [PubMed] [Google Scholar]
- 40.Tenney D J, Hurlburt W W, Micheletti P, Bifano M, Hamatake R K. The UL8 component of the herpes simplex virus helicase-primase complex stimulates primer synthesis by a subassembly of the UL5 and UL52 components. J Biol Chem. 1994;269:5030–5035. [PubMed] [Google Scholar]
- 41.Tenney D J, Sheaffer A K, Hurlburt W W, Bifano M, Hamatake R. Sequence-dependent primer synthesis by the herpes simplex virus helicase-primase complex. J Biol Chem. 1995;270:9129–9136. doi: 10.1074/jbc.270.16.9129. [DOI] [PubMed] [Google Scholar]
- 42.Thompson R L, Cook M L, Devi-Rao G B, Wagner E K, Stevens J G. Functional and molecular analyses of the avirulent wild type herpes simplex virus type 1 strain KOS. J Virol. 1986;58:203–211. doi: 10.1128/jvi.58.1.203-211.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson R L, Wagner E K, Stevens J G. Physical location of a herpes simplex virus type-1 gene function(s) specifically associated with a 10 million-fold increase in HSV neurovirulence. Virology. 1983;131:180–192. doi: 10.1016/0042-6822(83)90544-5. [DOI] [PubMed] [Google Scholar]
- 44.Venkatesan M, Silver L L, Nossal N G. Bacteriophage T4 gene 41 protein, required for the synthesis of RNA primers, is also a DNA helicase. J Biol Chem. 1982;257:12426–12434. [PubMed] [Google Scholar]
- 45.Weller S K. Herpes simplex virus DNA replication and genome maturation. In: Cooper G M, Temin R G, Sugden B, editors. The DNA provirus: Howard Temin’s Scientific Legacy. Washington, D.C: ASM Press; 1995. pp. 189–213. [Google Scholar]
- 46.Weller S K, Spadaro A, Schaffer J E, Murray A W, Maxam A M, Schaffer P A. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol Cell Biol. 1985;5:930–942. doi: 10.1128/mcb.5.5.930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu L, Weller S K. The six conserved helicase motifs of the UL5 gene product, a component of the herpes simplex virus type 1 helicase-primase, are essential for its function. J Virol. 1992;66:469–479. doi: 10.1128/jvi.66.1.469-479.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu L, Weller S K. The UL5 gene of herpes simplex virus type 1: isolation of a lacZ insertion mutant and association of the UL5 gene product with other members of the helicase-primase complex. J Virol. 1992;66:458–468. doi: 10.1128/jvi.66.1.458-468.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]