

Abstract
The cytoplasmic tail of the measles virus (MV) fusion (F) protein is often altered in viruses which spread through the brain of patients suffering from subacute sclerosing panencephalitis (SSPE). We transferred the coding regions of F tails from SSPE viruses in an MV genomic cDNA. Similarly, we constructed and transferred mutated tail-encoding regions of the other viral glycoprotein hemagglutinin (H) gene. From the mutated genomic cDNAs, we achieved rescue of viruses that harbor different alterations of the F tail, deletions in the membrane-distal half of the H tail, and combinations of these mutations. Viruses with alterations in any of the tails spread rapidly through the monolayer via enhanced cell-cell fusion. Double-tail mutants had even higher fusion competence but slightly decreased infectivity. Analysis of the protein composition of released mutant viral particles indicated that the tails are necessary for accurate virus envelope assembly and suggested a direct F tail-matrix (M) protein interaction. Since even tail-altered glycoproteins colocalized with M protein in intracellular patches, additional interactions may exist. We conclude that in MV infections, including SSPE, the glycoprotein tails are involved not only in virus envelope assembly but also in the control of virus-induced cell fusion.
The surface of enveloped viruses is covered with protrusions, or spikes, composed of multimeric glycoproteins which bind to cellular receptors and are instrumental in fusing the viral membrane with the cellular membrane. The spike glycoproteins are connected to the internal virion proteins through their cytoplasmic tails, and interactions between the tails and the internal virion components trigger the formation of budding particles of many viruses (20, 40, 42). Nevertheless, for some viruses, tailless mutants can be produced (15, 16, 23, 25).
Several enveloped viruses propagate both by virus release and reinfection and by cell-cell fusion. In a few retroviruses, including human immunodeficiency virus, the glycoprotein tails regulate the cell-cell fusion process (12, 14, 33, 51, 53). For the propagation and pathogenicity of the paramyxovirus measles virus (MV), virus-induced cell-cell fusion is also a prominent property. In the brains of patients who died from the MV-induced syndrome subacute sclerosing parencephalitis (SSPE), MV propagates apparently without budding, probably by cell-cell fusion (reviewed in reference 1). In fact, MV may spread mainly in a cell-associated form in human tissues. Even tissue culture-adapted MV remains about 90% cell associated (47); therefore, virus stocks are usually prepared from cell extracts.
The MV envelope protein complex, which is instrumental both for virus attachment and cell fusion, is composed of two integral membrane proteins, the hemagglutinin (H) and fusion (F) proteins, and a membrane-associated protein, the matrix (M) protein (49). The type II transmembrane protein H mediates virus-cell attachment by binding to the cell surface protein CD46 (11, 29) and is an essential cofactor for fusion (9, 50). The H protein transmembrane segment is preceded by a 34-amino-acid cytoplasmic tail.
The type I transmembrane protein F is synthesized as an inactive precursor, F0, which is cleaved to generate the disulfide-linked F2 and F1 subunits. Cleavage of the F0 precursor occurs near a hydrophobic region, the so-called fusion peptide. After virus release, possibly following the contact of viral particles with the MV receptor (3), this peptide may insert in the target membrane, initiating virus-cell fusion. Another type of MV-induced membrane fusion is fusion of MV-infected cells with neighboring cells, resulting in the formation of syncytia. The F1 subunit is anchored in the membrane through a hydrophobic segment which is followed by a 33-amino-acid cytoplasmic tail.
The M protein lines the inner surface of the membrane and may play a key role in assembly by concentrating the F and H proteins as well as the ribonucleocapsid, the viral replicative unit, at sites of virus assembly (8, 31).
MV is a strongly cell-associated virus. Released infectious particles are pleomorphic and have low infectivity (21, 24, 28). Current models of MV assembly and release rely mainly on the study of related paramyxoviruses (26, 31). Biochemical and microscopic investigations of Sendai virus (SeV) suggested that the tails of both glycoproteins enhance M protein-membrane association (35, 36). However, part of these findings could not be reproduced (45).
Direct biochemical analysis of the MV M protein-glycoprotein tail interaction is difficult because the M protein strongly associates with cellular membranes even in the absence of the glycoproteins (7), as for the M proteins of Sendai and influenza viruses (45, 52). In MV, an F tail-M protein interaction was postulated on the basis of observations made on assembly-defective viruses replicating in the brains of SSPE patients. In all SSPE-derived F genes analyzed so far, mutations in the regions coding for the cytoplasmic domain were detected; the corresponding F tails were shortened, elongated, or strongly altered (37). Conversely, M protein expression was frequently strongly impaired or abolished (1, 10).
We have used a novel genetic approach (32) to analyze the function of the cytoplasmic tails of the MV glycoproteins. We rescued nine MV mutants with altered glycoprotein tails and characterized their mode of propagation and intracellular assembly and the protein composition of released virus particles.
MATERIALS AND METHODS
Antibodies.
The following antibodies were used: anti-MV goat polyclonal serum (courtesy of S. Udem), anti-Fcyt rabbit anti-peptide serum (13), anti-F rabbit polyclonal serum (courtesy of T. Varsanyi), anti-Hcyt rabbit anti-peptide serum (3), anti-H rabbit polyclonal serum (courtesy of C. Örvell), anti-H mouse monoclonal ascites (39), and anti-M mouse monoclonal ascites (39). The antibody directed against the F cytoplasmic tail (Fcyt) was raised against a peptide corresponding to the 14 carboxyl-terminal amino acids of this protein, NH2-(C)PDLTGTSKSYVRSL-COOH. The H antibody (Hcyt) was raised against a peptide corresponding to the 14 H protein amino-terminal residues, NH2-SPQRDRINAFYKDN(C)-COOH. A cysteine (C) was attached to the peptide termini to allow coupling to keyhole limpet hemocyanin for rabbit immunization.
Plasmid constructs.
Plasmids peFHL and peH5 (Fig. 1A) are shuttle vectors for subcloning in the full-length genomic plasmid p(+)MV (32). Plasmid peH5 contains a single ClaI site in the 5′ untranslated region of the H protein and a single EcoRI site at the border between the transmembrane domain and the ectodomain. These sites were previously introduced by silent point mutations (5).
FIG. 1.

Structure of the MV genome and of the plasmids used for virus rescue, and amino acid sequence of the cytoplasmic tails of the MV glycoproteins. (A) Structure of p(+)MV and of the plasmid intermediates used in the construction of mutant viruses. Plasmid p(+)MV encoding the MV Edmonston B strain antisense genome is shown at the top (32). Solid boxes represent the reading frames of the six MV cistrons. The T7 promoter and relevant restriction sites are indicated. The structures of the plasmids used for subcloning are shown below. Shaded boxes represent gene segments coding for the altered F and H tails. The nucleotide numbering is that used for EMBL accession no. Z66517. (B) Predicted amino acid sequence of the standard (std) and altered MV glycoprotein tails. Boldface letters represent residues conserved within five members of the morbillivirus genus. Residues in the SSPE-derived F protein tails differing from that of the Edmonston strain are indicated by lowercase letters. TM, transmembrane; aa, amino acids.
The H tail deletion plasmids peHcΔ8, peHcΔ14, and peHcΔ24 were constructed by subcloning a ClaI-EcoRI PCR fragment into peH5 (Fig. 1A, bottom). PCR was performed with peH5 DNA as the template; forward primers 5′-CCATCGATAATGGCCTTCTACAAAGATAACC, 5′-CCATCGATAATGAGCCATCCCAAGGGAAGTAGG, and 5′-CCATCGATAATGAACAGAGAACATCTTATGATT (restriction sites in italics), respectively; and a reverse primer annealing downstream from the region encoding the H transmembrane domain.
To construct plasmid peHcSeV, fusion PCR was performed. The SeV HN tail-encoding region was amplified on the pGem4-SVHN (48) template with primers 5′-CCATCGATAATCATGGATGGTGATAGGGG and 5′-GCAAAACATAAGGGGTGTCAACTTTACTTGA. Primer 5′-GACACCCCTTATGTTTTGCTGGC and a primer annealing downstream of the region coding for the H transmembrane domain were used to amplify the MV H transmembrane encoding region. In the fusion step, the isolated PCR fragments with an overlapping sequence of 19 nucleotides (underlined) were mixed and amplified with the external primers, also used for the first amplification round. The resulting fragment was digested with ClaI and EcoRI and then subcloned into peH5.
Plasmids peFHLP, peFHLF, and peFHLI were described previously (37). peF(cSeV)HL was generated by subcloning a PstI-PacI PCR fragment encoding the SeV F cytoplasmic tail into peFHL. PCR was performed with pGem4-SVF0 (48) as the template and primers 5′-AAAACTGCAGACTCAAAAGGTCAATGC and 5′-CCCTTAATTAATATACAGATCTCAACGGAT (restriction sites in italics).
Plasmids p(+)MV-HcΔ8, p(+)MV-HcΔ14, p(+)MV-HcΔ24, and p(+)MV-HcSeV, containing the full-length antigenomic copy of the H-tail mutant MV genomes, were constructed by three-way ligations of a PacI-SpeI fragment containing the mutated H gene from plasmid peHcΔ8, peHcΔ14, peHcΔ24, or peHcSeV, respectively, with a SpeI-SacII fragment and a SacII-PacI fragment of p(+)MV.
Plasmids p(+)MV-Fc+28, p(+)MV-FcΔ16, p(+)MV-FcΔ24, and p(+)MV-FcSeV coding for the F tail mutant MV genomes were constructed by three-way ligations of a NarI-PacI fragment carrying the mutated F gene coding region from plasmid peFHLP, peFHLF, peFHLI, or peF(cSeV)HL, respectively, with a PacI-SacII fragment and a SacII-NarI fragment of p(+)MV.
Plasmids p(+)MV-HcΔ14/Fc+28, p(+)MV-HcΔ14/FcΔ24, and p(+)MV-HcΔ14/FcSeV, containing the MV tail double-mutant genomes were constructed by ligation of a PacI-SacII fragment of p(+)MV-HcΔ14 with the corresponding SacII-PacI fragment of p(+)MV-Fc+28, p(+)MV-FcΔ24, and p(+)MV-FcSeV, respectively.
All generated full-length plasmids coding for a mutated antigenomic copy of the MV genome conform to the rule-of-six (17). The sequences were confirmed by dideoxynucleotide sequencing, and the nucleotide numbering is that used for EMBL accession no. Z66517.
Cells, viruses, and virus assays.
Cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal calf serum (DMEM-5) for Vero (African green monkey kidney) cells, 10% fetal calf serum (DMEM-10) for HeLa T4 (human cervical carcinoma) and 293 (human embryonic kidney) cells, or DMEM-10 containing 1 mg of G418 per ml for the helper cell line 293-3-46.
The Edmonston B strain based standard MV and all mutant derivatives were rescued, propagated, and purified basically as described previously (32). Propagation and purification of the recombinant vaccinia virus expressing the T7 RNA polymerase (vTF7-3) were performed as described previously (6). The biological activities of the viruses were determined by a plaque assays as described previously (32) or by 50% end-point dilution assays performed as follows. Serial dilutions (10-fold) of virus samples were made in DMEM-5, and 50 μl of each dilution was used to infect eight replica 6-mm wells containing 7,500 Vero cells in 50 μl of DMEM-5 overnight at 37°C. The next day, 150 μl of DMEM-5 was added, and 4 days postinfection, the wells were stained with 0.1% crystal violet in 20% ethanol. Virus-induced cytopathic effects were scored, and the log10 50% tissue culture infective dose (TCID50) was calculated by the method of Kärber (18).
Fusion assays.
Cell fusion assays were performed basically as described previously (6). Briefly, HeLa T4 cells were infected with vTF7-3 at a multiplicity of infection (MOI) of 5 and subsequently colipofected with 3.5 μg of an H protein-expressing plasmid and with 1.5 μg of an F plasmid. Syncytium formation was quantified 20 h posttransfection.
Metabolic labelling, immunoprecipitation, and endo H digestion.
Vero cells (106 cells) in 35-mm wells were infected with standard MV or MV mutants at an MOI of 0.5 for 2 h at 37°C. At 16 to 20 h postinfection, the cells were starved for 30 min in Met- and Cys-free DMEM, pulsed for 1 h in the presence of 100 μCi of Tran35S-label (ICN), and chased for the indicated time with DMEM-10. Alternatively, the same number of HeLa T4 cells were infected with vTF7-3 and lipofected as described above.
Immunoprecipitation with a polyclonal goat anti-MV antibody was performed as described previously (9). For endo-β-N-acetylglucosaminidase H (endo H) analysis, immunoprecipitated samples were adjusted to 75 mM sodium citrate (pH 5.6) and incubated in the presence or absence of 1 mU of endo H (Boehringer Mannheim) for 4 h at 37°C.
Immunoblotting.
Vero cells infected with MV mutants as described above were harvested 20 h postinfection in lysis buffer (50 mM Tris [pH 8.0], 62.5 mM EDTA, 1% Nonidet P-40, 0.4% deoxycholate). Samples from equivalent numbers of cells were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the separated proteins were transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked with 1% bovine serum albumin–1% skim milk powder in TBST (10 mM Tris [pH 8.0], 150 mM NaCl, 0.05% Tween 20) for 2 h at room temperature and then incubated with different specific rabbit anti-MV protein antibodies (anti-Fcyt at 1:4,000 or anti-Hcyt at 1:3,000) overnight at 4°C. The proteins were visualized after incubation with horseradish peroxidase-conjugated swine anti-rabbit immunoglobulin G (IgG) (1:3,000; Dakopatts) for 1 h at room temperature by using the enhanced chemiluminescence (Amersham) system.
FACScan analysis.
HeLa T4 cells (5 × 105 cells) were infected with vTF7-3 and lipofected with H-expressing plasmids as described above. H protein surface expression was determined 24 h posttransfection. Cells in DMEM-10 were incubated in a total volume of 50 μl with a 1:100 dilution of a monoclonal anti-H antibody for 1 h at 4°C, washed, and incubated with a 1:50 dilution of R-phycoerythrin-conjugated rabbit anti-mouse Ig (Serotec) for 30 min at room temperature. Flow cytometry was performed after fixation of the cells in phosphate-buffered saline (PBS) containing 1% paraformaldehyde.
One-step growth analysis.
Vero cells (2 × 105 cells) in 15-mm wells were infected with MV mutants at an MOI of 3 for 2 h at 37°C. Inocula were collected to control virus adsorption. The cells were washed twice with PBS, overlaid with 500 μl of DMEM-5, and incubated at 32.5°C to optimize virus release (47). Every 12 h, cell-free virus and cell-associated virus were collected and stored at −30°C. The cell-free samples were prepared by clarifying supernatants of infected cells for 2 min at 8,000 rpm in an Eppendorf model 5415C centrifuge. Cell-associated virus was recovered by scraping infected cells into 500 μl of DMEM-5. The infectivity of the samples was determined by TCID50 measurement.
Isolation of viral particles.
Vero cells (107 cells) were infected at an MOI of 0.1 for 2 h at 37°C. At 20 h postinfection, the cells were starved for 30 min in Met- and Cys-free DMEM. Continuous labelling was performed in the presence of 1 mCi of Tran35S-label in Met- and Cys-free DMEM supplemented with 1/20 volume of DMEM-10 at 32.5°C. For velocity centrifugation, culture supernatants were collected 2 days after infection, clarified, and pelleted in an SW41 rotor at 30,000 rpm for 2 h through 20% sucrose onto a 60% cushion prepared in TNE buffer (10 mM Tris [pH 7.5], 100 mM NaCl, 1 mM EDTA [pH 7.5]). Purified particles were layered on top of a 20 to 60% step gradient and centrifuged for 18 h in an SW41 rotor at 35,000 rpm, and 10 fractions of equal volume were collected. Viral particles were pelleted from diluted fractions in a TLA 45 rotor for 1 h at 45,000 rpm, resuspended in lysis buffer, and directly subjected to SDS-PAGE.
Confocal microscopy.
Vero cells (105 cells) on glass coverslips were infected at an MOI of 0.1 for 3 h at 37°C. After infection, the cells were shifted to 32.5°C. At 50 h postinfection, the cells were permeabilized and fixed for 5 min in methanol at −20°C, blocked in PBSB (0.5% bovine serum albumin in PBS) for 10 min, and then incubated consecutively with the following antibodies diluted in PBS containing 0.5% Triton X-100: monoclonal anti-M (1:200), tetramethylrhodamine-5-isothiocyanate-conjugated goat anti-mouse IgG (Cappel; 1:50), polyclonal serum against H or F, respectively (1:100 each), and donkey anti-rabbit IgG coupled to fluorescein isothiocyanate (Chemicon; 1:100). The coverslips were mounted with the antifadant Citifluor AF1 (Cancer Research Campaign), and immunoreactivity was visualized by confocal microscopy (Bio-Rad MRC-600 scanner) in conjunction with a Zeiss Axiophot fluorescence microscope in 0.5-μm optical sections. Image stacks, confirmed by single excitation to prevent spillover artifacts, were obtained in double-excitation mode and recombined with Imaris and Selima image-processing software (Bitplane AG, Technopark Zurich, Switzerland).
RESULTS
To analyze the function of the envelope protein tails, we truncated, elongated, or substituted them. In the F gene, cDNAs of natural mutants isolated from SSPE patients were available (37), whereas H gene deletion mutants were produced by PCR-based mutagenesis. Also, two mutants in which the SeV glycoprotein tails replaced the homologous MV domains were constructed.
A schematic diagram of the cytoplasmic tail mutants is presented in Fig. 1B. The Fc+28 protein has a tail elongated by 28 amino acids due to a mutation of the stop codon incurred in the brain of SSPE patient P (37). The FcΔ16 and FcΔ24 proteins have premature stop codons due to nonsense mutations in the brains of patients F and I, respectively (10, 37). In the FcSeV protein, we substituted the 33-amino-acid MV F protein tail by the 43-residue SeV F protein tail, which is not sequence related. In addition, three H proteins with tail deletions of different lengths, HcΔ8, HcΔ14, and HcΔ24, were constructed. Finally, in the HcSeV protein, we substituted the 34-residue MV H tail with the 35-amino-acid SeV tail.
Intracellular transport of tail-altered glycoproteins.
The intracellular transport of the altered proteins was characterized in a vaccinia virus T7 RNA polymerase (VV-T7) expression system (9). Transfected cells were pulse-labelled for 1 h with [35S]Met plus [35S]Cys, and the proteins were immunoprecipitated with a polyclonal anti-MV serum after a 3-h chase. As expected, the immunoprecipitated F proteins differed in their mobility (Fig. 2A) and were resolved into several bands. The upper bands correspond to distinct glycosylation stages of the F0 precursor (6, 13). The approximately 40-kDa band represents the nonglycosylated F1 subunit. The observed mobilities correlated well with the predicted size of all the proteins. As observed previously for standard F0 precursors (13), treatment with endo H showed that most oligosaccharides on all F0 precursors had high sensitivity toward this enzyme (Fig. 2A, lanes +), indicating that F0 endo H resistance cannot be used to monitor F protein transport. That the relative transport efficiency of the different F proteins is similar is suggested by the comparable extents of F0 protein cleavage (Fig. 2A).
FIG. 2.
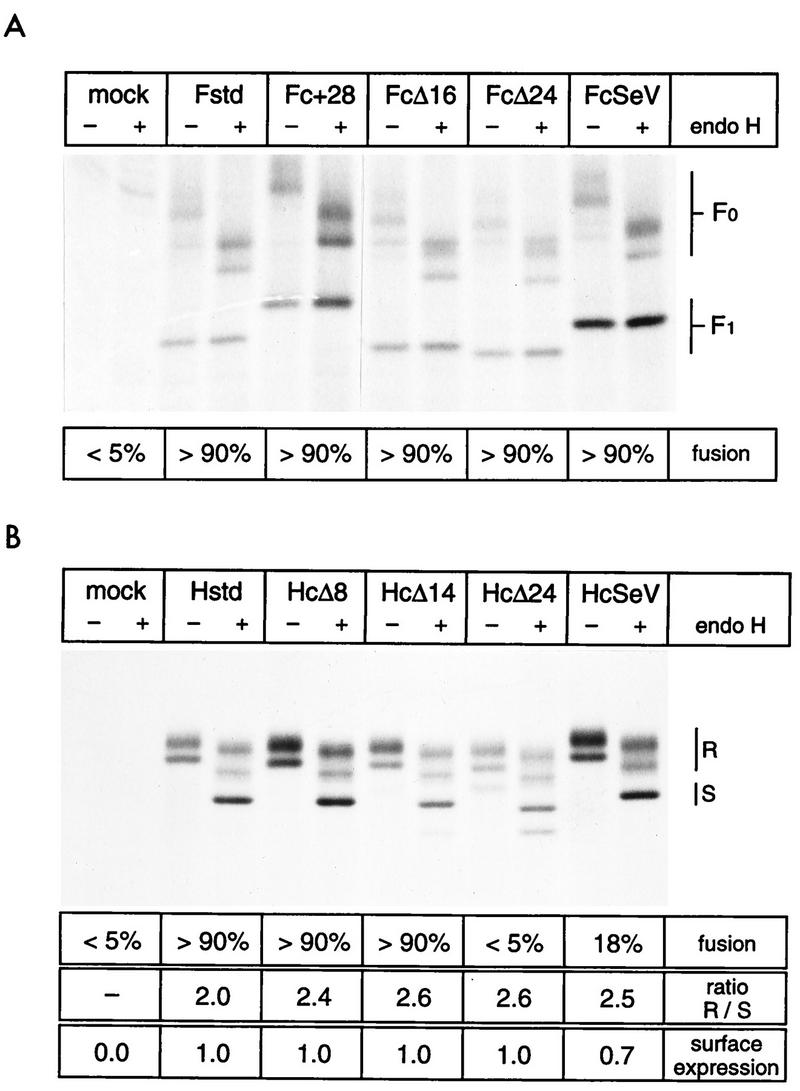
Intracellular transport and fusogenic activity of glycoproteins with altered tails. (A) F proteins with altered tails. HeLa T4 cells were infected with a recombinant vaccinia virus expressing the T7 polymerase and subsequently lipofected with the plasmids as indicated above each lane. At 20 h posttransfection, the cells were metabolically labelled for 1 h, chased for 3 h, and lysed, and clarified supernatants were used for immunoprecipitation with an MV-specific antiserum. Precipitated proteins were digested with (+) or without (−) endo H and then separated by reducing SDS-PAGE (8% polyacrylamide). The positions of F0 and F1 are indicated on the right. Below the gel, fusion activities are indicated as a percentage of the nuclei found in syncytia. The extent of fusion was determined after cotransfection of the indicated plasmid with a plasmid coding for standard H protein by counting the nuclei found in syncytia and in nonfused cells. (B) H proteins with altered tails. The methods are the same as for panel A. R, endo H-resistant and partially resistant forms; S, endo H-sensitive forms. The exact nature of the minor faster-migrating band in the HcΔ24 lanes is not known. At the bottom the percentage of nuclei found in syncytia, the ratio of endo H-resistant to -sensitive forms (R/S) as determined by densitometric analyses of autoradiographs, and the surface expression level of the H proteins as determined by FACScan analysis are indicated. Surface expression was normalized to the standard H protein expression.
For the H protein, the ratio of endo H-resistant to -sensitive molecules can be used to estimate transport (9). This ratio was similar for the standard and all altered H proteins (Fig. 2, bottom). Moreover, the oligomeric state of the altered and standard H proteins was analyzed by nonreducing SDS-PAGE: more than 95% of all H proteins were in the dimeric form after 3 h of chase (data not shown). Thus, intracellular transport was not significantly affected by the H protein tail alterations.
The membrane proximal residues of the H tail are necessary for fusion-helper function.
The function of the altered envelope proteins was tested by a cell-cell fusion assay (9). F protein-mediated membrane fusion of human or primate cells is dependent on coexpression of the H protein (50). Coexpression of altered F proteins with standard H protein induced fusion to a similar extent to that found with the standard F protein (Fig. 2A, bottom), confirming that intracellular transport was maintained and indicating that fusion function was not impaired.
The fusion assay for the H constructs indicated that the fusion-helper efficiency of HcΔ8 and HcΔ14 was comparable to that of standard H protein (more than 90% of the nuclei in syncytia) whereas HcΔ24 and HcSeV manifested either no or a significantly reduced fusion-helper activity, respectively (Fig. 2B, bottom). Lower fusion-helper activity can result either from inefficient fusion-helper function or from decreased levels of cell surface expression. The latter was subjected to FACScan analysis with a monoclonal antibody directed against the H ectodomain. We found that HcΔ24 and HcSeV were expressed at levels approaching 100% or more than 70% of that of the standard H protein, respectively. Therefore, surface expression does not account at all (HcΔ24) or not entirely (HcSeV) for the drop in the fusogenic activity of the proteins. The fact that HcSeV and HcΔ24 but not HcΔ14 lost most of their fusion-helper function indicates that residues proximal to the membrane are crucial for the fusion process.
Viruses with altered glycoprotein tails can be rescued.
We then attempted rescue of MVs that harbor envelope proteins with altered cytoplasmic tails by using a recently developed reverse genetic system (32). We constructed full-length antigenomic cDNAs that carry the same mutations as the T7 expression plasmids described above. We transfected these plasmids or the standard plasmid p(+)MV (Fig. 1A, top) in helper cells. With two exceptions, syncytia were observed 3 to 5 days after transfection, suggesting virus rescue.
Plasmids p(+)MV-HcΔ24 and p(+)MV-HcSeV did not support virus rescue. The most likely explanation for the failure is the reduced fusion-helper activity of the altered proteins HcΔ24 and HcSeV. This may prevent not only efficient cell-cell fusion but also virus-cell fusion and thus impede virus propagation.
To investigate whether viruses with alterations in both envelope protein tails could be rescued, we constructed cDNAs in which the plasmid encoding the shortest H tail (HcΔ14) was combined with plasmids coding for three altered F tails (Fc+28, FcΔ24, or FcSeV). All three plasmids supported virus rescue.
The MV-specific proteins expressed in cells infected with each of the nine rescued mutant viruses were analyzed by immunoprecipitation of pulse-labelled proteins (Fig. 3). Altered F proteins migrated slight differently from their standard counterparts, whereas the difference in the mobility of the H proteins was less pronounced due to the poorer resolution of the gel in this molecular weight range. The reason for the N protein expression imbalance in MV-HcΔ8 is not known.
FIG. 3.

Protein produced by nine rescued mutant viruses in Vero cells, immunoprecipitated by an MV-specific antiserum. Vero cells were infected with the cytoplasmic tail mutants as indicated above the lanes. At 16 h postinfection, the cells were pulsed for 1 h in the presence of Tran35S-label and chased for 4 h. Immunoprecipitated proteins were subjected to SDS-PAGE (8% polyacrylamide). The positions of H, N, F1, and M proteins are indicated on the left.
To definitively discriminate between standard and altered proteins, we analyzed extracts of infected cells by immunoblotting based on sera raised against the terminal regions of the cytoplasmic tails. These sera (Fcyt or Hcyt) were expected to react only with the standard F and H proteins as well as Fc+28. The identity of all the proteins was confirmed. The Fcyt antibody reacted only with F proteins derived from infections with MV-HcΔ8, MV-HcΔ14, MV-Fc+28, MV-HcΔ14/Fc+28, and standard MV, whereas the Hcyt antibody recognized only H proteins from infection with MV-Fc+28, MV-FcΔ16, MV-FcΔ24, MV-FcSeV, and standard MV (data not shown). We also verified by pulse-chase analysis that the intracellular processing of the mutated envelope proteins was comparable to that of the corresponding standard protein and equivalent to that observed in the VV-T7 system (data not shown). In summary, all rescued viruses produced proteins of the expected size and characteristics. No indication for the selection of revertants could be observed. The fact that not even F tail revertants, which may have arisen via single point mutations, were observed suggests that tail-mutated viruses do not have significant selective disadvantages in cultured cells.
Mutant viruses reach high titers.
To gain insight into the mode of propagation of the rescued viruses, we performed a one-step growth analysis (MOI, 3). Figure 4 shows the growth curves of the relevant single and double mutants (without the F and H mutants with the shortest deletions, MV-FcΔ16 and MV-HcΔ8). Cell-free and cell-associated viruses were harvested at 12-h intervals.
FIG. 4.

Time course of cell-associated and cell-free virus production in Vero cells infected with standard and mutant viruses. Vero cells were infected at an MOI of 3, and virus titers were determined by 50% end-point dilution at the indicated time points postinfection (p.i.). The titer of cell-associated infectivity is indicated by solid triangles, and that of released virus is indicated by open triangles. The continuous lines join the average points of two representative experiments. The CPE corresponds to the extent of fusion. +, 5 to 25% of nuclei were found in syncytia; ++, 26 to 50% of nuclei were found in syncytia; +++, 51 to 75% of nuclei were found in syncytia; ++++, ≥76% of nuclei were found in syncytia.
In the standard MV infection, the titers of cell-associated virus reached a plateau 48 h postinfection (2.0 × 106 TCID50/ml). In the medium, a lag phase of about 36 h was followed by a rapid increase in the virus titer, which reached its maximum (1.1 × 106 TCID50/ml) 72 h after infection, when most cells had died. This pattern was common to all mutants, with minor differences. In particular, all mutants reached cell-associated titers between 105 and 106 TCID50/ml, approaching those of standard virus. The titers of released viruses lagged 12 to 24 h behind those of cell-associated viruses and reached lower end values than in standard MV infections. In contrast to other viruses, the F tail exchange mutant MV-FcSeV grew reproducibly slower but reached similar end titers after incubation times of longer than 72 h (data not shown). Interestingly, the double mutant MV-HcΔ14/FcSeV achieved a higher titer than its F tail-altered progenitor MV-FcSeV. This may be explained either by a compensatory effect of the H tail deletion in MV-HcΔ14/FcSeV or by an undefined lesion in the MV-FcSeV genome, which was eliminated when the P-M-F insert was transferred to the HcΔ14 genomic plasmid background.
In summary, these results suggest that the glycoprotein tails may not be required for virus release in Vero cells. However, it must be considered that MV budding is inefficient and that substantial particle release may occur even in standard infections at cell death, without proper budding.
Viruses with altered glycoprotein tails gain cell fusion competence.
We then asked if alteration of the glycoprotein tails had other effects on viral propagation, as may have been expected from the characteristics of SSPE-derived MVs. Indeed, microscopic observation of the morphology of cells infected with the mutants revealed a markedly different series of events from that for standard virus. Figure 5 shows the cytopathic effects (CPE) monitored 12 h (Fig. 5A to E), 36 h (Fig. 5F to J), or 60 h (Fig. 5K to O) after infection with different MVs. Standard virus (Fig. 5B, G, and L) caused nearly no CPE during the first 36 h of infection. Then, within the next 12 h, small syncytia formed, rapidly rounded up, and later detached. Syncytia induced by MV-Fc+28 (Fig. 5C, H, and M) and MV-FcΔ24 (Fig. 4, CPE panel) grew marginally faster than those induced by standard infection, whereas syncytia induced by MV-FcSeV mutant grew slowly, correlating with slower virus growth (not shown).
FIG. 5.
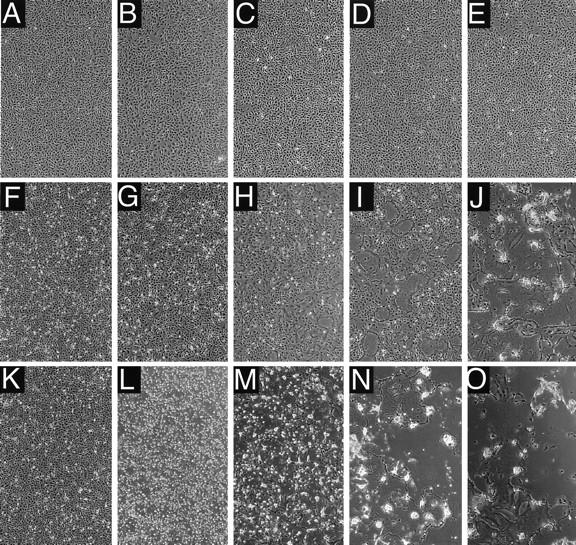
CPEs in Vero cells infected with standard virus or with one of three mutant viruses. Vero cells were infected (MOI, 3) with standard MV (B, G, and L), MV-Fc+28 (C, H, and M), MV-HcΔ14 (D, I, and N), or MV-HcΔ14/Fc+28 (E, J, and O) or left uninfected (A, F, and K). CPEs were monitored by phase-contrast microscopy 12 h (A to E), 36 h (F to J), and 60 h (K to O) after infection.
All the double mutants consistently induced cell fusion more rapidly and more extensively than standard virus (shown for MV-HcΔ14/Fc+28 in Fig. 5E, J, and O and in the CPE panels in Fig. 4). At 36 h postinfection, almost all the cells were fused in very large syncytia (>100 nuclei), which progressively detached and lysed. Viruses with mutations only in the H tail had an intermediate form of CPE, that is, extensive cell fusion but with slower kinetics (MV-HcΔ14 [Fig. 5D, I, and N]). These data indicate that alterations of the glycoprotein tails, especially deletion of the membrane-distal 14 residues of the H tail, result in enhanced cell-to-cell fusion.
Intracellular M-F-H protein colocalization does not require intact glycoprotein tails.
We recently observed (8) that infection of cells with an M protein-deficient MV leads not only to faster cell-to-cell propagation but also to a homogeneous distribution of the viral glycoproteins over the entire cell surface, rather than to their concentration in patches as in MV standard infections. To investigate the effect of alterations of the glycoprotein cytoplasmic tails on viral envelope assembly, the intracellular localization of the envelope proteins was characterized by confocal microscopy.
We compared a standard MV infection (Fig. 6A to C and G to I) with an infection with the shortest viable H protein tail mutant, MV-HcΔ14 (Fig. 6D and J), the F protein tail exchange mutant MV-FcSeV (Fig. 6E and K), and the double mutant MV-HcΔ14/FcSeV (Fig. 6F and L). In a standard infection, the F protein (Fig. 6A), the H protein (Fig. 6G), and the M protein (Fig. 6B and H) were concentrated in bright patches. Superimposition of the two image pairs revealed colocalization of the glycoproteins with M protein (Fig. 6C and I). Internal patches (Fig. 6C) and plasmalemma-proximal patches (Fig. 6C) were observed. Extensive colocalization of the glycoproteins with M protein was observed in cells infected with every mutant virus, including MV-HcΔ14 (Fig. 6D and J), MV-FcSeV (Fig. 6E and K), and the double tail mutant MV-HcΔ14/FcSeV (Fig. 6F and L). This indicates that assembly of the viral envelope can occur even when the glycoproteins have altered tails.
FIG. 6.
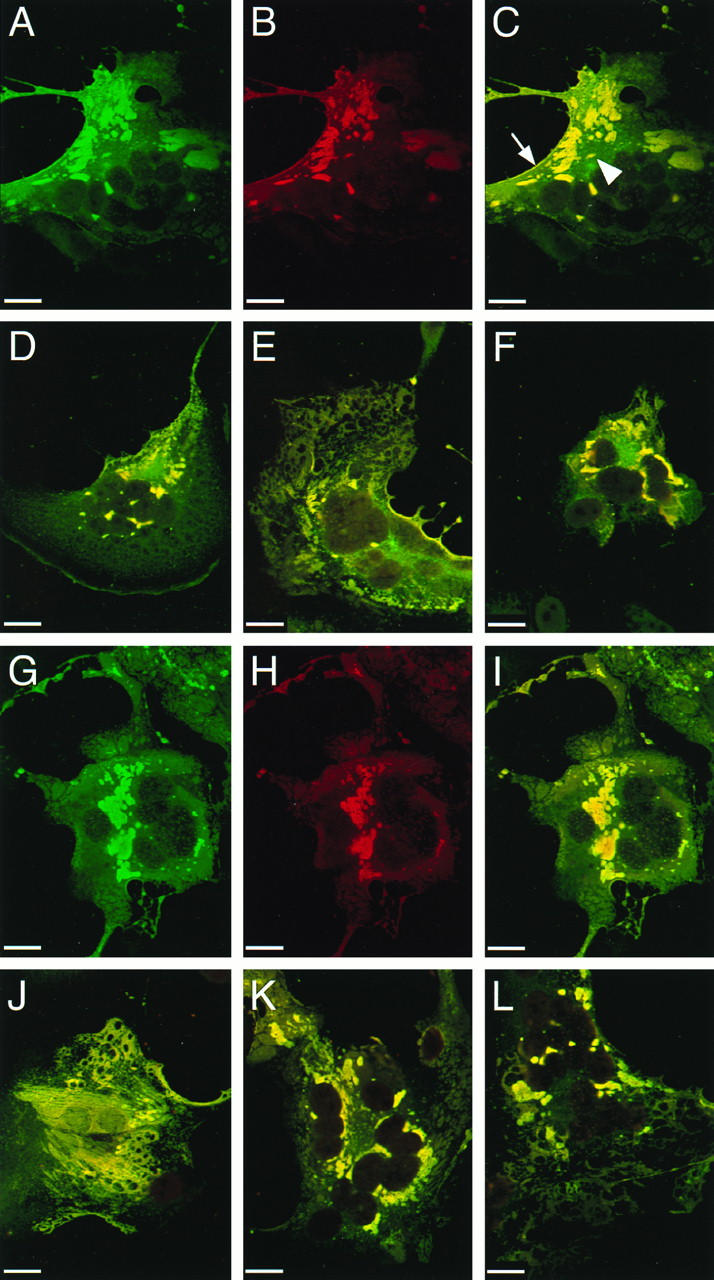
Localization of F, H, and M proteins in Vero cells infected with standard virus or with one of three mutant viruses. Infected Vero cells were permeabilized 50 h after infection and reacted with F, H, or M protein-specific antibodies. Immunoreactivity was monitored by confocal microscopy. Micrographs represent a 0.5-μm optical section of a syncytium induced either by infection with standard MV (A to C and G to I), MV-HcΔ14 (D and J), MV-FcSeV (E and K), or MV-HcΔ14/FcSeV (F and L). Image stacks were obtained in double excitation mode and shown for standard MV infection: F (A) or H protein (G) in green and M protein (B and H) in red. Recombining the two single images revealed colocalization of either F and M protein (C to F) or H and M protein (I to L) in yellow. For the mutant viruses, only the recombined single images are shown. The arrow indicates colocalization at the cell surface, whereas the arrowhead indicates intracellular colocalization of the F and M proteins. Bar, 20 μm.
The tails determine envelope protein incorporation efficiency.
To further characterize the effects of F and H protein tail alterations on virus assembly, we analyzed the composition of released MV particles. To obtain sufficient particle yields, supernatants of infected cells had to be collected late in infection, when cell lysis was already important. It is thus unknown how many of the analyzed particles were virions which completed budding and how many were released from dying cells. Particles were prepared by pelleting onto a sucrose cushion and subsequent purification by equilibrium centrifugation.
Figure 7 shows the protein composition and sucrose gradient migration characteristics of particles from standard MV (Fig. 7A), MV-HcΔ14 (Fig. 7B), MV-FcSeV (Fig. 7C), and MV-HcΔ14/FcSeV (Fig. 7D). In the standard MV infection, the five major structural MV proteins (labelled from the top H, P, N, F1, and H) represented the large majority of the protein detected and were concentrated in fraction 7 and in the two neighboring fractions. A sixth strong band, migrating slightly above F1 (labelled X), may represent cellular actin (47).
FIG. 7.

Protein composition of standard and mutant virus particles. Standard MV (A), MV-HcΔ14 (B), MV-FcSeV (C), and MV-HcΔ14/FcSeV (D) were grown in Vero cells in the presence of Tran35S-label. Supernatant virus was first pelleted by sedimentation onto a 60% sucrose cushion and then purified by equilibrium centrifugation in a 20 to 60% sucrose step gradient. Virus particles from 10 gradient fractions were pelleted, disrupted in lysis buffer, and directly analyzed by SDS-PAGE (8% polyacrylamide). Marker proteins in lanes (a) of panels A to C were immunoprecipitated proteins from a standard MV infection, whereas immunoprecipitated proteins from MV-HcΔ14/FcSeV infected cells were applied in panel D. Lanes (b) show proteins immunoprecipitated from mock-infected cells. The individual fractions are marked at the top (1 to 10 from top to bottom). The positions of the H (or HcΔ14), P, N, X, F1 (or FcSeV1), and M proteins are indicated. X may be cellular actin (47).
In the infections with the tail-altered MV, the viral protein peak was distributed more broadly over fractions 5 to 7, and higher levels of nonviral proteins were detected. Cellular proteins were particularly abundant in particles from MV-FcSeV (Fig. 7C) and from MV-HcΔ14/FcSeV (Fig. 7D).
Moreover, although the viral glycoproteins were produced and transported at similar ratios in infected cells (Fig. 3), differences in the envelope protein composition of the mutant particles were observed. In standard MV particles (Fig. 7A), the N/H/F1/M signal ratio determined by densitometry was approximately 1:1:1:1. In MV-HcΔ14 particles, it was 10:3:5:5 (Fig. 7B), reflecting a slightly less efficient incorporation of all viral envelope proteins, in particular of H. A protein ratio in MV-FcSeV particles was difficult to determine because of the high background of cellular proteins (Fig. 7C), but selective loss not only of F protein but also of M protein was evident. Finally, in MV-HcΔ14/FcSeV the amount of cellular proteins was greatest and that of viral envelope proteins was smallest. We attempted to analyze mutant virus particles by electron microscopy, but the analysis of standard MV particles (43) confirmed their extremely pleomorphic nature (21, 24, 28), rendering a prospective comparative study difficult to interpret. The above data indicate that the glycoprotein tails are important determinants of virus particle assembly and that the F tail may determine the efficiency of M protein incorporation in the envelope.
DISCUSSION
Our results indicate that in MV infections an important function of the glycoprotein tails is to regulate cell fusion. Accurate MV envelope assembly also depends on the glycoprotein tails; the envelopes of tail-altered viruses are adulterated with cellular proteins, but these viruses remain infectious.
Glycoprotein tails and envelope assembly.
We observed that glycoproteins with altered tails are incorporated in the MV envelope less efficiently than are standard glycoproteins and that the envelopes of tail-altered MVs are adulterated with cellular proteins. The presence of the homotypic glycoprotein tails was previously shown to contribute to the specificity of envelope assembly in several viruses (22, 27, 30, 38). It is thus possible that adulteration of the MV envelope is a consequence of the absence of the homologous interactions between the glycoproteins and internal proteins, as demonstrated for a retroviral envelope (46).
All mutant viruses retained high levels of infectivity, indicating that adulteration of their envelope did not cause major problems at cell entry. This may not be so surprising, since even the well-structured vesicular stomatitis virus (VSV) particles can tolerate insertion of foreign glycoproteins (up to 35% of the own envelope glycoprotein weight) without losing much infectivity (19, 38).
An important observation is that the F tail determined not only the efficiency of F protein incorporation into the envelope but also that of the M protein. This may be due to a direct interaction, since it was recently shown that a hybrid VSV glycoprotein with an F tail directed the incorporation of the MV M protein in the envelope of the MV-VSV chimera whereas a standard VSV glycoprotein did not (44).
Since even tail-altered glycoproteins colocalized with M protein in intracellular patches, additional interactions may exist. These interactions could be direct, involving the transmembrane domains of the two glycoproteins, or indirect, involving membrane rafts (41) in which the two viral glycoproteins may be enriched.
Glycoprotein tails and cell fusion.
The F tail and/or the M protein are invariably altered in the brains of SSPE patients. This observation led to the suggestion that impairment of the F tail-M protein interaction may favor MV propagation in brain cells (1). In line with the above suggestion, we demonstrate here that the modification of the F or H glycoprotein tail, respectively, in the background of a standard genome results in increased fusion competence of the mutated viruses. Double tail mutants have the highest fusion competence, which is comparable to that of a rescued M-less virus (8).
Why are MVs with altered F tails, but not those with altered H tails, found in the brains of SSPE patients? The reason lies in the structure of the F and H genes. In the F coding region, single point mutations introducing premature stop codons can lead to the formation of proteins with shorter tails (37). In the H coding region, two combined point mutations (one eliminating the initiator methionine codon and a second introducing a new one) or a deletion would be needed to produce a tail-altered H protein. Thus, MV variants with truncated F tails are likely to appear much more frequently than those with altered H tails.
The enhanced cell fusion of SSPE-derived MV strains and that of the glycoprotein tail mutants produced in this study can be explained by considering a regulatory role of the M protein on fusion function. M may lock the F and H protein oligomers in a fusion-inefficient conformation, which may be necessary to limit fusion of intracellular membranes once the fusion peptide has been liberated. Upon virus release, an external stimulus may result in a conformational change of the M-F-H complex, which then becomes fusion competent.
A fusion-refractory state of the envelope protein complex has been previously postulated based on morphological observations of particles released by another paramyxovirus, Newcastle disease virus La Sota strain. Immature, noninfectious Newcastle disease virus La Sota particles possess a regular arrangement of envelope protein spikes (34) which may reflect the M protein organization at the inner surface of the lipid bilayer (4). It was proposed that the regular arrangement of the spike proteins coincides with a fusion-refractory conformation. Upon maturation this regular arrangement disappears and fusion-competence is established (34).
The cytoplasmic tails of the envelope proteins of certain retroviruses have also been shown to regulate fusion efficiency. In particular, the expression of truncated forms of simian immunodeficiency virus envelope proteins in CD4-positive cells caused enhanced syncytium formation (33, 51, 53). Moreover, removal of a short peptide of the Moloney murine leukemia virus and of a type D retrovirus during viral maturation renders the envelope protein competent for fusion (2, 14). We suggest that regulation of fusion competence via the envelope protein tails is a widespread mechanism, especially relevant for the propagation of certain cell-associated enveloped viruses.
ACKNOWLEDGMENTS
We thank Roland Naef for help with confocal microscopy, Martin A. Billeter for critical reading of the manuscript and continuous support, and Clas Örvell, Tamas Varsanyi, and Erling Norrby for generous gifts of antibodies. Plasmids pGem4-SVF0 and pGem4-SVHN were kind gifts of Laurent Roux.
This work was supported by grants 31-29343.90 (START) and 31-45900.95 from the Swiss National Science Foundation.
REFERENCES
- 1.Billeter M A, Cattaneo R. Molecular biology of defective measles virus persisting in the human central nervous system. In: Kingsbury D W, editor. The paramyxoviruses. New York, N.Y: Plenum Press; 1991. pp. 323–345. [Google Scholar]
- 2.Brody B A, Rhee S S, Sommerfelt M A, Hunter E. A viral protease-mediated cleavage of the transmembrane glycoprotein of Mason-Pfizer monkey virus can be suppressed by mutations within the matrix protein. Proc Natl Acad Sci USA. 1992;89:3443–3447. doi: 10.1073/pnas.89.8.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buchholz C J, Schneider U, Devaux P, Gerlier D, Cattaneo R. Cell entry by measles virus: long hybrid receptors uncouple binding from membrane fusion. J Virol. 1996;70:3716–3723. doi: 10.1128/jvi.70.6.3716-3723.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buechi M, Bächi T. Microscopy of internal structures of Sendai virus associated with the cytoplasmic surface of host membranes. Virology. 1982;120:349–359. doi: 10.1016/0042-6822(82)90036-8. [DOI] [PubMed] [Google Scholar]
- 5.Cathomen, T. 1991. Unpublished data.
- 6.Cathomen T, Buchholz C J, Spielhofer P, Cattaneo R. Preferential initiation at the second AUG of the measles virus F mRNA: a role for the long untranslated region. Virology. 1995;214:628–632. doi: 10.1006/viro.1995.0075. [DOI] [PubMed] [Google Scholar]
- 7.Cathomen, T., and R. Cattaneo. 1995. Unpublished data.
- 8.Cathomen, T., D. Spehner, R. Naef, R. Drillien, M. A. Billeter, and R. Cattaneo. Unpublished data. [DOI] [PMC free article] [PubMed]
- 9.Cattaneo R, Rose J K. Cell fusion by the envelope glycoproteins of persistent measles viruses which caused lethal human brain disease. J Virol. 1993;67:1493–1502. doi: 10.1128/jvi.67.3.1493-1502.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cattaneo R, Schmid A, Eschle D, Baczko K, ter Meulen V, Billeter M A. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 1988;55:255–265. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dörig R E, Marcil A, Chopra A, Richardson C D. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 12.Gabuzda D H, Lever A, Terwilliger E, Sodroski J. Effects of deletions in the cytoplasmic domain on biological functions of human immunodeficiency virus type 1 envelope glycoproteins. J Virol. 1992;66:3306–3315. doi: 10.1128/jvi.66.6.3306-3315.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu A, Cathomen T, Cattaneo R, Norrby E. Influence of N-linked oligosaccharide chains on the processing, cell surface expression and function of the measles virus fusion protein. J Gen Virol. 1995;76:705–710. doi: 10.1099/0022-1317-76-3-705. [DOI] [PubMed] [Google Scholar]
- 14.Januszeski M M, Cannon P M, Chen D, Rozenberg Y, Anderson W F. Functional analysis of the cytoplasmic tail of Moloney murine leukemia virus envelope protein. J Virol. 1997;71:3613–3619. doi: 10.1128/jvi.71.5.3613-3619.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin H, Leser G P, Lamb R A. The influenza virus hemagglutinin cytoplasmic tail is not essential for virus assembly or infectivity. EMBO J. 1994;13:5504–5515. doi: 10.1002/j.1460-2075.1994.tb06885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin H, Leser G P, Zhang J, Lamb R A. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997;16:1236–1247. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaelin K. Ph.D. thesis. Zurich, Switzerland: University of Zurich; 1995. [Google Scholar]
- 18.Kärber G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch Exp Pathol Pharmakol. 1931;162:480–483. [Google Scholar]
- 19.Kretzschmar E, Buonocore L, Schnell M J, Rose J K. High-efficiency incorporation of functional influenza virus glycoprotein into recombinant vesicular stomatitis viruses. J Virol. 1997;71:5982–5989. doi: 10.1128/jvi.71.8.5982-5989.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S, Owen K E, Choi H K, Lee H, Lu G G, Wengler G, Brown D T, Rossmann M G, Kuhn R J. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure. 1996;4:531–541. doi: 10.1016/s0969-2126(96)00059-7. [DOI] [PubMed] [Google Scholar]
- 21.Lund G A, Tyrrell D L, Bradley R D, Scraba D G. The molecular length of measles virus RNA and the structural organization of measles nucleocapsids. J Gen Virol. 1984;65:1535–1542. doi: 10.1099/0022-1317-65-9-1535. [DOI] [PubMed] [Google Scholar]
- 22.Mebatsion T, Conzelmann K K. Specific infection of CD4(+) target cells by recombinant rabies virus pseudotypes carrying the HIV-1 envelope spike protein. Proc Natl Acad Sci USA. 1996;93:11366–11370. doi: 10.1073/pnas.93.21.11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mebatsion T, König M, Conzelmann K K. Budding of rabies virus particles in the absence of the spike glycoprotein. Cell. 1996;84:941–951. doi: 10.1016/s0092-8674(00)81072-7. [DOI] [PubMed] [Google Scholar]
- 24.Miller C A, Raine C S. Heterogeneity of virus particles in measles virus. J Gen Virol. 1979;45:441–453. doi: 10.1099/0022-1317-45-2-441. [DOI] [PubMed] [Google Scholar]
- 25.Mitnaul L J, Castrucci M R, Murti K G, Kawaoka Y. The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication. J Virol. 1996;70:873–879. doi: 10.1128/jvi.70.2.873-879.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison T, Portner A. Structure, function, and intracellular processing of the glycoproteins of paramyxoviridae. In: Kingsbury D W, editor. The paramyxoviruses. New York, N.Y: Plenum Press; 1991. pp. 347–382. [Google Scholar]
- 27.Naim H Y, Roth M G. Basis for selective incorporation of glycoproteins into the influenza virus envelope. J Virol. 1993;67:4831–4841. doi: 10.1128/jvi.67.8.4831-4841.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakai T, Shand F L, Howatson A F. Development of measles virus in vitro. Virology. 1969;38:50–67. doi: 10.1016/0042-6822(69)90127-5. [DOI] [PubMed] [Google Scholar]
- 29.Naniche D, Varior-Krishnan G, Cervoni F, Wild T F, Rossi B, Rabourdin-Combe C, Gerlier D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owens R J, Rose J K. Cytoplasmic domain requirement for incorporation of a foreign envelope protein into vesicular stomatitis virus. J Virol. 1993;67:360–365. doi: 10.1128/jvi.67.1.360-365.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peeples M E. Paramyxovirus M proteins. Pulling it all together and taking it on the road. In: Kingsbury D W, editor. The paramyxoviruses. New York, N.Y: Plenum Press; 1991. pp. 427–456. [Google Scholar]
- 32.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dötsch C, Christiansen G, Billeter M A. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritter G D J, Mulligan M J, Lydy S L, Compans R W. Cell fusion activity of the simian immunodeficiency virus envelope protein is modulated by the intracytoplasmic domain. Virology. 1993;197:255–264. doi: 10.1006/viro.1993.1586. [DOI] [PubMed] [Google Scholar]
- 34.Russell P H, Almeida J D. A regular subunit pattern seen on non-infectious Newcastle disease virus particles. J Gen Virol. 1984;65:1023–1031. doi: 10.1099/0022-1317-65-6-1023. [DOI] [PubMed] [Google Scholar]
- 35.Sanderson C M, McQueen N L, Nayak D P. Sendai virus assembly: M protein binds to viral glycoproteins in transit through the secretory pathway. J Virol. 1993;67:651–663. doi: 10.1128/jvi.67.2.651-663.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanderson C M, Wu H H, Nayak D P. Sendai virus M protein binds independently to either the F or the HN glycoprotein in vivo. J Virol. 1994;68:69–76. doi: 10.1128/jvi.68.1.69-76.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmid A, Spielhofer P, Cattaneo R, Baczko K, ter Meulen V, Billeter M A. Subacute sclerosing panencephalitis is typically characterized by alterations in the fusion protein cytoplasmic domain of the persisting measles virus. Virology. 1992;188:910–915. doi: 10.1016/0042-6822(92)90552-z. [DOI] [PubMed] [Google Scholar]
- 38.Schnell M J, Buonocore L, Kretzschmar E, Johnson E, Rose J K. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc Natl Acad Sci USA. 1996;93:11359–11365. doi: 10.1073/pnas.93.21.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheshberadaran H, Norrby E, McCullough K C, Carpenter W C, Örvell C. The antigenic relationship between measles, canine distemper and rinderpest viruses studied with monoclonal antibodies. J Gen Virol. 1986;67:1381–1392. doi: 10.1099/0022-1317-67-7-1381. [DOI] [PubMed] [Google Scholar]
- 40.Simons K, Garoff H. The budding mechanisms of enveloped animal viruses. J Gen Virol. 1980;50:1–21. doi: 10.1099/0022-1317-50-1-1. [DOI] [PubMed] [Google Scholar]
- 41.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 42.Skoging U, Vihinen M, Nilsson L, Liljestrom P. Aromatic interactions define the binding of the alphavirus spike to its nucleocapsid. Structure. 1996;4:519–529. doi: 10.1016/s0969-2126(96)00058-5. [DOI] [PubMed] [Google Scholar]
- 43.Spehner, D., T. Cathomen, R. Cattaneo, and R. Drillien. 1996. Unpublished data.
- 44.Spielhofer, P., T. Bächi, T. Fehr, G. Christiansen, R. Cattaneo, K. Kaelin, M. A. Billeter, and H. Y. Naim. Chimeric measles viruses with a foreign envelope. J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 45.Stricker R, Mottet G, Roux L. The Sendai virus matrix protein appears to be recruited in the cytoplasm by the viral nucleocapsid to function in viral assembly and budding. J Gen Virol. 1994;75:1031–1042. doi: 10.1099/0022-1317-75-5-1031. [DOI] [PubMed] [Google Scholar]
- 46.Suomalainen M, Garoff H. Incorporation of homologous and heterologous proteins into the envelope of Moloney murine leukemia virus. J Virol. 1994;68:4879–4889. doi: 10.1128/jvi.68.8.4879-4889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Udem S A. Measles virus: conditions for the propagation and purification of infectious virus in high yield. J Virol Methods. 1984;8:123–136. doi: 10.1016/0166-0934(84)90046-6. [DOI] [PubMed] [Google Scholar]
- 48.Vidal S, Mottet G, Kolakofsky D, Roux L. Addition of high-mannose sugars must precede disulfide bond formation for proper folding of Sendai virus glycoproteins. J Virol. 1989;63:892–900. doi: 10.1128/jvi.63.2.892-900.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wild T F, Buckland R. Functional aspects of envelope-associated measles virus proteins. In: ter Meulen V, Billeter M A, editors. Measles virus. Berlin, Germany: Springer-Verlag KG; 1995. pp. 51–64. [DOI] [PubMed] [Google Scholar]
- 50.Wild T F, Malvoisin E, Buckland R. Measles virus: both the haemagglutinin and fusion glycoproteins are required for fusion. J Gen Virol. 1991;72:439–442. doi: 10.1099/0022-1317-72-2-439. [DOI] [PubMed] [Google Scholar]
- 51.Wilk T, Pfeiffer T, Bosch V. Retained in vitro infectivity and cytopathogenicity of HIV-1 despite truncation of the C-terminal tail of the env gene product. Virology. 1992;189:167–177. doi: 10.1016/0042-6822(92)90692-i. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Lamb R A. Characterization of the membrane association of the influenza virus matrix protein in living cells. Virology. 1996;225:255–266. doi: 10.1006/viro.1996.0599. [DOI] [PubMed] [Google Scholar]
- 53.Zingler K, Littman D R. Truncation of the cytoplasmic domain of the simian immunodeficiency virus envelope glycoprotein increases Env incorporation into particles and fusogenicity and infectivity. J Virol. 1993;67:2824–2831. doi: 10.1128/jvi.67.5.2824-2831.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]