

Abstract
Many viruses induce profound changes in cell metabolism and function. Here we show that vaccinia virus induces two distinct forms of cell movement. Virus-induced cell migration was demonstrated by an in vitro wound healing assay in which infected cells migrated independently into the wound area while uninfected cells remained relatively static. Time-lapse microscopy showed that the maximal rate of migration occurred between 9 and 12 h postinfection. Virus-induced cell migration was inhibited by preinactivation of viral particles with trioxsalen and UV light or by the addition of cycloheximide but not by addition of cytosine arabinoside or rifampin. The expression of early viral genes is therefore necessary and sufficient to induce cell migration. Following migration, infected cells developed projections up to 160 μm in length which had growth-cone-like structures and were frequently branched. Time-lapse video microscopy showed that these projections were formed by extension and condensation of lamellipodia from the cell body. Formation of extensions was dependent on late gene expression but not the production of intracellular enveloped (IEV) particles. The requirements for virus-induced cell migration and for the formation of extensions therefore differ from each other and are distinct from the polymerization of actin tails on IEV particles. These data show that poxviruses encode genes which control different aspects of cell motility and thus represent a useful model system to study and dissect cell movement.
Vaccinia virus (VV) is a member of the Poxviridae, a group of large, double-stranded DNA viruses that replicate in the cytoplasm of the host cell (20). Poxvirus infection induces extensive changes to cell metabolism and structure (8) which are collectively termed the viral cytopathic effect (CPE). Morphological changes include early cell rounding (3), formation of cytoplasmic inclusion bodies and vacuoles, changes in the actin cytoskeleton (5, 9, 15), and, with some virus strains, syncytium formation. Although VV encodes proteins that can interact with actin, such as p11 (14) and profilin (17), the role of these proteins in the virus life cycle is not understood, and deletion of the virus profilin gene was reported previously to be without phenotype (5). The molecular basis for virus-induced CPE remains poorly defined.
VV morphogenesis is complex, and two types of infectious virus particle are made (20). Intracellular mature virus (IMV) is formed by envelopment of the viral genome in modified membranes derived from the intermediate compartment (26). Some of these particles are then wrapped with cisternae from early endosomes or the trans-Golgi network (25, 29) to form intracellular enveloped virus (IEV). IEV particles induce actin polymerization to form an actin tail which propels them to the cell surface (9, 10). When IEV particles reach the cell surface, their outer membrane fuses with the plasma membrane to form extracellular enveloped virus (EEV), some of which is retained on the external surface of the cell as cell-associated enveloped virus (6). Actin tail formation is not essential for movement of IEV to the cell surface because a virus mutant lacking the A34R gene produces more EEV (7, 19) but fails to form actin tails (31).
Cell migration plays a crucial role in many normal and pathological processes including embryogenesis, wound healing, inflammation, and metastasis (16). The process of cell migration can be divided into three phases (16), each of which is mediated by a specific member of the rho subfamily of ras-related GTPases (2, 23). The first phase is the outward projection of cellular membranes by formation of filopodia and then lamellipodia. These two events are dependent upon the action of rac and cdc42 proteins, respectively (2). The second phase is the formation of new adhesion sites at the leading edge of the cell. Again, cdc42 and rac proteins are thought to be involved (2). The third phase is dependent upon rho proteins and involves the detachment and retraction of the trailing edge of the cell. A slight modification of the migration cycle is observed in neuronal cells during axon elongation. In this case, rac, cdc42, and rho proteins are involved in growth cone extension, but the cell body remains static as projections develop from specific points of the plasma membrane. For cells growing in confluent monolayers, movement is restricted by cell-cell contacts which must be disrupted for movement to occur. Such disruption occurs during tumor metastasis and is the subject of intensive investigation (12).
In this report, we demonstrate that VV infection induces cell motility and affects all three phases of cell migration. In addition, it induces extensive morphological differentiation involving formation of long cellular projections. Specifically, early VV proteins induce cell-cell dissociation and cell migration, while late viral proteins induce long-branched projections which form by elongation from the cell body. Together, these data demonstrate that VV can control several different aspects of cell motility and thus provides a useful model system to study these processes.
MATERIALS AND METHODS
Cells and viruses.
BS-C-1 cells were grown in minimum essential medium (Gibco) supplemented with 10% fetal bovine serum (FBS). The sources of viruses are as described elsewhere (1). Viruses were grown in BS-C-1 cells, and their infectivity was determined by plaque assay on BS-C-1 cells (18).
Reagents.
Anti-VV sera were obtained from rabbits previously infected with the WR strain of VV. Cytosine β-d-arabinofuranoside (Ara-C), rifampin, cycloheximide, trioxsalen (4,5,8-trimethylpsoralen), cytochalasin B, colchicine, taxol (Paclitaxol; Sigma), phalloidin, and fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit and anti-mouse immunoglobulins were obtained from Sigma (Poole, United Kingdom). FITC-conjugated rabbit anti-rat immunoglobulin G was purchased from Serotec (Oxford, United Kingdom). Mouse monoclonal antibody to human paxillin was obtained from Transduction Laboratories (Lexington, Ky.), and rat monoclonal antibody (YL1/2) to tubulin was obtained from Porton (Salisbury, United Kingdom). N1-Isonicotinoyl-N2-3-methyl-4-chloro-benzoylhydrazine was obtained from Hoechst (Frankfurt, Germany).
Preparation of conditioned medium from VV-infected cells.
Cultures of BS-C-1 cells were infected with VV strain WR at 10 PFU/cell. Culture medium was harvested 3 days after infection and was centrifuged at 3,000 rpm in a Beckman GPR centrifuge for 10 min at 4°C. Virus particles were removed from the resulting supernatant by centrifugation at 16,500 rpm in an SW-41 rotor for 60 min at 4°C. Finally, the clarified medium was treated with trioxsalen-UV light as described below to inactivate any residual virus particles.
Trioxsalen inactivation of VV.
The procedure was adapted from a published method (13). Ten microliters of 0.1-mg/ml trioxsalen in dimethyl sulfoxide was added to 1 ml of virus diluted in minimum essential medium–2.5% FBS. The mixture was irradiated with long-wave UV light for 4 min on ice. The distance between the sample and the UV source was fixed at 4.5 cm. Following inactivation, no infectious particles could be detected by plaque assay.
Microscopy.
Phase-contrast microscopy was performed with an Olympus CK2 inverted microscope. To distinguish infected and uninfected cells, monolayers were washed in phosphate-buffered saline (PBS), fixed in acetone-methanol (1:1) at −20°C for 10 min, and stained with a rabbit anti-VV serum diluted 1/500 in PBS–10% FBS. Following two washes in PBS, cells were stained with an FITC-conjugated goat anti-rabbit immunoglobulin (Sigma) diluted 1/40 in PBS–10% FBS. Fluorescent images were recorded with a Nikon Diaphot 2000 microscope. Time-lapse microscopy was performed manually with an Olympus CK2 microscope. For analysis of the cytoskeleton, cells were infected with VV at 10 PFU/cell and then processed as described elsewhere (2).
In vitro wound healing assay.
Confluent monolayers of BS-C-1 cells were scored in a grid pattern by using a yellow tip to generate wounds devoid of adherent cells and washed twice with PBS before addition of virus. Unless otherwise stated, migration assays were performed at 37°C for 24 h.
Quantification of cell migration and number of cellular extensions.
The number of cells within the wound area was determined after infection with VV at 5 PFU/cell. Separate wound areas (n = 4) were defined by the cell distribution at the start of infection and then photographed every hour for 24 h, and the number of cells within each wound area was counted from projected images. The distance travelled by individual cells was determined by projecting images of cell distribution after each hour of infection and marking the location of the nucleus and the leading edge of particular cells relative to their location at the start of infection. The morphology of individual infected cells was recorded at 2-h intervals, and the number of cell extensions was determined.
RESULTS
Plaques generated by VV infection contain cells with motile features.
Plaques formed by VV infection of BS-C-1 cells were examined 36 h postinfection (hpi) by phase-contrast microscopy (Fig. 1). Within each plaque, there are some cells which appear rounded but also cells with motile features including extended lamellipodia and fine long processes greater than the length of uninfected cells. These observations suggest that VV-induced CPE may be not a simple transition from adherent to rounded and refractile cells but a more complex process involving changes in host cell motility and morphology. This idea was investigated further by an in vitro wound healing assay and time-lapse microscopy.
FIG. 1.
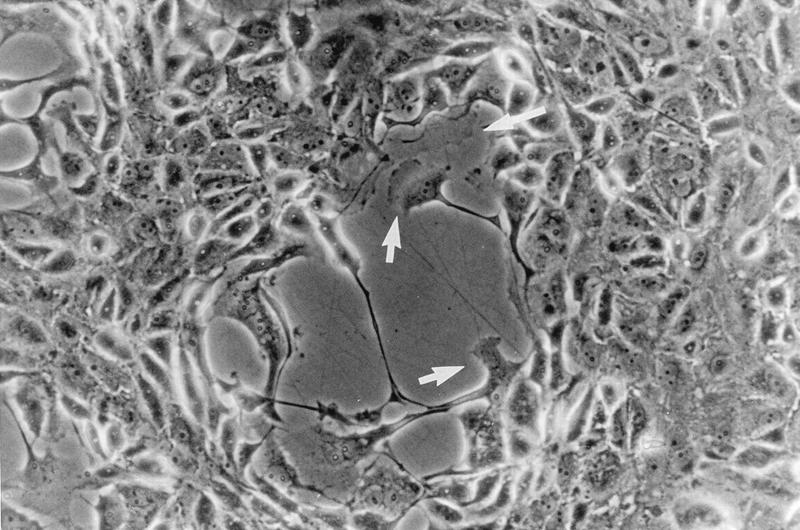
VV-induced plaques contain cells with motile features. Monolayers of BS-C-1 cells were infected with VV strain WR at 0.01 PFU/cell, and the plaque morphology was examined at 36 hpi by phase-contrast microscopy. Arrows indicate cells with a ruffled edge.
VV infection enhances host cell movement.
Wounds were made in BS-C-1 cell monolayers, and the migration of uninfected or infected cells into these wound areas was measured as described in Materials and Methods. After 24 h, the wound size in uninfected cells (Fig. 2B) was marginally smaller than that directly after wound formation (Fig. 2A) due to the cells encroaching slowly as a wave into the wound area. In contrast, in infected cultures individual cells migrated into the wound (Fig. 2C), showing that VV infection enhances independent cell migration. Infection with other VV strains (Copenhagen, Wyeth, Lister, IHD-J, and Tian-Tan) and cowpox virus (strain Brighton Red) also induced cell movement (data not shown). Movement of COS-1 cells into a wound was also observed (data not shown).
FIG. 2.
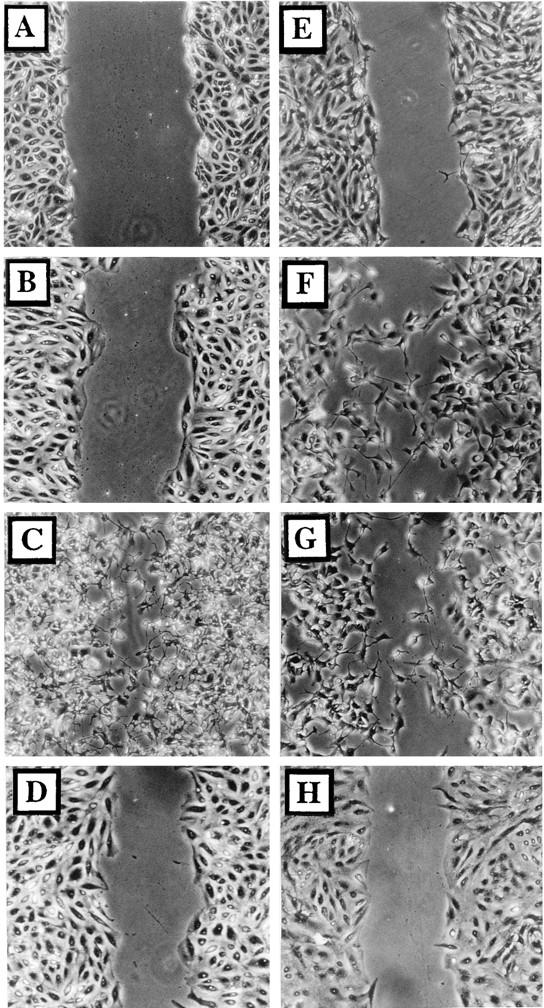
Infected cells become motile. Wounded BS-C-1 monolayers were photographed under phase contrast directly after wounding (A) or 24 h later (B to H). Cells were either mock infected (B), infected with VV at 5 PFU/cell (C, D, F, and G), or infected with VV which had been inactivated with trioxsalen and UV light at 5 PFU/cell (E). Cells shown in panel H were incubated in VV-conditioned medium for 24 h. Ara-C (40 μg/ml) (F), rifampin (100 μg/ml) (G), or cycloheximide (300 μg/ml) (D) was included throughout infection.
Early viral genes are necessary and sufficient to induce cell migration.
Cell migration might have been induced by soluble virus proteins released from infected cells, the binding or entry of virus particles, or the expression of virus proteins within the infected cell. To address these possibilities, cells were infected with VV that had been inactivated by trioxsalen-UV light treatment (Fig. 2E) or with medium from infected cells that had been similarly treated (Fig. 2H). In both cases, cell movement was not induced, suggesting that virus protein synthesis was needed within the infected cell. Consistent with this, infections performed in the presence of 300 μg of cycloheximide per ml induced no cell movement (Fig. 2D), but infection with 40 μg of Ara-C (Fig. 2F), an inhibitor of virus DNA replication and intermediate and late protein synthesis, per ml or 100 μg of rifampin (Fig. 2G), an inhibitor of IMV morphogenesis, per ml each induced cell migration. In addition, a mutant VV which lacked the B5R gene and is defective in IEV formation (11) still induced cell movement (data not shown). Late protein synthesis and virus formation are therefore not required, and early virus proteins are necessary and sufficient to induce cell migration.
To show directly that it was infected and not uninfected cells that moved into the wound, confluent wounded monolayers were infected with 0.05 PFU/cell to ensure a mixed population of infected and uninfected cells along the border of the wound area. At 24 hpi, cells were fixed and stained with anti-VV serum (Fig. 3B and D) to distinguish infected from uninfected cells. Only infected cells migrated into the wound area, while uninfected cells remained relatively static along the wound border (Fig. 3A and C).
FIG. 3.

Only infected cells are motile. Wounded monolayers of BS-C-1 cells were infected with virus strain WR at 0.05 PFU/cell. At 24 hpi, cells were fixed and stained with rabbit anti-VV serum as described in Materials and Methods (B and D). Also shown are phase-contrast images (A and C) of matching epifluorescence photographs (B and D). Arrows indicate infected cells within the wound area.
Kinetics of VV-induced cell migration.
The progression of VV-induced cell migration was measured by time-lapse microscopy as described in Materials and Methods (Fig. 4). Up to 5 hpi, there was no significant change in the profile of the wound area, but by 6 to 9 hpi, the boundaries of the wound area became distorted and many rounded cells were detected within the infected monolayer (data not shown). Between 10 and 12 hpi, lamellipodia were seen extending into the wound area (Fig. 4B), and migration of cells progressed up to 21 hpi (Fig. 4C to E). Quantification of cell movement showed that cells started to enter the wound area slowly from 6 hpi, and their rate of movement increased by 9 hpi, was maximal between 10 and 14 hpi, and slowed between 15 and 24 hpi (data not shown). These data showed that cells enter the wound by migration from the wound boundary and not by reattachment of detached cells.
FIG. 4.

Kinetics of VV-induced cell migration. A wounded monolayer of BS-C-1 cells was infected with VV at 5 PFU/cell and photographed at 9 (A), 12 (B), 15 (C), 18 (D), and 21 (E) hpi.
VV-induced cell projections appear late during infection after migration.
Examination of motile cells late during infection revealed the formation of long cellular processes (examples are seen in Fig. 1 to 4). Similar extensions were seen in other infected cells such as RK13 cells and COS-1 cells (data not shown). To enable individual cells to be better examined and to eliminate the effects of cell-cell contact, cells were seeded at low density and infected while isolated (Fig. 5A to D). The percentage of cells with projections is shown in Fig. 5E. Commonly, there were several projections (often branched) radiating from each cell, resulting in a stellate appearance.
FIG. 5.
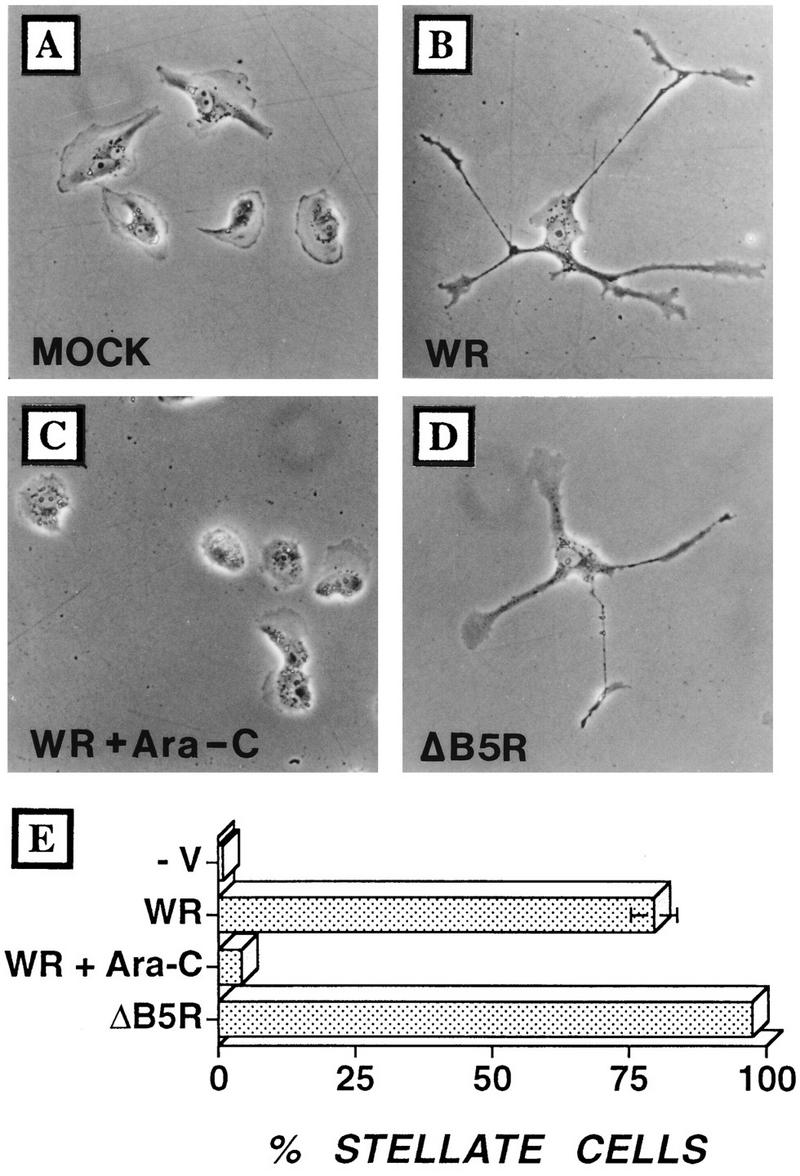
Infected cells develop multiple branched projections. BS-C-1 cells were seeded at low density to obtain isolated cells and then infected with VV at 5 PFU/cell. (A) Mock infection; (B) infection with VV; (C) infection with strain WR plus Ara-C; (D) infection with ΔB5R. Cells were photographed by phase-contrast microscopy at 24 hpi. (E) In different experiments (n = 4), between 100 and 120 cells were analyzed at 24 hpi and the proportion of cells showing three or more projections was calculated. Standard error bars are shown for each sample but are visible only for WR-infected cells.
Cells infected in the presence of Ara-C failed to develop multiple, long processes by 18 hpi (Fig. 5C), indicating a requirement for DNA replication and late protein synthesis. To examine whether virus-induced cell migration and projection formation are distinct from the mechanism of actin polymerization on IEV particles, cells were infected with a B5R deletion mutant virus (ΔB5R), which is severely restricted in IEV formation (11), or with wild-type VV in the presence of N1-isonicotinoyl-N2-3-methyl-4-chloro-benzoylhydrazine, an inhibitor of IEV particle formation. VV mutants lacking p37 (gene F13L) (6) or gp22-24 (gene A34R) (19), which fail to make actin tails (31), were also examined. In the absence of actin tails, both cell migration and projections were induced, suggesting that these are distinct processes (Fig. 5D and data not shown). A VV mutant lacking profilin (gene A42R) (5) also induced both cell migration and projection formation (data not shown).
The kinetics of cell movement and projection formation were compared by recording the position and morphology of three cells following infection (Fig. 6). Migration of cells was measured by recording the position of the leading edge of the cell and the cell nucleus. Migration was initiated by membrane extension between 4 and 10 hpi, and this was followed by movement of the cell body and nucleus from 10 to 15 hpi. From 15 to 24 hpi, movement of cell membranes and nucleus slowed considerably. This difference in the time of virus-induced cell migration and induction of cell processes suggested that they are sequential phases of VV-induced CPE. This was consistent with the requirement for late proteins for projection formation (Fig. 5C) but not cell movement (Fig. 2F). Formation of IEV particles was not required for either process.
FIG. 6.

Kinetics of cell movement and projection formation. BS-C-1 cells were grown and infected with VV as described for Fig. 3. The morphology and location of cells were recorded every 2 h until 24 hpi. (A) Distance moved for the leading edge of the cell (open squares) or the cell nucleus (closed circles) (n = 3). (B) Number of projections per cell (n = 3).
Cell projections are formed by extending lamellipodia.
The formation of projections was examined in more detail by time-lapse video microscopy. Figure 7 shows the rate and sequence of changes in morphology of a single cell between 13 and 16 hpi and is representative of changes seen in other cells. By 13 hpi, this cell already possessed multiple extensions (Fig. 7A); however, two new extensions developed simultaneously during the next 1 h and 45 min with a lamellipodium at the leading edge of each projection (Fig. 7B and C). The extension of one projection (arrow in Fig. 7A) is shown at higher magnification at 27-min intervals starting at 13 hpi (Fig. 7D to K). Initially, the projection was a broad lamellipodium which extended from the cell (Fig. 7E). After 81 min, membrane extension slowed considerably (Fig. 7G) and the structure progressively condensed laterally to form a fine projection (Fig. 7K).
FIG. 7.
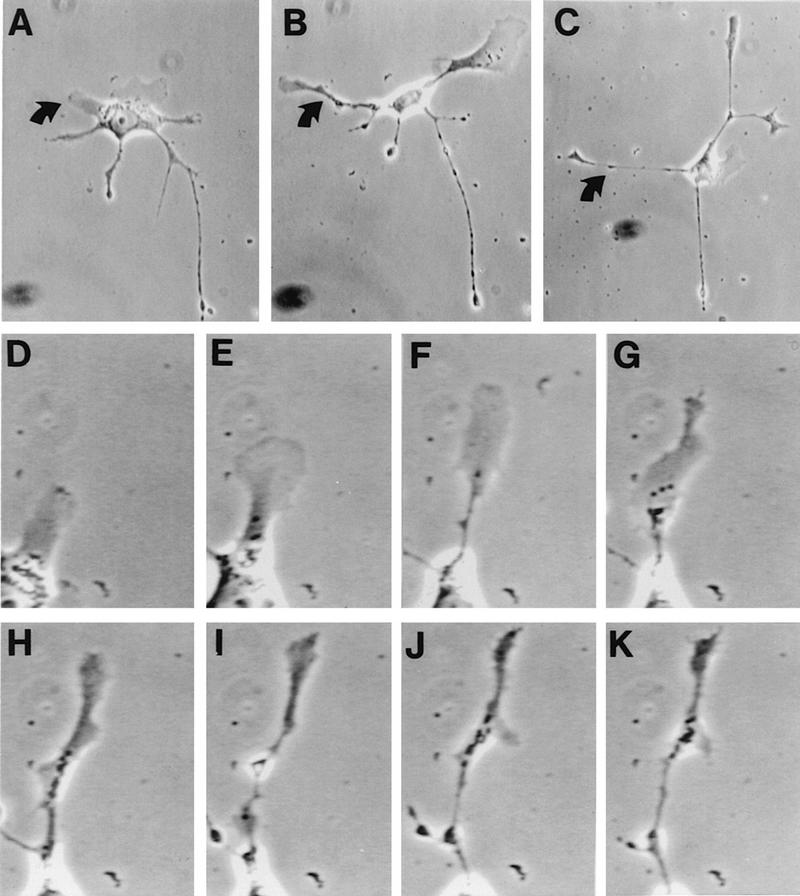
Video microscopy of projection formation. Shown is the formation of virus-induced cell extensions. BS-C-1 cells were grown and infected with VV strain WR as described for Fig. 4. From 13 to 16 hpi, cell morphology was monitored by video microscopy at 3-min intervals. (A to C) Single cell morphology at 13, 13.75, and 14.75 hpi, respectively. (D to I) Formation of the extension marked by arrows in panels A to C. The photograph shown in panel D was taken at 13 h and 9 min postinfection, and each subsequent photograph was taken 27 min after that shown in the previous panel (panel K = 16 h and 9 min postinfection). Magnification, ca. ×135 (A and B), ca. ×106 (C), and ca. ×451 (D to K).
As neuronal extension is dependent upon microtubules and actin filaments (28), we analyzed the distribution of tubulin and F-actin in the growth cones of virus-induced projections. Within growth cones, microtubules projected into the lamellipodium (Fig. 8A) and occasionally terminated at the focal adhesion points (Fig. 8C). F-actin was observed as a peripheral web and as fine filaments associated with paxillin-positive adhesion sites (Fig. 8D to F). Focal adhesion sites were found around the leading edge of growth cones but were not observed along condensed regions of the virus-induced projections (Fig. 8B and E).
FIG. 8.
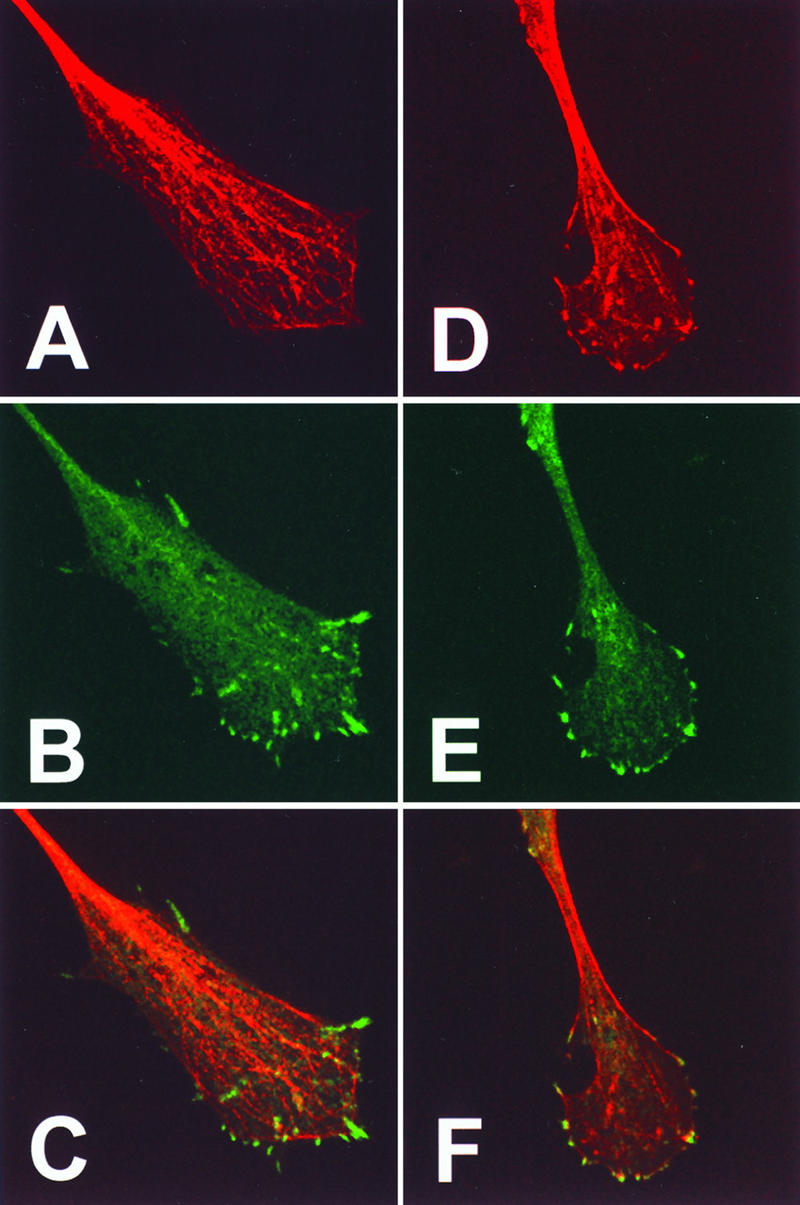
Distribution of microtubules, actin, and paxillin within the growth cone of virus-induced projections. BS-C-1 cells were infected with VV at 10 PFU/cell and at 18 hpi were fixed and processed for immunofluorescence microscopy. Cells were stained with antitubulin (A) or with antipaxillin (B and E). Filamentous actin was visualized with phalloidin (D). (C and F) Merged images of panels A and B or D and E, respectively. Bar in panel F, 10 μm.
To relate previous reports of VV-induced cell rounding to the new observations of cell migration, we performed time-lapse microscopy on populations of isolated infected cells. By 8 hpi, 81% of cells were rounded; however, 10 h later, 85% of cells were flattened and spread (Fig. 9A). This was consistent with the presence of many rounded cells in wound healing assays just before the period of migration (data not shown). Cell rounding is, therefore, a transient phase of VV-induced CPE which precedes cell migration. The involvement of actin- and tubulin-containing filaments in the transition between the different phases of VV-induced CPE was analyzed by addition of 2 μM cytochalasin B, which disrupts actin filaments, or 50 μM colchicine or 1 μM taxol, to disrupt or stabilize tubulin filaments. Addition of cytochalasin B at 8 hpi prevented the loss of the rounded cell phenotype and inhibited any further progression of CPE (Fig. 9). In contrast, addition of either colchicine or taxol did not prevent recovery from cell rounding (Fig. 9A) but did inhibit the formation of virus-induced projections (Fig. 9B). Together, these data show that microtubules and actin filaments mediate virus-induced changes within the host cell.
FIG. 9.

Involvement of actin filaments or microtubules in recovery from cell rounding and formation of cell projections. BS-C-1 cells were seeded at low density to obtain isolated cells and were then infected with VV at 5 PFU/cell. The number of spread cells was determined 8 hpi for mock-infected (−V) or WR strain-infected (+V) cells. Cytochalasin B (2 μM; CB), taxol (1 μM; T), or colchicine (50 μM; C) was added to infected cells at 8 hpi, and the number of spread cells (A) or stellate cells (B) was determined at 18 hpi.
DISCUSSION
This paper describes two types of cell movement induced by VV infection. Firstly, early VV proteins induce cell-cell dissociation and then cell migration. In this respect, VV-induced CPE resembles events observed during development, wound healing, and metastasis (16). Secondly, late viral proteins induce the formation of long cellular projections. Video microscopy showed that these projections are formed by sequential elongation and condensation of lamellipodia from the cell body. This process is reminiscent of the extension of neuronal projections: cytoskeletal components within the growth cones of virus-induced projections closely resemble those described for extending neurons (28); moreover, the elongation of virus-induced projections and that of neurons both require reorganization of microtubules (27).
The sequence of events during VV-induced CPE is cell rounding, cell flattening, cell migration, and projection formation. Cell rounding (3) required the expression of early genes because it was not induced with virus particles inactivated with trioxsalen and UV light (30). Our results confirm these observations but also show that cell rounding is a reversible process which precedes virus-induced migration. The changes in cytoskeletal organization which accompany cell rounding may be similar to the cytoskeletal reorganization required for migration of uninfected cells (16). Reversion of cell rounding requires actin filaments but not microtubules; after recovery from cell rounding, infected cells migrate, and this process requires early but not late virus proteins. After migration, the development of long cellular extensions requires late proteins and microtubules but not IEV particles. The requirement for microtubules in the formation of virus-induced projections is consistent with previous reports that microtubules are needed for neuronal elongation and formation of Rab-8-induced projections (22).
Concerning the mechanism by which VV induces cell movement, it was possible that, because this process would require actin polymerization, it was directly linked to the ability of VV to induce actin polymerization on IEV particles (9). However, the data presented here show that these processes have different requirements. Actin tail formation is dependent on IEV particles (9), since it is blocked by N1-isonicotinoyl-N2-3-methyl-4-chloro-benzoylhydrazine and in cells infected with a mutant virus which lacks the p37 EEV protein (6). In contrast, expression of only early genes is needed for cell movement, and projection formation requires late protein synthesis but not formation of IEV particles. Interestingly, IEV formation is not sufficient for actin tail formation because a mutant virus lacking the A34R EEV glycoprotein is unable to make actin tails (31) despite making more EEV than does wild-type virus (19). Cells infected with mutant viruses which have lost gene A34R or B5R or the VV profilin gene (A42R) all move and form projections. Therefore, neither actin tails, EEV particles, nor the virus profilin is required for either form of VV-induced cell movement.
Although virus-induced motility is independent of actin tail formation, it is possible that the same VV actin-nucleating proteins accumulate within the plasma membrane. This could occur naturally during virus maturation as the outer IEV membrane containing the actin-nucleating proteins and associated actin tail is transferred to the plasma membrane (10). Alternatively, in the absence of actin tail formation these actin-nucleating proteins might go directly to the plasma membrane, and several EEV proteins have been found on the plasma membrane (25).
Finally, is cell movement beneficial to the virus? One possibility is that cell movement helps the virus infection to spread in vivo. Virus spread is mediated by EEV rather than IMV in cell culture and in vivo (21), but most enveloped virus remains associated with the infected cell (6). In this situation, the movement or extension of infected cells would increase local spread of the virus and might impart a significant evolutionary advantage. Some evidence for migration of poxvirus-infected cells in vivo has been reported elsewhere (24). Following a focal dermal infection with ectromelia virus, single infected cells were observed >100 μm from the point of infection, and the authors concluded that this may represent migration of the infected cells. Alternatively, these cells might have been infected by virus leaking from the site of inoculation or represent naturally motile cells such as lymphocytes or macrophages. Until now, only viruses which transform cells, such as Rous sarcoma virus, have been shown to induce cell movement and gain an invasive phenotype (4).
Whatever the advantage to VV is, the system described here illustrates another way in which virus infection can control cell behavior and provides a model in which genetics can be used to study complex process such as cell migration, lamellipodium extension, and formation of branch points within cell projections.
ACKNOWLEDGMENTS
We thank Raffa Blasco and Bernard Moss for VVs lacking p37 or profilin, Inga Reckmann for excellent technical help, and Vic Small for critical reading of the manuscript.
We thank the Medical Research Council and The Wellcome Trust for grant support.
REFERENCES
- 1.Alcamí A, Smith G L. A soluble receptor for interleukin-1β encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell. 1992;71:153–167. doi: 10.1016/0092-8674(92)90274-g. [DOI] [PubMed] [Google Scholar]
- 2.Allen W E, Jones G E, Pollard J W, Ridley A J. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. J Cell Sci. 1997;110:707–720. doi: 10.1242/jcs.110.6.707. [DOI] [PubMed] [Google Scholar]
- 3.Bablanian R, Baxt B, Sonnabend J A, Esteban M. Studies on the mechanisms of vaccinia virus cytopathic effects. II. Early cell rounding is associated with virus polypeptide synthesis. J Gen Virol. 1978;39:403–413. doi: 10.1099/0022-1317-39-3-403. [DOI] [PubMed] [Google Scholar]
- 4.Behrens J, Vakaet L, Friis R, Winterhager E, Van Roy F, Mareel M M. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/β-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blasco R, Cole N B, Moss B. Sequence analysis, expression, and deletion of a vaccinia virus gene encoding a homolog of profilin, a eukaryotic actin-binding protein. J Virol. 1991;65:4598–4608. doi: 10.1128/jvi.65.9.4598-4608.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blasco R, Moss B. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J Virol. 1992;66:4170–4179. doi: 10.1128/jvi.66.7.4170-4179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blasco R, Sisler J R, Moss B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J Virol. 1993;67:3319–3325. doi: 10.1128/jvi.67.6.3319-3325.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buller R M L, Palumbo G J. Poxvirus pathogenesis. Microbiol Rev. 1991;55:80–122. doi: 10.1128/mr.55.1.80-122.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cudmore S, Cossart P, Griffiths G, Way M. Actin-based motility of vaccinia virus. Nature. 1995;378:636–638. doi: 10.1038/378636a0. [DOI] [PubMed] [Google Scholar]
- 10.Cudmore S, Reckmann I, Griffiths G, Way M. Vaccinia virus: a model system for actin-membrane interactions. J Cell Sci. 1996;109:1739–1747. doi: 10.1242/jcs.109.7.1739. [DOI] [PubMed] [Google Scholar]
- 11.Engelstad M, Smith G L. The vaccinia virus 42-kDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology. 1993;194:627–637. doi: 10.1006/viro.1993.1302. [DOI] [PubMed] [Google Scholar]
- 12.Gumbiner B M. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 13.Hanson C V, Riggs J L, Lennette E H. Photochemical inactivation of DNA and RNA viruses by psoralen derivatives. J Gen Virol. 1978;40:345–358. doi: 10.1099/0022-1317-40-2-345. [DOI] [PubMed] [Google Scholar]
- 14.Hiller G, Weber K. A phosphorylated basic vaccinia virus virion polypeptide of molecular weight 11,000 is exposed on the surface of mature particles and interacts with actin-containing cytoskeletal elements. J Virol. 1982;44:647–657. doi: 10.1128/jvi.44.2.647-657.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hiller G, Weber K, Schneider L, Parajsz C, Jungwirth C. Interaction of assembled progeny pox viruses with the cellular cytoskeleton. Virology. 1979;98:142–153. doi: 10.1016/0042-6822(79)90533-6. [DOI] [PubMed] [Google Scholar]
- 16.Lauffenburger D A, Horwitz A F. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 17.Machesky L M, Cole N B, Moss B, Pollard T D. Vaccinia virus expresses a novel profilin with a higher affinity for polyphosphoinositides than actin. Biochemistry. 1994;33:10815–10824. doi: 10.1021/bi00201a032. [DOI] [PubMed] [Google Scholar]
- 18.Mackett M, Smith G L, Moss B. The construction and characterization of vaccinia virus recombinants expressing foreign genes. In: Glover D M, editor. DNA cloning: a practical approach. Vol. 2. Oxford, United Kingdom: IRL Press; 1985. pp. 191–211. [Google Scholar]
- 19.McIntosh A A G, Smith G L. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J Virol. 1996;70:272–281. doi: 10.1128/jvi.70.1.272-281.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moss B. Poxviridae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. Vol. 2. New York, N.Y: Lippincott-Raven Press; 1996. pp. 2637–2671. [Google Scholar]
- 21.Payne L G. Significance of extracellular enveloped virus in the in vitro and in vivo dissemination of vaccinia virus. J Gen Virol. 1980;50:89–100. doi: 10.1099/0022-1317-50-1-89. [DOI] [PubMed] [Google Scholar]
- 22.Peränen J, Auvinen P, Virta H, Wepf R, Simons K. Rab8 promotes polarized membrane transport through reorganization of actin and microtubules in fibroblasts. J Cell Biol. 1996;135:153–167. doi: 10.1083/jcb.135.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ridley A J, Comoglio P M, Hall A. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho proteins in MDCK cells. Mol Cell Biol. 1995;15:1110–1122. doi: 10.1128/mcb.15.2.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts J A. Histopathogenesis of mousepox. II. Cutaneous infection. Br J Exp Pathol. 1962;43:462–468. [PMC free article] [PubMed] [Google Scholar]
- 25.Schmelz M, Sodeik B, Ericsson M, Wolffe E J, Shida H, Hiller G, Griffiths G. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J Virol. 1994;68:130–147. doi: 10.1128/jvi.68.1.130-147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sodeik B, Doms R W, Ericsson M, Hiller G, Machamer C E, van-’t-Hof W, van-Meer G, Moss B, Griffiths G. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J Cell Biol. 1993;121:521–541. doi: 10.1083/jcb.121.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka E, Ho T, Kirschner M W. The role of microtubule dynamics in growth cone motility and axonal growth. J Cell Biol. 1995;128:139–155. doi: 10.1083/jcb.128.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 29.Tooze J, Hollinshead M, Reis B, Radsak K, Kern H. Progeny vaccinia viruses and human cytomegalovirus particles utilize early enosomal cisternae for their envelopes. Eur J Cell Biol. 1993;60:163–178. [PubMed] [Google Scholar]
- 30.Tsung K, Yim J H, Marti W, Buller R M L, Norton J A. Gene expression and cytopathic effect of vaccinia virus inactivated by psoralen and long-wave UV light. J Virol. 1996;70:165–171. doi: 10.1128/jvi.70.1.165-171.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolffe E, Katz E, Weisberg A, Moss B. The A34R glycoprotein gene is required for induction of specialized actin-containing microvilli and efficient cell-to-cell transmission of vaccinia virus. J Virol. 1997;71:3905–3915. doi: 10.1128/jvi.71.5.3904-3915.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]