

Abstract
Innate immunity, cell death and inflammation underpin many aspects of health and disease. Upon sensing pathogens, pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), or alarmins, the innate immune system activates inflammatory cell death, such as pyroptosis and PANoptosis. These genetically-defined, regulated cell death pathways not only contribute to the host defense against infectious disease, but also promote pathological manifestations leading to cancer and inflammatory diseases. Our understanding of the underlying mechanisms has grown rapidly in recent years. However, how dying cells, cell corpses, and their liberated cytokines, chemokines, and inflammatory signalling molecules are further sensed by innate immune cells, and their contribution to further amplify inflammation, trigger antigen presentation, and activate adaptive immunity, is less clear. Here, we discuss how pattern-recognition and PANoptosome sensors in innate immune cells recognize and respond to cell-death signatures. We also highlight molecular targets of the innate immune response for potential therapeutic development.
Keywords: Pattern-recognition receptors, TLRs, NLRs, AIM2, CLRs, cGAS, STING, ZBP1, RLRs, pyroptosis, inflammasomes, necroptosis, PANoptosis, PANoptosomes
Healthy living cells do not cause inflammation, whereas cells, which have been infected, stressed, or on the verge of lytic cell death have the capacity to trigger inflammation. Cell death modalities that trigger inflammation are defined as inflammatory cell death. Inflammatory cell death can either be accidental, that is caused by physical or traumatic disruption of the plasma membrane, or regulated, that is executed by the activation of genetically-defined germline-encoded receptors and/or signalling proteins. A well-characterized regulated cell death pathway is inflammasome-dependent pyroptosis, defined by the activation of inflammatory caspases, human caspase-1, -4, -5, or mouse caspase-1 and -11 1, 2, leading to membrane-pore formation and lytic cell death driven by gasdermin proteins 3, 4. PANoptosis is another innate immune cell death pathway initiated by innate immune sensors and driven by caspases and RIPKs through PANoptosomes 5. PANoptosomes are multi-protein complexes assembled by innate immune sensor(s) in response to pathogens, pathogen- or damage-associated molecular patterns (PAMPs or DAMPs), cytokines or homeostatic changes that drive PANoptosis. This pathway is predominantly activated under physiologically-relevant infectious and inflammatory disease conditions. Necroptosis is an alternate lytic cell death pathway that occurs in the absence of caspase-8, driven by receptor-interacting serine-threonine kinase 3 (RIPK3) 6–9 and the effector mixed lineage kinase domain-like psuedokinase (MLKL) 10, 11. The kinetics of cell death pathways differ among cell types and between host mammalian species. For example, human and mouse innate immune cells, such as macrophages, express an array of innate immune sensors and cell death proteins and are more susceptible to innate immune lytic cell death compared to non-immune cells such as fibroblasts that express a more limited subset of cell death proteins, suggesting a more prominent role for necroptosis in these cells12.
The pro-inflammatory effects of innate immune inflammatory cell death are principally driven by cytokines and intracellular components that are actively or passively secreted as a cell dies. During lytic cell death, mammalian cells undergo balloon-like swelling, followed by physical permeabilization and rupture of the plasma membrane. Plasma membrane rupture during innate immune lytic cell death promotes the non-selective release of endogenous components 14. Liberated cytoplasmic components are no longer concealed from immunological detection by pattern-recognition receptors (PRRs), and are therefore, potentially immunostimulatory and capable of triggering signalling pathways that initiate and amplify the inflammatory response 15.
PRRs are important innate immune sensor proteins and include the Toll-like receptors (TLRs), NOD-like receptors (NLRs), AIM2-like receptors (ALRs), C-type lectin receptors (CLRs), and retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs). There also exist expanding families of cytoplasmic nucleic-acid sensors, including cGAS-like receptors (cGLRs) and Zα-domain-like receptors (ZLRs), containing the founding members cGAS and Z-DNA binding protein 1 (ZBP1), respectively 16–18. These PRRs collectively sense a variety of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). PAMPs include any pathogen-derived components, toxins, and virulence factors that drive activation of the immune response and can be released from infected cells during cell death. DAMPs, or alarmins, are used to describe endogenous or newly-synthesized molecules released by damaged or dying cells. Examples of DAMPs include ATP, histones, metabolites, nuclear and mitochondrial DNA, RNA, S100 proteins, and uric acid, among others 19. The actual or relative contribution of many DAMPs and their cognate PRRs in driving inflammation under physiological conditions is still unclear. For example, ATP induces inflammation in cell culture experiments 20–22; however, the pro-inflammatory contribution of ATP in mice is only physiologically observed under specific inflammatory conditions, such as thermal injury to the liver 23, but not in response to exogenously-injected necrotic cells 24. It is possible that not all DAMPs are physiologically pro-inflammatory and that the actual contribution might be obscured by functional redundancy amongst DAMPs released by dying cells and amongst PRRs from the host.
In addition to classical DAMPs, cytokines and chemokines are also secreted by mammalian cells during cell death and can activate immune responses 25, 26, such as the pro-inflammatory cytokines interleukin (IL)-1α, a classical alarmin, and IL-1β and IL-18, secreted by pyroptotic cells to recruit and activate leukocytes 27, 28. Immune sensing of PAMPs and DAMPs and immune responses to cytokines released during cell death therefore have important pathophysiological consequences that shape the course of infection and immunity, and the development of cancer, sepsis, and autoinflammatory diseases 29, 30, providing a rationale to further understand how PAMPs and DAMPs are recognized by the immune system, how cytokine signalling drives pathogenesis, and how therapeutic intervention in these processes might be used in the treatment of diseases (Fig. 1).
Fig. 1. Dying or dead cells release PAMPs, DAMPs, and cytokines that contribute to innate immunity and disease pathogenesis.

Following infection or sterile injury such as trauma, dying or dead cells release a plethora of pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and cytokines that activate pattern-recognition receptors (PRRs) and other receptors such as cytokine receptors in responding immune cells. These cells respond by liberating more PAMPs (in the case of infection), DAMPs, or cytokines that contribute to the manifestation, or in some cases attenuation, of localized and systemic diseases. A subset of cytokines called chemokines induce the recruitment of additional immune cells to the site of injury, amplifying the inflammatory response. Collectively, PAMPs, DAMPs, and cytokines fuel the inflammation circuitry, resulting in the progression and perpetuation of inflammatory diseases.
In this review, we provide an overview on immune-sensing of cell death and the PAMPs, DAMPs, as well as cytokines associated with cell death through the perspective of PRRs. We also consider the cell biology and physiological and pathological consequences of the connections between innate immunity and cell death.
Toll-like receptors
TLRs are a family of transmembrane PRRs located on the cell surface or on the endosomal membrane that recognize a range of PAMPs and DAMPs. Ten human TLRs and twelve mouse TLRs have been identified 31. These TLRs activate the transcription factors NF-κB and STAT3 that drive the production of inflammatory cytokines and type I IFNs and stimulate many biological processes (Fig. 2a) 31. While the typical response of TLR engagement is pro-inflammatory and pro-survival signalling, TLR signalling can also induce cell death.
Fig. 2. Necrotic cells release DAMPs that activate Toll-like receptors and inflammasomes.

a, TLR2 and TLR4 share the immune sensing of many DAMPs. TLR2 can additionally recognize nucleosomes and versican, whereas TLR4 can additionally recognize DAMPs such as oxidized low-density lipoprotein-immune complexes (OxLDL-IC). TLR3 can sense RNA from necrotic cells, whereas TLR7 might sense single-stranded RNA (ssRNA) and TLR9 might sense single-stranded DNA (ssDNA). TLR9 can also sense DNA complexed with the antimicrobial peptide LL37 or the nuclear protein HMGB1, or DNA-containing immune complexes, and DNA from dying or dead hepatocytes or chemotherapy-killed cancer cells, mitochondrial DNA (mtDNA), and neutrophil-derived DNA. b, Priming via TLRs (also known as Step 1) induces transcription of the genes encoding NLRP3 and pro-IL-1β. Necrotic cells release DAMPs which trigger several common events that activate NLRP3 (also known as Step 2). The most widely accepted mechanism is the efflux of potassium ions (K+) from within the cell. In many cases, NLRP3 interacts with the kinase NEK7 in order to assemble an inflammasome complex. AIM2 binds directly to and is activated by double-stranded DNA, which is readily released by dying or dead cells. Both inflammasome sensor proteins form a separate inflammasome complex of 0.5 to 1 micron in diameter comprised of the inflammasome adaptor protein ASC and the cysteine protease caspase-1. Caspase-1 is activated to induce the proteolytic cleavage of pro-IL-1β and pro-IL-18, and the pyroptosis-inducing protein gasdermin D (GSDMD). The bioactive N-terminal fragment of GSDMD assembles into pores on the plasma membrane, through which these pores mediate the secretion of bioactive IL-1β and IL-18 and hundreds of other smaller DAMPs, including IL-1α and galectin-1 that can escape through the diameter of the GSDMD pores. The GSDMD pores eventually drive ninjurin-1 (NINJ1) activation and oligomerization, leading to physical plasma membrane rupture, allowing the release of lactate dehydrogenase (LDH), HMGB1, and other larger DAMPs. Sufficient tears on the plasma membrane eventually lead to pyroptosis. NLRP3 inflammasome specks are released by pyroptotic cells to further perpetuate inflammation in the extracellular environment. These specks are also phagocytosed by neighbouring macrophages to induce a second round of NLRP3 activation.
TLR2 and TLR4
TLR2 and TLR4 are sensors of PAMPs that recognize lipoproteins and LPS, respectively; additionally both TLRs can detect DAMPs (Fig. 2a). In mouse models of atherosclerosis, for example, genetic deletion of TLR2 in low-density lipoprotein receptor-deficient (Ldlr–/–) mice 32, and genetic deletion of TLR4 in apolipoprotein E-deficient (Apoe–/–) mice 33, reduces the development of disease, hinting that these TLRs might recognize DAMPs. Many candidate DAMPs that can activate TLR2 and TLR4 have emerged, but whether these DAMPs originate specifically from dying or dead cells is not known. The glycosaminoglycan hyaluronan (also known as hyaluronic acid), found in the extracellular matrix, pericellular region, and blood, activates both TLR2 and TLR4 in individuals with acute lung injury, triggering the production of inflammatory cytokines and maintaining epithelial cell integrity during acute lung injury in mice 34, 35. Histones released by dying or dead cells can directly interact with TLR2 and TLR4, leading to the production of TNF and IL-6 in mouse dendritic cells and in mice 36.
Although TLR2 and TLR4 recognize many of the same DAMPs, they can also exhibit specificity. In human and mouse CD4+ T cells, histones activate TLR2, but not TLR4, and promote the differentiation of CD4+ T cells into inflammatory TH17 cells that can potentially convey pathological effects 37. Nucleosomes which carry histones, DNA, and other proteins are also thought to be sensed by TLR2 but not TLR4 38. Additionally, TLR2 but not TLR4 can sense the hyaluronan-binding proteoglycan versican, which is secreted by the Lewis lung carcinoma cell line 39. In myeloid cells, activation of TLR2 by versican drives TNF-dependent metastasis in mice injected with the Lewis lung carcinoma cell line 39. TLR4 recognizes free fatty acids to induce pro-inflammatory cytokine production in macrophages, adipocytes, and the liver in mice, leading to insulin resistance, a precursor to diabetes 40. TLR4 is also a sensor of calprotectin proteins S100A8 and S100A9 (also known as MRP8 and MRP14, respectively) released by phagocytes, and these proteins are elevated in patients with severe sepsis 41. TLR4 and its accessory protein MD2 form a signalling-competent receptor complex, which can bind directly to S100A8, with both S100A8 and S100A9 amplifying TLR4-dependent TNF production in response to LPS- and E. coli-induced endotoxemia in mice 41. The extracellular matrix molecules fibronectin extra domain A and tenascin-C, which are elevated in patients with the skin condition scleroderma, can activate TLR4 in fibroblasts, triggering cutaneous fibrosis and/or delayed fibrosis resolution and healing in mice and human skin organoids 42, 43.
Heme is another DAMP released during cell death and red blood cell (RBC) lysis in many infectious diseases as well as inflammatory and hemolytic diseases. In hereditary spherocytosis and sickle cell anemia, heme drives a feed-forward loop of chronic inflammation and tissue damage to cause disease 44–46. TLR2 and TLR4 are activated by heme 47, 48, and signal through MyD88 to drive the innate inflammatory response to heme plus PAMP stimulation 49, 50. Taken together, it is clear that activation of these TLRs by DAMPs is largely detrimental to the host.
TLR3, TLR7, and TLR9
The endosomal TLRs, TLR3, TLR7, and TLR9, function in sensing nucleic acids. TLR3 is a sensor of double-stranded RNA (dsRNA) that can be activated by RNA from necrotic neutrophils in macrophages, inducing inflammation and lethality in mice with tissue injury from gut ischemia and polymicrobial sepsis 51. TLR9, a sensor of CpG DNA, can bind to DNA released by dead cells, specifically DNA complexed with the endogenous antimicrobial peptide LL37 52 or the nuclear protein HMGB1 released by necrotic cells 53. When HMGB1 is complexed with DNA or DNA-containing immune complexes, TLR9 functions in partnership with the HMGB1-binding receptor RAGE to induce the secretion of TNF and IFN-γ in plasmacytoid dendritic cells and the expansion of autoreactive B cells 54. The combined effects of these two cell types are thought to contribute to the development of the autoimmune condition systemic lupus erythematosus in mice 54. HMGB1 amplifies the ability of CpG DNA to activate TLR9 in mouse macrophages and dendritic cells, possibly mediated by an interaction between HMGB1 and TLR9 55. A further study revealed that DNA binds directly to RAGE, such that RAGE delivers DNA to TLR9 on the endosome, effectively lowering the amount of DNA required to activate TLR9 56.
In a mouse model of liver injury, treatment with the analgesic drug acetaminophen (also known as paracetamol) leads to apoptotic hepatocyte death, freeing DNA to activate TLR9 in sinusoidal endothelial cells, driving inflammasome activation and liver inflammation 57. In another sterile inflammatory model using mice lacking the phospholipase D3 (PLD3) and PLD4, which spontaneously develop inflammatory liver damage due to the accumulation of ssDNA and ssRNA, genetic deletion or antibody blockade of TLR9 or TLR7 partially prevents pathological inflammation and spontaneous fatal hemophagocytic lymphohistiocytosis (HLH) 58.
TLR9 can also sense circulating mitochondrial DNA suspected to be released by mechanical trauma of cells. Mitochondrial DNA or RBC-bound mitochondrial DNA is increased in patients with trauma and during sepsis and pneumonia in mice and humans 59–61. Mitochondrial DNA enhances the ability of CpG-DNA to activate TLR9 in human neutrophils, leading to the production of IL-8 59. TLR9 expressed on RBCs can also capture circulating DNA, which mediates clearance of RBCs by macrophage phagocytosis, and induction of local and systemic cytokine production in mice 61. TLR9 sensing can also be beneficial, when surviving cancer cells recognize DNA from chemotherapy-killed cancer cells and promote inflammation and anti-tumour responses 62. Therefore, endogenous nucleic acids can induce an inflammatory response through endosomal TLRs, with evidence demonstrating that the source of the nucleic acids can be from necrotic cells and mitochondria (Fig. 2a).
Inflammasomes and PANoptosomes
Inflammasomes are a family of cytosolic multi-component signalling complexes activated by inflammasome sensors that, in most cases, recruit the adaptor protein called PYCARD (also known as ASC), followed by the inflammatory caspase, caspase-1. Activation of caspase-1 leads to the proteolytic cleavage of the pro-inflammatory cytokines pro-IL-1β and pro-IL-18, and the membrane pore-forming protein gasdermin D which mediates pyroptosis (Fig. 2b) 3, 4.
Multiple members of the PRR-sensing family, including NLRs, ALRs, and others, play a role in regulating or acting as the sensor for the formation of inflammasomes to drive pyroptosis. In addition to inflammasomes, many of the same PRRs may also regulate or assemble unique cell-death complexes called PANoptosomes, which mediate PANoptosis 5. In PANoptosis, in addition to gasdermin D-mediated IL-1β and IL-18 release, other gasdermins, including gasdermin E, and other executioners such as MLKL and ninjurin-1 (NINJ1) 13, also drive cell death and the release of hundreds of other proteins to perpetuate inflammation 63, including the DAMP galectin-1 64 and the cytokine IL-1α 65 from mouse macrophages, and S100A8 or S100A9 from human and mouse neutrophils 66–69. PANoptosis is implicated in several inflammatory diseases, including Aicardi-Goutieres syndrome 70, 71, cerebral ischemia 72, cytokine storm 25, HLH 25, 50 and sepsis 17, as well as in bacterial 69, 73, viral 17, 69, 74, and fungal 75 infections. Thus, determining the relative contribution of PAMPs, DAMPs, and cytokines in licensing PANoptosis is an active and emerging area of research. A holistic understanding of how PANoptosis can be blocked in systemic inflammatory diseases such as COVID-19, or activated during tumour development, is expected to provide therapeutic advances in the treatment of many chronic health conditions.
NLRP3 inflammasome and PANoptosome
NLRP3 is the most versatile inflammasome sensor protein identified to date, and it is often called the global sensor of PAMPs and DAMPs (Fig. 2b) 76. Oligomeric inflammasome structures called ‘specks’ of 0.5 to 1 micron in diameter form in the cytoplasm of an inflammasome-activated cell; these specks are readily visualised under confocal microscopy and are comprised of ASC arranged in filaments of 8 to 15 nm in diameter 77, 78. NLRP3 specks are also released from the cell corpse and activate caspase-1 outside the cell 79, 80. These specks have been used as serum biomarkers for a group of autoinflammatory diseases known as cryopyrin-associated periodic syndromes or autoimmune diseases in humans, and are seen engulfed by neighbouring macrophages where the specks trigger a second round of NLRP3 inflammasome activation in the recipient cell 79, 80. Therefore, NLRP3 specks are DAMPs and powerful perpetuators of inflammasome-mediated inflammation.
NLRP3 is highly responsive to lytic dead cell corpses 81 and DAMPs associated with cell death, notably ATP 20–23, β-amyloids 82, 83, biglycan 84, endogenous crystals 85, 86, free fatty acids 87, histones 88, mitochondrial DNA 89–91, RNA 92, 93, and S100A8 or S100A9 (Fig. 2b) 94. NLRP3 does not directly bind to DAMPs, in general, and the responsiveness of NLRP3 to these DAMPs has been demonstrated largely under in vitro conditions. In a sterile inflammatory model in mice, NLRP3 has been reported to sense ATP released from necrotic neutrophils caused by thermal injury to the liver, promoting localized inflammation and adherence of circulating neutrophils to the liver sinusoids 23. ATP released by chemotherapy-induced dying cells also activates the NLRP3 inflammasome–IL-1β pathway in dendritic cells and drives anti-tumour cytotoxic CD8+ T cell responses in mice 95. However, there is no evidence that ATP drives inflammation in mice undergoing drug-induced liver injury or injection of necrotic cells 24, suggesting that either the concentration of ATP necessary to trigger inflammation is not achieved or that NLRP3 has no role in these experimental models. Additionally, in response to NLRP3 triggers such as ATP and bacterial PAMPs, it is likely that NLRP3-dependent but caspase-1-, gasdermin-D-independent lytic cell death can occur through PANoptosis. In this context, the activation of caspases and executioners associated with PANoptosis would occur, and a PANoptosome complex containing ASC, caspase-8 and RIPK3 would form, positioning NLRP3 as a PANoptosome sensor activating PANoptosis. Furthermore, beyond its crucial roles in inflammasome-dependent pyroptosis and as a PANoptosome sensor, NLRP3 is also integral to several other PANoptosomes (Fig. 3), as discussed below.
Fig. 3. PAMPs, DAMPs, and cytokines activate PANoptosomes.
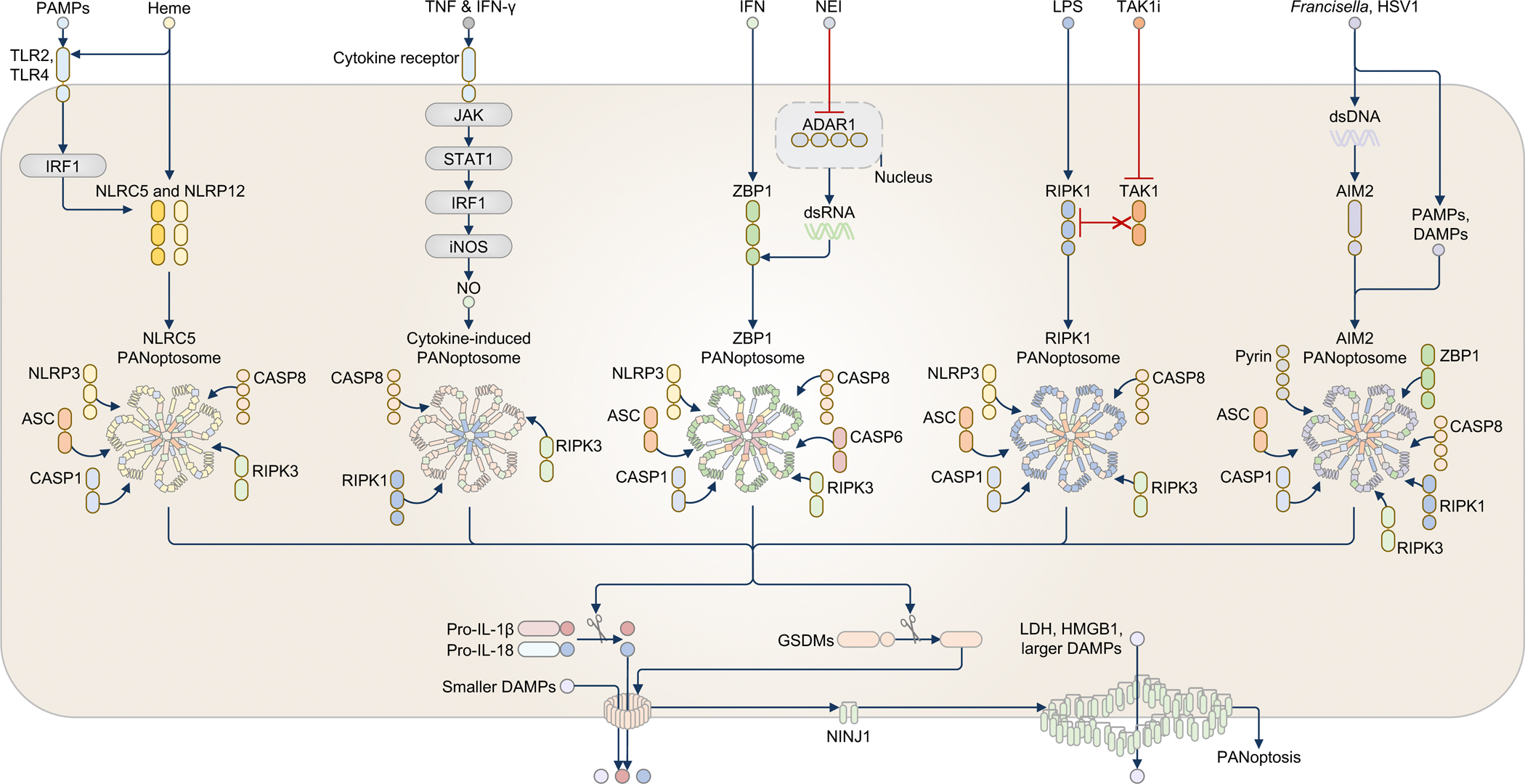
Heme in combination with PAMPs or TNF (not shown) triggers the activation of the NLRC5-PANoptosome, which also contains NLRP12. PAMPs induce the activation of the transcription factor IRF1 to upregulate the NLRC5 and NLRP12 proteins. Heme then induces PANoptosome formation by recruiting NLRP3, ASC, caspase-1 (CASP1), caspase-8 (CASP8), and RIPK3, leading to PANoptosis. The cytokine combination of TNF and interferon (IFN)-γ triggers a second PANoptosome. TNF and IFN-γ upregulate the production of nitric oxide (NO) via JAK, STAT1, IRF1, and inducible nitric oxide synthase (iNOS), triggering a cytokine-induced PANoptosome requiring CASP8, RIPK1, and RIPK3 to drive PANoptosis. The ZBP1-PANoptosome functions in sensing cytokines and DAMPs, as well as infections. In response to the combination of IFN and a nuclear export inhibitor (NEI), IFN upregulates the expression of ZBP1 and ADAR1, and NEI restricts ADAR1 to the nucleus, allowing ZBP1 to freely sense accumulated dsRNA and assemble a PANoptosome. The ZBP1-PANoptosome contains NLRP3, ASC, CASP1, CASP8, caspase-6 (CASP6), and RIPK3. The ZBP1-PANoptosome can also be activated by Influenza A virus infection (not shown). The RIPK1-PANoptosome senses the combination of the PAMP LPS and DAMPs in the form of TAK1 inhibition (TAK1i). TAK1i unleashes RIPK1, allowing LPS or other PAMPs or DAMPs to induce the assembly of the RIPK1-PANoptosome comprised of RIPK1, NLRP3, ASC, CASP1, CASP8, and RIPK3. The RIPK1-PANoptosome can also be activated by Yersinia infection (not shown). In response to PAMPs and DAMPs, such as those released during Francisella or herpes simplex virus 1 (HSV1) infection (not shown), dsDNA derived from these pathogens serves as a PAMP to activate the AIM2-PANoptosome. Other PAMPs carried by these pathogens and possibly DAMPs resulting from these infection events are likely required to drive the assembly of the AIM2-PANoptosome because transfection of dsDNA alone cannot recapitulate this response. The AIM2-PANoptosome comprises AIM2, Pyrin, ZBP1, ASC, CASP1, CASP8, RIPK1, and RIPK3. PANoptosomes activate or induce the proteolytic cleavage of many substrates, including gasdermin proteins, cytokines, and caspases, resulting in inflammatory lytic cell death and the secretion of cytokines and DAMPs. The symbols in the legend denote the colors and shapes used to depict each molecule; the number of shapes in a molecule reflects the general domain structure and Is not intended to suggest the number of molecules present.
NLRP3 also senses needle-shaped monosodium urate crystals 85, formed by uric acid in joint tissues leading to gout. The ability of the NLRP3 inflammasome to drive the secretion of IL-1β contributes to this pathological manifestation. In mice, genetic deletion of the gene encoding IL-1R reduces neutrophil influx in monosodium-urate-induced peritonitis 85. In humans, anti-IL-1 biologic therapies are highly effective in the treatment of gout 96. Monosodium-urate-induced lytic cell death in macrophages is independent of NLRP3 and gasdermin D 97, 98, implying that other DAMPs would still be released even if NLRP3 is pharmacologically inhibited. The effectiveness of NLRP3 blockade in the treatment of gout in humans is not yet clear, but a phase 2b trial has shown efficacy of an oral NLRP3 inhibitor Dapansutrile in reducing joint pain in patients with gout flares 99.
NLRP3 also has an emerging role in sensing and responding to DAMPs in platelets. S100A8 and S100A9 are neutrophilic DAMPs comprising 45% of the total cytosolic protein content in human neutrophils 100, and their levels are elevated in patients with severe sepsis 101. S100A8 or S100A9, released by human and mouse neutrophils via gasdermin D pores, can trigger the assembly of the NLRP3 inflammasome in mouse platelets 94. These platelets then undergo pyroptosis and amplify the inflammatory feedback loop by liberating oxidized mitochondrial DNA and triggering more S100A8 or S100A9 release from neutrophils 94.
AIM2 inflammasome and PANoptosome
AIM2 is a cytoplasmic inflammasome sensor that binds to double-stranded DNA and has been well-characterized in the context of infection 69, 102–104, inflammatory skin and intestinal diseases 105–107 and cancer 108, 109. In infection, the exact source of DNA leading to AIM2 activation is presumed to be derived from microbes or the microbiome; however, identification of a role for AIM2 in the host defense against the RNA virus, Influenza A Virus (IAV), which does not carry DNA, suggests host DNA might be involved 110. Indeed, IAV infection causes the extracellular release of DNA into the lung fluid of mice 110, 111, and the ion channel activity of the IAV M2 protein and the mitochondrial localization of the IAV PB1-F2 protein induce the release of mitochondrial DNA and oxidized DNA into the cytoplasm of macrophages and extracellularly from macrophages 112.
Further evidence that AIM2 can sense host DNA from dying cells came from a study showing that self-DNA released into the gastrointestinal tract of mice, due to treatment with the cytotoxic drug irinotecan, is captured by exosomes and delivered into intestinal cells to activate AIM2 113. In another mouse model, DNA is taken up by leukocytes in the injured kidneys caused by ureteral obstruction 114. Sensing of DNA by AIM2 is supported by evidence using mice lacking AIM2, which have abrogated irinotecan-induced intestinal inflammation and diarrhoea 113, reduced influenza-induced lung inflammation, morbidity and mortality 111, and attenuated renal injury, fibrosis, and inflammation 114.
In addition to forming an inflammasome, AIM2 also assembles a PANoptosome in response to physiologically relevant triggers such as HSV1 and Francisella tularensis 69 (Fig. 3). In this complex, ZBP1 and Pyrin are recruited in conjunction with ASC and caspase-1, as well as caspase-8, RIPK1 and RIPK3 69. Activation of PANoptosis requires infection with the live pathogen, which releases PAMPs and DAMPs together; in contrast, treatment with the bacterial PAMP mimic poly(dA:dT) alone induces inflammasome activation rather than PANoptosome formation 69. These findings highlight key distinctions between PANoptosis and pyroptosis in physiological settings in response to dying cells and liberated cytokines and DAMPs.
ZBP1-PANoptosome
The ZBP1-PANoptosome was the first PANoptosome identified, and it was discovered to form in response to IAV infection 17 (Fig. 3). It also plays a key role in the pathophysiology of IFN therapy during coronavirus infection 115, and the ZBP1-PANoptosome was also found to form upon combined stimulation with IFN and a nuclear export inhibitor (NEI) 116. In this case, NEIs, such as KPT-330, prevent the export of ADAR1 from the nucleus. Given that IFN upregulates the expression of ZBP1 and its negative regulator ADAR1, evidence suggests that the NEI restricts the activity of ADAR1 and indirectly promotes the assembly of the ZBP1-PANoptosome, which includes NLRP3, ASC, caspase-1, caspase-6, caspase-8, and RIPK3 5, 74, 116. This combination of IFN and KPT-330 promotes ZBP1-mediated PANoptosis and attenuates tumour growth in vivo in mice 116.
NLRC5- and NLRP12-PANoptosome
NLRC5 and NLRP12 were recently discovered to be key sensors in a PANoptosome complex (Fig. 3). NLRP12 was identified as a sensor of specific combinations of PAMPs and DAMPs, such as heme and LPS, Pam3, or TNF 49. Subsequently, the NLRC5-PANoptosome was identified to activate PANoptosis in response to NAD+ depletion, a common occurrence during disease 50. In this complex, there is coordinated activity among multiple NLRs, including NLRC5, NLRP12 and NLRP3, suggesting NLR networks similar to those seen in plants also form in mammals 50. NLRC5- and NLRP12-dependent PANoptosis depends on the transcription factor IRF1, which upregulates NLRC5 and NLRP12 in response to heme and PAMPs or TNF 49, 50, 117. Then, NLRP3, ASC, caspase-1, caspase-8 and RIPK3 are recruited to the PANoptosome, leading to PANoptosis. Nlrp12–/– and Nlrc5–/– mice are resistant to acute kidney injury and lethality during drug-induced hemolysis combined with LPS exposure 49, 50. In addition, NLRC5 deficiency confers protection in colitis and HLH models 50. Thus, inhibition of NLRC5 or NLRP12 could be therapeutically beneficial in the context of highly diverse stimuli, including pathogen-mediated hemolysis or other red blood cell disorders.
RIPK1-PANoptosome
The RIPK1-PANoptosome was initially shown to form downstream of LPS signalling in TAK1-deficient macrophages 118 (Fig. 3). This LPS-mediated cell death requires RIPK1 but not its kinase activity, implicating its scaffolding function in the formation of a multiprotein complex 118. Indeed, in the absence of TAK1, RIPK1 forms a complex with NLRP3, ASC, caspase-1, caspase-8 and RIPK3 to mediate PANoptosis 73. While TAK1 promotes cell survival in response to stimuli such as LPS or TNF, TAK1 is also a cell-intrinsic negative regulator of PANoptosis via inhibition of RIPK1 118, 119. Inhibition of TAK1 in vivo induces a sepsis-like state 118, 119, which indicates that constitutively active PANoptotic processes can generate unregulated and potentially lethal systemic inflammation.
Cytokine-induced PANoptosome
TNF and IFN-γ are a specific cytokine combination that is frequently elevated in inflammatory diseases 25. TNF and IFN-γ upregulate the production of nitric oxide in macrophages via JAK, STAT1 and IRF1 signalling, inducing the formation of a PANoptosome which is responsible for cell death and lethality during SARS-CoV-2 infection, sepsis, HLH and cytokine shock 25, 26, 115 (Fig. 3). However, the specific sensor required to promote the formation of this PANoptosome remains unknown. Nevertheless, this cytokine-induced PANoptosome has potential therapeutic implications for the treatment of cancer 116, 120, as TNF and IFN-γ trigger PANoptotic cell death in melanoma and leukaemia as well as colon and lung tumour cells 121.
C-type lectin receptors
C-type lectin receptors (CLRs) are a large class of cell-surface or secreted PRRs carrying one or more C-type lectin recognition domains 122. They primarily recognize carbohydrate structures and provide host defense against pathogens, but CLRs also bind to and phagocytose dead cells, sense DAMPs, and trigger or inhibit the production of inflammatory cytokines, antigen presentation and T cell responses (Fig. 4). Certain CLRs bind to endogenous ligands expressed on live mammalian cells 123, 124, but other CLRs sense DAMPs that are liberated from dying or dead cells.
Fig. 4. Necrotic cells release DAMPs that activate C-type lectin receptors.

MINCLE binds to spliceosome-associated protein 130 (SAP130) and associates with the signalling receptor FcRγ and signals via SYK and CARD9 to drive the production of TNF and chemokines, inducing neutrophil recruitment. MINCLE activation by SAP130 also promote pro-tumorigenic immunosuppression in pancreatic cancer. MINCLE also recognizes β-glucosylceramide and activates CARD9, leading to cytokine and chemokine production, increased co-stimulation, and activation of adaptive immunity. Clec1a binds to the histidine-rich glycoprotein, mediating phagocytosis of bacteria. Clec1a also binds to TRIM21 and inhibits cross-presentation, promoting tumour growth. Clec2d is a sensor of DNA-free histones (not shown) and DNA-bound histones. Clec2d delivers DNA to TLR9 within the endosome to trigger cytokine production, enhancing liver injury and lethality. Clec9a binds to actin filaments and F-actin, activating SYK and promoting the delivery of the necrotic cell debris to the recycling endosome and cross presentation to CD8+ T cells. Further, Clec9a activates the tyrosine phosphatase SHP-1 and inhibits the production of the chemokine CXCL2, thereby preventing neutrophil recruitment, necrotizing pancreatitis, and pathology during infection with Candida albicans. Clec12a senses monosodium urate crystals from dead cells and inhibits SYK-dependent cytokine and chemokine production, reducing neutrophil influx. Clec12a also mediates the production of type I interferons (IFNs) by enhancing RIG-I signalling. Dectin-1 binds to N-glycan structures found on the cell-surface of live tumour cells and presumably also dead tumour cells, and via the membrane-associated protein MS4A4A, activates IRF5 and induces the cell-surface expression of IRF3-dependent NK-activating molecule (INAM) on dendritic cells. INAM mediates enhanced contact between dendritic cells and NK cells, promoting NK-cell anti-tumour immunity. Dectin-1 also senses galectin-9 found on the cell-surface of tumour cells and promotes immunosuppression and the growth of pancreatic ductal adenocarcinoma.
MINCLE
Human and mouse MINCLE (also known as Clec4e) are expressed on macrophages and dendritic cells, and recognize pathogenic fungal Malassezia and bacterial Mycobacterium species, as well as dead cells and DAMPs 125. MINCLE binds to the small nuclear ribonucleoprotein SAP130, and potentially to related proteins SAP49, SAP145, and SAP155 125. In response to SAP130, MINCLE associates with the immunoreceptor tyrosine-based activation motifs (ITAM)-containing adaptor FcRγ and signals via the adaptor proteins SYK and CARD9 to drive the production of TNF and chemokine CXCL2 (also known as MIP-2) in macrophages, leading to the recruitment of neutrophils to the site of injury in mice 125.
Additionally, MINCLE can recognize the metabolite glycolipid β-glucosylceramide released from damaged mammalian cells 126. β-glucosylceramide is normally found in the endoplasmic reticulum or Golgi and is unlikely to be exposed to cell-surface-associated MINCLE during homeostasis. In response to LPS stimulation or bacterial infection, increased production of β-glucosylceramide can occur 127, 128, and subsequent cell death offers a physiological scenario by which β-glucosylceramide can be liberated and interact with MINCLE on neighbouring cells. In response to β-glucosylceramide, MINCLE activates CARD9, driving the secretion of TNF and MIP-2, and inducing the expression of costimulatory molecules CD40, CD80 and CD86 and MHC class II on mouse dendritic cells 126.
Inhibition of MINCLE using a neutralizing DNA aptamer or an antibody reduces the severity of chemically-induced inflammatory bowel disease in mice 129, suggesting that targeting MINCLE in inflammatory diseases might be beneficial. However, comparison between wild-type and Mincle–/– mice identified no role for MINCLE in two other experimental models of cell-death-induced inflammation: injection with necrotic EL4 tumour cells resulting in a neutrophilic inflammatory response and treatment with acetaminophen causing liver cell necrosis 24. In the context of cancer development, pancreatic ductal adenocarcinoma cells expressing recombinant SAP130 injected into wild-type mice grew faster than in mice lacking MINCLE 130, indicating that MINCLE activation by SAP130 is detrimental and promotes oncogenesis in pancreatic cancer.
Clec1a
Clec1a is expressed on human and mouse dendritic cells, macrophages, monocytes, neutrophils and endothelial cells 131–133. Clec1a binds directly to the plasma protein histidine-rich glycoprotein 134, and the E3 ubiquitin ligase TRIM21 expressed on the surface of necrotic cells 135. Clec1a has different roles depending on the disease context and cell types. In response to infection in the presence of histidine-rich glycoprotein, Clec1a mediates phagocytosis of bacteria in human neutrophils 136. In response to human or mouse cells killed by UV or X-ray radiation, Clec1a binds to TRIM21 expressed on dead cells and inhibits cross-presentation by conventional type 1 dendritic cells to CD8+ T cells, promoting tumour growth in mice 135. Blockade of Clec1a using a Clec1a fusion protein prolongs the survival of mice injected with MC38 adenocarcinoma 135, indicating a disease-promoting role of Clec1a.
Clec2d
Clec2d is found on the plasma membrane and endosomes of mouse macrophages and can sense histones 137. Histones are released by activated neutrophils as part of neutrophil extracellular traps, or they can be found in either a DNA-bound form in a nucleosome or in an octameric protein form free of DNA 138. Clec2d senses both DNA-free histones and DNA-bound histones, including histone variants H1, H2A, H2B, H3, and H4, by recognizing the N-terminal tail regions of all histones and the C-terminal tail region of H1 137. Unlike most CLRs, Cled2d cannot induce signalling on its own, because it lacks an ITAM 137. On activation by DNA-bound histones, Clec2d delivers DNA to TLR9 within the endosome to trigger cytokine production, which enhances the liver injury and lethality induced by acetaminophen overdose in mice 137.
Clec9a
Clec9a (also known as DNGR-1) is expressed on mouse CD8α+ dendritic cells and human dendritic cells, and it recognizes dead mammalian cells undergoing secondary necrosis following UV treatment 139. Human and mouse Clec9a bind directly to actin filaments secreted by necrotic cells 140, 141, with modest affinity for F-actin 142. The binding between Clec9a and F-actin, however, is strengthened by multivalent interactions defined by the ligand-binding domain of Clec9a interacting with three actin subunits at the interface of two actin protofilaments 142.
In addition, the F-actin-associated motor protein, myosin II, also exposed in necrotic cells, promotes the binding between Clec9a and F-actin 143. This pathway of dead-cell recognition activates SYK signalling 140, but does not result in cytokine production in CD8α+ dendritic cells 139. Instead, Clec9a promotes the delivery of necrotic cell debris to the recycling endosome, leading to phagosomal rupture where the antigens are cross-presented to CD8+ T cells 144, 145. Further, Clec9a activates the tyrosine phosphatase SHP-1 and inhibits the production of the chemokine CXCL2 by conventional type I dendritic cells during systemic infection with the fungus Candida albicans 146. Sensing of early damage by Clec9a, presumably via actin release caused by C. albicans infection, reduces CXCL2-mediated neutrophil recruitment, necrotizing pancreatitis, and pathology 146.
Clec12a
Clec12a can directly bind dead mammalian cells leading to crosslinking between Clec12a proteins and inhibition of SYK 147. SYK is activated by cell-surface receptors carrying ITAM or ITAM-like motif; however, Clec12a carries an immunoreceptor tyrosine-based inhibition motif (ITIM), which indicates that the functional activity of Clec12a is anti-inflammatory. Clec12a senses the monosodium urate crystals of dead cells in humans and mice 147. Mice lacking Clec12a are hypersusceptible to neutrophil influx following injection of monosodium urate crystals or X-ray irradiation to induce thymocyte killing 147. In response to X-ray irradiation in mice, Clec12a can also drive type I interferon (IFN) responses by enhancing RNA-sensing RIG-I signalling pathways 148. The physiological RIG-I ligands in this case are not known, nor are the potentially broader effects of Clec12a in other nucleic-acid-sensing pathways.
cGAS-like receptors
cGAS is a cytoplasmic PRR of the cGLR family that recognizes DNA 149. Following activation, cGAS generates the second messenger cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), which binds to and activates the adaptor protein STING 150. STING recruits and activates the kinase TBK1 and promotes the activation of the transcription factor IRF3 and type I IFN production. cGAS directly senses DNA from infectious agents and mislocalized self-DNA within the same cell (Fig. 5).
Fig. 5. Necrotic cells release DAMPs that activate the cytosolic DNA-sensing and RNA-sensing pathways.

In phagocytic cells, cGAS recognizes DNA from dying or dead cells. DNA that results from DNase II deficiency (not shown), apoptotic cells, erythroid precursor cells or mitochondria accumulates within the cytoplasm of phagocytes. cGAS catalyzes cGAMP production for STING activation to trigger the production of inflammatory cytokines via NF-κB and type I interferons (IFNs) via IRF3/7, resulting in embryonic lethality and inflammatory arthritis. DNA from dying tumour cells and tumour-derived mitochondrial DNA taken up by dendritic cells is also sensed by cGAS, leading to IFN-β production and cross-presentation to CD8+ T cells, inducing anti-tumour immunity. Due to a loss of the mitochondrial RNA helicase SUV3 and polynucleotide phosphorylase PNPase (not shown), mitochondrial double-stranded RNA (dsRNA) from inside the cell or from dying or dead cells accumulates and escapes into the cytoplasm to activate MDA5, and via MAVS, triggers the production of inflammatory cytokines and type I IFNs. RIG-I can sense cytoplasmic RNA (not shown), but there is currently no evidence suggesting that RIG-I can sense RNA directly from dead cells. Defects in adenosine-to-inosine RNA editing induced by deficiencies or mutations in ADAR1 also lead to accumulation of endogenous RNA that is sensed by MDA5.
The recent model proposed for DNase II deficiency-mediated disease provides the strongest evidence that DNA from dying neighbouring cells also activates cGAS. DNase II deficiency in mice leads to the lysosomal accumulation of undigested DNA, driving TNF and IFN-β responses, embryonic lethality and autoimmune diseases 151, 152. The disease-initiating DNA has been reported to come from apoptotic cells 153, 154, erythroid precursor cells 155, or within the nucleus of DNase II-deficient cells 156. Self-DNA can also accumulate within the cytoplasm of cells lacking the DNA nuclease TREX1, but the evidence that the self-DNA comes from another dying cell is limited. Earlier studies showed that genetic deletion of TLR3, TLR9 or MyD88 and TRIF in DNase II−/− mice does not prevent embryonic lethality and disease 154, but deletion of the type I IFN receptor 1, IFNAR1, prevents embryonic lethality but not polyarthritis 151, 152, suggesting the involvement of other DNA-sensing PRRs and additional cytokines. Later studies demonstrated that deletion of either STING or cGAS in DNase II−/− mice leads to reduced cytokine production and prevents both embryonic lethality and inflammatory arthritis 157, 158, corroborating with in vitro experiments showing that necrotic cells trigger STING-dependent production of IFN-β in mouse macrophages 157. Consistent with the role of cGAS in catalysing cGAMP production for STING activation, cGAMP does not accumulate in the fetal liver of DNase II−/− mice lacking cGAS 158.
The cGAS-STING pathway can also activate NF-κB, leading to the production of TNF and IL-6. This additional signalling capacity may in part explain why DNase II−/−Ifnar1−/− mice 151, 152 or DNase II−/− mice carrying a mutant form of STING (called STINGS365A/S365A), which cannot induce type I IFN signalling but retains the ability to induce NF-κB activation 159, still develop polyarthritis. Neutralizing antibodies against TNF or the IL-6 receptor alleviate polyarthritis in DNase II−/−STINGS365A/S365A mice 159. Additional genetic deletion of STING, AIM2 or the TLR-chaperon protein UNC93B1 in DNase II−/−Ifnar1−/− mice also reduces the severity of polyarthritis 160, 161. Unravelling the complexity of PRRs has allowed the development and fine-tuning of therapeutic targeting for autoinflammatory disease instigated by self-DNA.
cGAS and STING are also implicated in sensing DNA from dying cells in tumorigenesis 162–165. Labelled DNA from tumour cells injected into mice are found inside dendritic cells 165, with cGAS, STING and IRF3 driving the induction of IFN-β in dendritic cells exposed to irradiated tumour cells or dying cells 162, 163. STING-dependent IFN-β production and cross-presentation are both inhibited by physical separation between mouse dendritic cells and irradiated tumour cells or by an actin polymerization inhibitor 162, supporting a model where phagocytosis of dead cells by dendritic cells is essential. This process drives dendritic-cell cross-presentation of tumour-associated antigens to CD8+ T cells, inducing anti-tumour immunity in mice 162, 165, 166. In response to antibodies blocking the immunoglobulin-like domain–containing molecule CD47, tumour-derived mitochondrial DNA is phagocytosed by dendritic cells to activate the cGAS-STING pathway that promotes tumour rejection in mice 167. This occurs through antibody-mediated interference of binding between CD47 on tumour cells and signal-regulatory protein alpha (SIRPα) on dendritic cells, activating a NOX2-dependent delay in acidification of the phagosome and degradation of engulfed mitochondrial DNA. This delay maintains the necessary DNA concentration for cGAS sensing 167. How genomic or mitochondrial DNA from tumour cells are released into the cytoplasm of recipient cells to activate the cGAS-STING pathway is still unknown. A different model suggests that constitutively-active cGAS in tumour cells catalyses cGAMP production, with cGAMP activating STING-dependent IFN-β production in neighbouring non-tumour cells, leading to anti-tumour responses by natural killer cells 168. However, a direct transfer of cGAMP between tumour cells and non-tumour cells licensing STING activation and immune responses in vivo remains to be tested, noting that cGAMP transfer in vivo is largely limited by the degradation of extracellular cGAMP by Ectodomain phosphatase/phosphodiesterase-1 (ENPP1) 169.
RIG-I-like receptors
RIG-I-like receptors (RLRs) are found in the cytoplasm, with the family member RIG-I sensing shorter dsRNA, and another family member MDA5 sensing longer dsRNA 170. Both receptors trigger the production of type I IFNs and pro-inflammatory cytokines via the adaptor protein MAVS. While RLRs are most associated with immune responses to viral pathogens, they also sense DAMPs in the form of endogenous RNA (Fig. 5). Mitochondrial dsRNA, which is normally rapidly degraded, can accumulate due to a loss of the mitochondrial RNA helicase SUV3 and polynucleotide phosphorylase PNPase 171. Mitochondrial dsRNA escapes into the cytoplasm to activate MDA5 and type I IFN production, which may contribute to the clinical manifestations of humans with mutations in PNPase 171. Additionally, defects in adenosine-to-inosine RNA editing induced by deficiencies or mutations in ADAR1 can also lead to the accumulation of endogenous RNA that can be sensed by MDA5 in humans and mice 172–174, and the loss of ADAR1 in some tumours sensitizes them to immunogenic cell death 116, 175, 176. While these studies point to the presence of mislocalized or accumulating self-RNA in triggering RNA-sensing mechanisms within the same cell, whether extracellular RNA liberated from dead cells can activate RLRs in neighbouring cells has not been reported. RNA can be released passively from dying or dead cells or via extracellular vesicles, but this RNA is degraded rapidly by extracellular RNAses 177. However, with functional defects in extracellular RNAses, it may be possible that extracellular RNA would accumulate to a quantity that can activate RLRs.
Conclusions and outlook
While inflammatory cell death is a key outcome of innate immune activation, it is a driving and perpetuating force of PRR sensing that results in chronic inflammation and pathology. Unravelling the connections between innate immune cell death and pathological inflammation is expected to be crucial for understanding fundamental cell biology that enables the identification of therapeutic strategies. The biological effects of DAMPs derived from dying or dead cells and their sensing by PRRs are largely characterized by the amplification of inflammation and immune cell recruitment, which can drive detrimental tissue damage, delayed healing, cytokine storm, metastasis and insulin resistance. These effects contribute to the development of cancer, diabetes, sepsis and other inflammatory conditions. However, some protective effects of PRR-sensing of cell death have been documented, including lowering the magnitude of inflammatory responses, induction of cross-presentation, and anti-tumour immunity. Therefore, PRRs can sense the same cell corpse, DAMPs and cytokines but generate converging or opposing outcomes. How PRRs precisely coordinate with one another holistically to produce a coherent cellular response to the same dying or dead cell has remained unclear. The answers may lie in spatially-distinct multi-protein complexes whose organization and execution of the various activities of PRRs, caspases, and RIPKs within a cell is orchestrated at a singular signalling hub, such as the PANoptosomes.
From a therapeutic perspective, strategies used to block PRRs that sense PAMPs, DAMPs and cytokines released during cell death can prevent inflammation and pathology. Inhibiting PANoptosome activation using anti-TNF and anti-IFN-γ antibodies prevents mortality during SARS-CoV-2 infection, sepsis, HLH, and cytokine shock 25. Furthermore, blocking activation of the NLRP12-PANoptosome protects against hemolytic disease models 49, and inhibiting the NLRC5-PANoptosome prevents PANoptosis activation in response to NAD+ depletion and reduces morbidity and mortality during hemolytic disease, colitis, and HLH models 50. Blocking DAMPs such as HMGB1 has also shown promising effects in reducing pathology and mortality during pneumococcal meningitis 178, stroke 179, Alzheimer’s disease 180, and several other CNS and inflammatory diseases in animal models. For therapeutic activation of PRRs to harness beneficial effects, activation of the ZBP1-PANoptosome using IFN and nuclear export inhibitors could be considered in anti-tumour therapies 116, and additional strategies to target PANoptosis through other PRRs should also be considered. Continued investigation to identify critical PRR-sensing pathways in responding to cell death is expected to pave the way for new diagnostic and prognostic biomarkers and therapeutic advancement for the treatment of diseases.
Acknowledgments
We acknowledge our colleagues whose work contributed to the advancement of immune sensing of cell death, and we apologize to those whose work could not be comprehensively cited due to space constraints. Research studies in our laboratories are supported by the US National Institutes of Health (grants AI101935, AI124346, AI160179, AR056296, and CA253095 to T-D.K.) and the American Lebanese Syrian Associated Charities (T-D.K.), and the National Health and Medical Research Council of Australia (Ideas Grant APP2002686 and Investigator Grant 2026910 to S.M.M.) and the CSL Centenary Fellowship (S.M.M.).
Footnotes
Competing financial interests
T-D.K. and S.M.M. have no interests to declare.
References
- 1.Brennan MA & Cookson BT Salmonella induces macrophage death by caspase-1-dependent necrosis. Molecular microbiology 38, 31–40 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Kayagaki N et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Christgen S et al. Identification of the PANoptosome: A Molecular Platform Triggering Pyroptosis, Apoptosis, and Necroptosis (PANoptosis). Front Cell Infect Microbiol 10, 237 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degterev A et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature chemical biology 1, 112–119 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Cho YS et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He S et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Zhang DW et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Sun L et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Zhao J et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A 109, 5322–5327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choudhury SM, Sarkar R, Karki R & Kanneganti TD A comparative study of apoptosis, pyroptosis, necroptosis, and PANoptosis components in mouse and human cells. PLoS One 19, e0299577 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kayagaki N et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Han JH et al. NINJ1 mediates inflammatory cell death, PANoptosis, and lethality during infection conditions and heat stress. Nat Commun 15, 1739 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rock KL & Kono H The inflammatory response to cell death. Annu Rev Pathol 3, 99–126 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y et al. cGLRs are a diverse family of pattern recognition receptors in innate immunity. Cell 186, 3261–3276 e3220 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuriakose T et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karki R & Kanneganti TD ADAR1 and ZBP1 in innate immunity, cell death, and disease. Trends Immunol 44, 201–216 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong T, Liu L, Jiang W & Zhou R DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 20, 95–112 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Perregaux D & Gabel CA Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 269, 15195–15203 (1994). [PubMed] [Google Scholar]
- 21.Solle M et al. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem 276, 125–132 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Mariathasan S et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006). [DOI] [PubMed] [Google Scholar]
- 23.McDonald B et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (New York, N.Y.) 330, 362–366 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Kataoka H, Kono H, Patel Z, Kimura Y & Rock KL Evaluation of the contribution of multiple DAMPs and DAMP receptors in cell death-induced sterile inflammatory responses. PLoS One 9, e104741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karki R et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 184, 149–168 e117 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simpson DS et al. Interferon-gamma primes macrophages for pathogen ligand-induced killing via a caspase-8 and mitochondrial cell death pathway. Immunity 55, 423–441 e429 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kono H, Karmarkar D, Iwakura Y & Rock KL Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol 184, 4470–4478 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Place DE & Kanneganti TD Cell death-mediated cytokine release and its therapeutic implications. J Exp Med 216, 1474–1486 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanneganti TD Intracellular innate immune receptors: Life inside the cell. Immunol Rev 297, 5–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Man SM & Jenkins BJ Context-dependent functions of pattern recognition receptors in cancer. Nat Rev Cancer 22, 397–413 (2022). [DOI] [PubMed] [Google Scholar]
- 31.Kawai T, Ikegawa M, Ori D & Akira S Decoding Toll-like receptors: Recent insights and perspectives in innate immunity. Immunity 57, 649–673 (2024). [DOI] [PubMed] [Google Scholar]
- 32.Mullick AE, Tobias PS & Curtiss LK Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest 115, 3149–3156 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michelsen KS et al. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proceedings of the National Academy of Sciences of the United States of America 101, 10679–10684 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Termeer C et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med 195, 99–111 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang D et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 11, 1173–1179 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Allam R et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 23, 1375–1388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson AS et al. Neutrophil extracellular traps and their histones promote Th17 cell differentiation directly via TLR2. Nat Commun 13, 528 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urbonaviciute V et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 205, 3007–3018 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim S et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102–106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi H et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116, 3015–3025 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vogl T et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13, 1042–1049 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Bhattacharyya S et al. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Sci Transl Med 6, 232ra250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhattacharyya S et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun 7, 11703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gozzelino R, Jeney V & Soares MP Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50, 323–354 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Martins R & Knapp S Heme and hemolysis in innate immunity: adding insult to injury. Curr Opin Immunol 50, 14–20 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Soares MP & Bozza MT Red alert: labile heme is an alarmin. Curr Opin Immunol 38, 94–100 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Figueiredo RT et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem 282, 20221–20229 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Min H, Choi B, Jang YH, Cho IH & Lee SJ Heme molecule functions as an endogenous agonist of astrocyte TLR2 to contribute to secondary brain damage after intracerebral hemorrhage. Mol Brain 10, 27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sundaram B et al. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell 186, 2783–2801 e2720 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sundaram B et al. NLRC5 senses NAD+ depletion, forming a PANoptosome and driving PANoptosis and inflammation. Cell (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cavassani KA et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med 205, 2609–2621 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lande R et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449, 564–569 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Scaffidi P, Misteli T & Bianchi ME Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Tian J et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8, 487–496 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Ivanov S et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 110, 1970–1981 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sirois CM et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med 210, 2447–2463 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imaeda AB et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest 119, 305–314 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gavin AL et al. Cleavage of DNA and RNA by PLD3 and PLD4 limits autoinflammatory triggering by multiple sensors. Nat Commun 12, 5874 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Q et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hotz MJ et al. Red Blood Cells Homeostatically Bind Mitochondrial DNA through TLR9 to Maintain Quiescence and to Prevent Lung Injury. Am J Respir Crit Care Med 197, 470–480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lam LKM et al. DNA binding to TLR9 expressed by red blood cells promotes innate immune activation and anemia. Sci Transl Med 13, eabj1008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tuomela J et al. DNA from dead cancer cells induces TLR9-mediated invasion and inflammation in living cancer cells. Breast Cancer Res Treat 142, 477–487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Phulphagar K et al. Proteomics reveals distinct mechanisms regulating the release of cytokines and alarmins during pyroptosis. Cell Rep 34, 108826 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Russo AJ et al. Intracellular immune sensing promotes inflammation via gasdermin D-driven release of a lectin alarmin. Nat Immunol 22, 154–165 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsuchiya K et al. Gasdermin D mediates the maturation and release of IL-1alpha downstream of inflammasomes. Cell Rep 34, 108887 (2021). [DOI] [PubMed] [Google Scholar]
- 66.Rogers C et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8, 14128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pruenster M et al. E-selectin-mediated rapid NLRP3 inflammasome activation regulates S100A8/S100A9 release from neutrophils via transient gasdermin D pore formation. Nat Immunol 24, 2021–2031 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Lee S et al. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 597, 415–419 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiao H et al. ADAR1 averts fatal type I interferon induction by ZBP1. Nature 607, 776–783 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hubbard NW et al. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature 607, 769–775 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan WT et al. Do pyroptosis, apoptosis, and necroptosis (PANoptosis) exist in cerebral ischemia? Evidence from cell and rodent studies. Neural Regen Res 17, 1761–1768 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malireddi RKS et al. RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons 4, 789–796 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zheng M, Karki R, Vogel P & Kanneganti TD Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell 181, 674–687 e613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Banoth B et al. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J Biol Chem 295, 18276–18283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sharma BR & Kanneganti TD NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol 22, 550–559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu A et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu Y et al. Cryo-electron tomography of NLRP3-activated ASC complexes reveals organelle co-localization. Nat Commun 14, 7246 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baroja-Mazo A et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15, 738–748 (2014). [DOI] [PubMed] [Google Scholar]
- 80.Franklin BS et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15, 727–737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Iyer SS et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 106, 20388–20393 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Halle A et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9, 857–865 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Masters SL et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol 11, 897–904 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Babelova A et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem 284, 24035–24048 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martinon F, Petrilli V, Mayor A, Tardivel A & Tschopp J Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Duewell P et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wen H et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12, 408–415 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang H et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol 191, 2665–2679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shimada K et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cabral A et al. Differential Binding of NLRP3 to non-oxidized and Ox-mtDNA mediates NLRP3 Inflammasome Activation. Commun Biol 6, 578 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhong Z et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kanneganti TD et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Kanneganti TD et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 281, 36560–36568 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Su M et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat Cardiovasc Res 1, 732–747 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ghiringhelli F et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15, 1170–1178 (2009). [DOI] [PubMed] [Google Scholar]
- 96.Dalbeth N et al. Gout. Nat Rev Dis Primers 5, 69 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Rashidi M et al. The Pyroptotic Cell Death Effector Gasdermin D Is Activated by Gout-Associated Uric Acid Crystals but Is Dispensable for Cell Death and IL-1beta Release. J Immunol 203, 736–748 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhong CS et al. Gout-associated monosodium urate crystal-induced necrosis is independent of NLRP3 activity but can be suppressed by combined inhibitors for multiple signaling pathways. Acta Pharmacol Sin 43, 1324–1336 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kluck V et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol 2, e270–e280 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Edgeworth J, Gorman M, Bennett R, Freemont P & Hogg N Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem 266, 7706–7713 (1991). [PubMed] [Google Scholar]
- 101.van Zoelen MA et al. Expression and role of myeloid-related protein-14 in clinical and experimental sepsis. Am J Respir Crit Care Med 180, 1098–1106 (2009). [DOI] [PubMed] [Google Scholar]
- 102.Rathinam VA et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11, 395–402 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fernandes-Alnemri T et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11, 385–393 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karki R et al. Concerted activation of the AIM2 and NLRP3 inflammasomes orchestrates host protection against Aspergillus infection. Cell Host Microbe 17, 357–368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dombrowski Y et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci Transl Med 3, 82ra38 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Naik S et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hu B et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Man SM et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 162, 45–58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wilson JE et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med 21, 906–913 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schattgen SA, Gao G, Kurt-Jones EA & Fitzgerald KA Cutting Edge: DNA in the Lung Microenvironment during Influenza Virus Infection Tempers Inflammation by Engaging the DNA Sensor AIM2. J Immunol 196, 29–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang H et al. AIM2 Inflammasome Is Critical for Influenza-Induced Lung Injury and Mortality. J Immunol 198, 4383–4393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moriyama M et al. Influenza Virus-Induced Oxidized DNA Activates Inflammasomes. iScience 23, 101270 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lian Q et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res 27, 784–800 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Komada T et al. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J Am Soc Nephrol 29, 1165–1181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karki R et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci Immunol 7, eabo6294 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Karki R et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep 37, 109858 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sharma BR, Karki R, Rajesh Y & Kanneganti TD Immune regulator IRF1 contributes to ZBP1-, AIM2-, RIPK1-, and NLRP12-PANoptosome activation and inflammatory cell death (PANoptosis). J Biol Chem 299, 105141 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Malireddi RKS et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med 217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Malireddi RKS et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med 215, 1023–1034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Karki R et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI insight 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Malireddi RKS et al. Inflammatory Cell Death, PANoptosis, Mediated by Cytokines in Diverse Cancer Lineages Inhibits Tumor Growth. Immunohorizons 5, 568–580 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Geijtenbeek TB & Gringhuis SI C-type lectin receptors in the control of T helper cell differentiation. Nat Rev Immunol 16, 433–448 (2016). [DOI] [PubMed] [Google Scholar]
- 123.Chiba S et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife 3, e04177 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Daley D et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med 23, 556–567 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yamasaki S et al. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol 9, 1179–1188 (2008). [DOI] [PubMed] [Google Scholar]
- 126.Nagata M et al. Intracellular metabolite beta-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proceedings of the National Academy of Sciences of the United States of America 114, E3285–E3294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Memon RA et al. Regulation of glycosphingolipid metabolism in liver during the acute phase response. J Biol Chem 274, 19707–19713 (1999). [DOI] [PubMed] [Google Scholar]
- 128.Brennan PJ et al. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol 12, 1202–1211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stephens M, Keane K, Roizes S, Liao S & Weid PV Mincle-binding DNA aptamer demonstrates therapeutic potential in a model of inflammatory bowel disease. Mol Ther Nucleic Acids 28, 935–947 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Seifert L et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 532, 245–249 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Colonna M, Samaridis J & Angman L Molecular characterization of two novel C-type lectin-like receptors, one of which is selectively expressed in human dendritic cells. Eur J Immunol 30, 697–704 (2000). [DOI] [PubMed] [Google Scholar]
- 132.Sobanov Y et al. A novel cluster of lectin-like receptor genes expressed in monocytic, dendritic and endothelial cells maps close to the NK receptor genes in the human NK gene complex. Eur J Immunol 31, 3493–3503 (2001). [DOI] [PubMed] [Google Scholar]
- 133.Thebault P et al. The C-type lectin-like receptor CLEC-1, expressed by myeloid cells and endothelial cells, is up-regulated by immunoregulatory mediators and moderates T cell activation. J Immunol 183, 3099–3108 (2009). [DOI] [PubMed] [Google Scholar]
- 134.Gao S et al. Histidine-Rich Glycoprotein Inhibits High-Mobility Group Box-1-Mediated Pathways in Vascular Endothelial Cells through CLEC-1A. iScience 23, 101180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Drouin M et al. CLEC-1 is a death sensor that limits antigen cross-presentation by dendritic cells and represents a target for cancer immunotherapy. Sci Adv 8, eabo7621 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Takahashi Y et al. Histidine-Rich Glycoprotein Stimulates Human Neutrophil Phagocytosis and Prolongs Survival through CLEC1A. J Immunol 206, 737–750 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lai JJ, Cruz FM & Rock KL Immune Sensing of Cell Death through Recognition of Histone Sequences by C-Type Lectin-Receptor-2d Causes Inflammation and Tissue Injury. Immunity 52, 123–135 e126 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Marsman G, Zeerleder S & Luken BM Extracellular histones, cell-free DNA, or nucleosomes: differences in immunostimulation. Cell Death Dis 7, e2518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sancho D et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458, 899–903 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ahrens S et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36, 635–645 (2012). [DOI] [PubMed] [Google Scholar]
- 141.Zhang JG et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity 36, 646–657 (2012). [DOI] [PubMed] [Google Scholar]
- 142.Hanc P et al. Structure of the Complex of F-Actin and DNGR-1, a C-Type Lectin Receptor Involved in Dendritic Cell Cross-Presentation of Dead Cell-Associated Antigens. Immunity 42, 839–849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schulz O et al. Myosin II Synergizes with F-Actin to Promote DNGR-1-Dependent Cross-Presentation of Dead Cell-Associated Antigens. Cell Rep 24, 419–428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zelenay S et al. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J Clin Invest 122, 1615–1627 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Canton J et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat Immunol 22, 140–153 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Del Fresno C et al. DNGR-1 in dendritic cells limits tissue damage by dampening neutrophil recruitment. Science (New York, N.Y.) 362, 351–356 (2018). [DOI] [PubMed] [Google Scholar]
- 147.Neumann K et al. Clec12a is an inhibitory receptor for uric acid crystals that regulates inflammation in response to cell death. Immunity 40, 389–399 (2014). [DOI] [PubMed] [Google Scholar]
- 148.Li K et al. The uric acid crystal receptor Clec12A potentiates type I interferon responses. Proceedings of the National Academy of Sciences of the United States of America 116, 18544–18549 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sun L, Wu J, Du F, Chen X & Chen ZJ Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wu J et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (New York, N.Y.) 339, 826–830 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kawane K et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443, 998–1002 (2006). [DOI] [PubMed] [Google Scholar]
- 152.Yoshida H, Okabe Y, Kawane K, Fukuyama H & Nagata S Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol 6, 49–56 (2005). [DOI] [PubMed] [Google Scholar]
- 153.Kawane K et al. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol 4, 138–144 (2003). [DOI] [PubMed] [Google Scholar]
- 154.Okabe Y, Kawane K, Akira S, Taniguchi T & Nagata S Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med 202, 1333–1339 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kawane K et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science (New York, N.Y.) 292, 1546–1549 (2001). [DOI] [PubMed] [Google Scholar]
- 156.Lan YY, Londono D, Bouley R, Rooney MS & Hacohen N Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep 9, 180–192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ahn J, Gutman D, Saijo S & Barber GN STING manifests self DNA-dependent inflammatory disease. Proceedings of the National Academy of Sciences of the United States of America 109, 19386–19391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gao D et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proceedings of the National Academy of Sciences of the United States of America 112, E5699–5705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Li T et al. TBK1 recruitment to STING mediates autoinflammatory arthritis caused by defective DNA clearance. J Exp Med 219 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Baum R et al. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II-deficient mice. J Immunol 194, 873–877 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jakobs C, Perner S & Hornung V AIM2 Drives Joint Inflammation in a Self-DNA Triggered Model of Chronic Polyarthritis. PLoS One 10, e0131702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Deng L et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Klarquist J et al. STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol 193, 6124–6134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhu Q et al. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. J Immunol 193, 4779–4782 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Woo SR et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ahn J, Xia T, Rabasa Capote A, Betancourt D & Barber GN Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 33, 862–873 e865 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xu MM et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity 47, 363–373 e365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Marcus A et al. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763 e754 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Carozza JA et al. Extracellular cGAMP is a cancer cell-produced immunotransmitter involved in radiation-induced anti-cancer immunity. Nat Cancer 1, 184–196 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Roers A, Hiller B & Hornung V Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 44, 739–754 (2016). [DOI] [PubMed] [Google Scholar]
- 171.Dhir A et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560, 238–242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pestal K et al. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 43, 933–944 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liddicoat BJ et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chung H et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 172, 811–824 e814 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ishizuka JJ et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Stok JE et al. RNA sensing via the RIG-I-like receptor LGP2 is essential for the induction of a type I IFN response in ADAR1 deficiency. EMBO J 41, e109760 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tosar JP, Witwer K & Cayota A Revisiting Extracellular RNA Release, Processing, and Function. Trends Biochem Sci 46, 438–445 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Masouris I et al. Inhibition of DAMP signaling as an effective adjunctive treatment strategy in pneumococcal meningitis. J Neuroinflammation 14, 214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Liu K et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J 21, 3904–3916 (2007). [DOI] [PubMed] [Google Scholar]
- 180.Fujita K et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci Rep 6, 31895 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]