

Abstract
Here we report the development of two independent assays which demonstrate for the first time that exogenous model RNA templates based on influenza virus virion RNA (vRNA) are transcribed in vitro to produce polyadenylated mRNA. We investigated the activities of mutated templates with known polymerase binding properties to test our model that polyadenylation occurs when a polymerase complex, which is bound to conserved 5′ sequences of vRNA, prevents read-through of the U track at which polyadenylation subsequently occurs by reiterative copying. Mutated templates with perturbed polymerase binding sites (i.e., a deletion mutant lacking the first 4 5′ residues and a U→A point mutant at the third residue) initiated transcription in the in vitro assay but failed to produce polyadenylated transcripts, whereas an A→U point mutant at the fourth residue, which retained polymerase binding properties similar to those of the wild type, produced polyadenylated transcripts. Our results show that nucleotides within the conserved 5′ sequence are required for polyadenylation and support the hypothesis that polymerase binding to 5′ sequences of the template is required for mRNA synthesis.
Each of the eight negative-stranded genome segments of influenza A virus is a template for transcription of two distinct types of positive-stranded RNA (reviewed in reference 12). Early in infection, capped and polyadenylated mRNA molecules are transcribed from virion RNA (vRNA). Later, full-length cRNA molecules are synthesized and act as templates for further synthesis of new vRNA genomes. Ribonucleoprotein (RNP) purified from virions can be transcribed in vitro to produce polyadenylated mRNA, demonstrating a host-independent mechanism (6, 20). Sequence analysis of vRNA showed a run of 5 to 7 uridine residues 17 nucleotides from the 5′ end of the template in all gene segments. This U5-7 track was directly adjacent to a predicted base-paired region of the panhandle, which was thought to form between the conserved segment termini that displayed partial inverted complementarity (2, 23). Mapping of the 3′ ends of mRNAs showed that polyadenylation occurs at a run of 5 to 7 A residues complementary to the U5-7 track (24). These observations led to a model for polyadenylation in which it was proposed that the influenza virus RNA polymerase is unable to melt the base-paired region of the panhandle; therefore, instead of copying to the end of the genome segment, it reiteratively copies the U5-7 track. Direct evidence for the existence of a vRNA panhandle was obtained by psoralen cross-linking experiments (8). vRNA molecules were found in circular configuration both in virions and infected cells. Consistent with the panhandle slippage model for polyadenylation, the circular forms were most abundant at times of greatest mRNA production (8).
More recent developments have allowed the study of transcription of influenza virus-like vRNA templates both in vitro and in vivo (7, 9, 11, 15–18, 25). In vivo studies showed that the conserved 5′- and 3′-terminal sequences were sufficient for the expression, replication, and packaging of genome segments (15). Initial in vitro transcription studies showed that added influenza virus-like templates containing only 3′ conserved sequences were sufficient for promoter recognition by the influenza virus polymerase complex derived from virions (18, 25). Further in vitro experiments identified a polymerase complex binding site in the 5′ strand of the vRNA panhandle (3, 27) and demonstrated that both the 5′ and 3′ arms of the panhandle were involved in transcription initiation and capped-primer utilization (3–5). The involvement of 5′ sequences suggested a new model for polyadenylation in which, after the initiation of transcription, the polymerase complex remains bound to 5′-terminal sequences and sterically blocks synthesis before the end of the template. Instead, reiterative copying of the U5-7 track results in poly(A) addition (3, 27). Although previous in vitro studies have shown that the polymerase complex also binds 5′ sequences of added cRNA molecules (5, 22) and that adenylyl (3′→5′) guanosine (ApG)-primed transcription can occur from a cRNA panhandle (22), endonuclease function was only poorly activated by a cRNA panhandle, suggesting a vRNA template-dependent mechanism for mRNA production (1).
The role of the panhandle and U5-7 track in polyadenylation was investigated in two in vivo studies by using the expression of a model chloramphenicol acetyltransferase (CAT) reporter gene (13, 14). Those studies confirmed that both the U5-7 track and the adjacent base-paired region of the panhandle were required. It has been our long-term aim to establish an in vitro reconstitution system in which polyadenylated mRNA can be transcribed from exogenous vRNA-like templates, allowing the precise molecular requirements for polyadenylation to be determined in isolation. Although nuclear extracts containing RNP assembled in vivo from transfected cDNAs can be transcribed in vitro to produce polyadenylated mRNA (19), no study of mutated templates by using this system has previously been reported.
In this report, we demonstrate that polyadenylated mRNA is synthesized in in vitro-reconstituted transcription reactions containing exogenous vRNA-like templates. Mutated templates differ in their ability to produce polyadenylated mRNA, depending on the presence of a functioning polymerase binding site in the 5′ conserved sequence. Our results show that nucleotides within the 5′ conserved sequence are required for polyadenylation and support the hypothesis that polymerase binding to these nucleotides is required for mRNA production.
MATERIALS AND METHODS
Influenza virus transcription reactions.
Influenza virus polymerase preparations were prepared as reported previously (25) by treating glycerol gradient-purified RNP from influenza A virus strain X-31 with micrococcal nuclease. Reaction mixtures typically contained 0.1 to 1 μg of RNA template and approximately 1 μg of RNP preparation (∼5 ng of polymerase proteins [26]). Reaction volumes ranged from 5 to 20 μl and contained 500 μM (each) GTP, CTP, and UTP, 1 mM ATP, and 0.5 mM ApG as primer in a buffer containing 50 mM Tris-HCl (pH 7.4), 50 mM KCl, 10 mM NaCl, 5 mM MgCl2, 5 mM dithiothreitol, and 10 U of placental RNase inhibitor. The nucleoside triphosphate (NTP) concentrations were adjusted as required for the incorporation of radiolabelled precursor into transcription products (see below). Reaction mixtures were incubated at 30°C for 3 h. Reaction products were either used directly or extracted with phenol-chloroform, precipitated with ethanol, and dissolved in water.
Influenza virus vRNA-like template RNA preparation.
The 717-nucleotide (nt) and 49-mer T7 RNA polymerase transcripts were made from BbsI- or BpuAI-linearized pBXPCAT1 (a gift from P. Palese) and derivatives (Fig. 1). The wild-type 717-nt RNA contains vRNA terminal sequences derived from segment 8 of influenza virus A/PR/8/34, linker sequences, and an antisense copy of the CAT gene (13). Mutated versions of pBXPCAT1 were made by an inverse PCR technique with Pfu DNA polymerase (21). Mutated regions and all relevant terminal sequence of mutated constructs were sequenced. Wild-type and mutated 49-mer constructs were transcribed by T7 RNA polymerase from internally deleted versions of pBXPCAT1 made by digesting pBXPCAT plasmids with XhoI and BglII, end filling with the Klenow fragment of DNA polymerase I, and religating. T7 transcription reaction mixtures (20 μl) typically contained 0.25 μg of linearized plasmid DNA, 25 U of T7 RNA polymerase, 10 U of placental RNase inhibitor, and 1 mM (each) NTPs in a buffer containing 40 mM Tris-HCl (pH 8.0), 8 mM MgCl2, 50 mM NaCl, 2 mM spermidine, and 10 mM dithiothreitol. Reaction mixtures were incubated for 20 min to 2 h at 37°C, treated with RNase-free DNase I to remove template DNA, extracted with phenol-chloroform, precipitated with ethanol, redissolved in water, and added without further treatment to influenza virus transcription reactions. The quantities of RNAs used in reactions were standardized either by examination of ethidium bromide-stained agarose gels loaded with 717-nt RNAs or by polynucleotide kinase labelling with [γ-32P]ATP for 49-mer templates.
FIG. 1.

RNA templates used in in vitro influenza virus transcription reactions. Vertical lines indicate the proposed base-paired region in the RNA fork model (3, 4). The U6 track, the proposed poly(A) site, is in bold. Nucleotides are numbered as primed numbers (3), starting from the 5′ end. The point mutations at positions 3′ and 4′ are indicated above those positions. (A) Sequence of 717-nt template. The sites for XhoI and BglII in plasmid pBXPCAT1 are indicated by arrows. The complements of the initiation and termination codons of the CAT gene are underlined and overlined, respectively. (B) Sequence of 49-mer template. The four residues in parentheses were absent in the deletion mutant.
[α-32P]ATP incorporation assay.
For 49-mer templates, the ATP concentration in the influenza virus transcription reaction mixture was reduced to 25 μM and included 2 to 4 μCi of [α-32P]ATP (3,000 Ci/mmol) per 5 to 10 μl of reaction mixture. After 3 h at 30°C, samples were diluted to 100 μl with 2.4 M ammonium acetate containing 10 μg of Escherichia coli tRNA as carrier, extracted with phenol-chloroform, and precipitated with ethanol. Pelleted RNA was dissolved in formamide loading dyes, heated to 99°C for 3 min, and electrophoresed through 16% polyacrylamide–7 M urea gels, which were autoradiographed or phosphorimaged for quantitative estimates. RNA size markers were made by T7 RNA polymerase transcription of suitably restricted plasmids in the presence of [α-32P]ATP. A graphical plot of the distance migrated against the lengths of size markers was used to estimate the length range of polyadenylated products by extrapolation. The poly(A) tail length was calculated from this range by subtracting 27 to account for template sequences.
RT-PCR with the 5′-GC-clamped T20 primer.
Material from an influenza virus transcription reaction mixture containing the 717-nt influenza virus-like RNA was added directly to a reverse transcriptase (RT) reaction mixture containing 50 pmol of 5′-GC-clamped T20 primer (5′ GCCCCGGGATCCT20), 200 μM (each) dNTPs, 10 U of placental RNase inhibitor, and 100 U of Moloney murine leukemia virus RT in a buffer containing 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 2.5 mM MgCl2, and 0.1% Triton X-100. After incubation at 40°C for 20 min, 50 pmol of a CAT-specific primer (5′ CGGTGAAAACCTGGCCTATTTCCCTAAAGGG) and 1.5 U of Taq DNA polymerase were added in the same buffer, doubling the initial 10-μl RT reaction mixture volume to 20 μl and halving the dNTP concentration to 100 μM. Reaction mixtures were thermal cycled (1 min at 94°C, 1 min at 60°C, and 2 min at 72°C) for 33 cycles. PCR mixtures were electrophoresed through 1.2% agarose in TAE (40 mM Tris base, 20 mM acetate, 1 mM EDTA) buffer, and products were visualized by ethidium bromide staining. For cloning, material from the broad band was eluted and DNA was purified by silica matrix binding. After end repair with T4 DNA polymerase and 5′ phosphorylation with polynucleotide kinase, DNA was ligated to HincII-cut and dephosphorylated pUC118. Clones were sequenced with an automated sequencer (Applied Biosystems). To observe the cRNA synthesized from 717-nt templates, 5-μl influenza virus transcription reaction mixtures which contained 10 μCi of [α-32P]CTP (800 Ci/mmol) in addition to 50 μM of unlabelled CTP were set up. Reaction products were extracted with phenol, precipitated with ethanol, denatured by heating in formamide, electrophoresed through 4% polyacrylamide–8 M urea gels, and autoradiographed. A 717-nt marker was made by 32P labelling the 3′ end of template RNA by using an oligonucleotide-directed Klenow labelling protocol (10). The intensities of bands were determined by phosphorimage analysis.
Oligo(dT)-cellulose separation of poly(A)+ and poly(A)− fractions.
Influenza virus transcription reaction products were extracted with phenol-chloroform precipitated with ethanol in the presence of E. coli tRNA carrier, and redissolved in 5 to 10 μl of water prior to oligo(dT)-cellulose separation. A commercial kit for mRNA isolation (Micro-FastTrack; Invitrogen) was modified so that both poly(A)+ and poly(A)− fractions were recoverable. One-fourth of a tablet of oligo(dT)20-30 cellulose was mixed with RNA in 0.2 ml of binding buffer (0.5 M NaCl, 10 mM Tris-HCl [pH 7.5]) and incubated with agitation at room temperature for 1 h. Oligo(dT)-cellulose was pelleted in a microcentrifuge for 10 s. The supernatant was the poly(A)− fraction. The pellet was washed twice in binding buffer (1 ml) at room temperature, followed by incubation in 0.2 ml of low-salt buffer (0.25 M NaCl, 10 mM Tris-HCl [pH 7.5]) at 37°C for 15 min and repelleting. After washing the pellet with an additional 0.2 ml of low-salt buffer, bound, poly(A)+ RNA was eluted with 0.2 ml of 10 mM Tris-HCl (pH 7.5) by incubating at 50°C for 15 min and repelleting. Poly(A)+ and poly(A)− RNA fractions were recovered by ethanol precipitation in the presence of 0.6 M ammonium acetate and 10 μg of E. coli tRNA.
RNase A digestion.
Transcription products labelled with [α-32P]ATP were extracted with phenol-chloroform and precipitated with ethanol prior to RNase A digestion. An aliquot equivalent to 1/10th of a transcription reaction was digested in a 5-μl volume with 0.002 μg of RNase A (Sigma) per ml for 30 min at 37°C in 10 mM Tris-HCl (pH 8.0)–1 mM EDTA. An equal volume of formamide was added before being heated to 99°C for 3 min and electrophoresed through a 16% polyacrylamide–7 M urea gel.
RESULTS
[α-32P]ATP incorporation assay for poly(A) detection.
The first polyadenylation assay used a 49-mer influenza virus vRNA-like template RNA (Fig. 1) and incorporation of labelled ATP to detect mRNA, since ATP is a more sensitive marker for polyadenylation than are the other radiolabelled NTPs. Transcription products corresponding to full-length template copies (cRNAs) and a higher-molecular-weight putative mRNA smear (band X) were observed (Fig. 2A, lane 2). The cRNA bands (see below) and putative mRNA smear were not observed when either the influenza virus polymerase preparation or template RNA was omitted from the transcription reaction (Fig. 2A, lanes 4 and 5). The relative yield of mRNA to cRNA bands was 4.9% ± 0.85%, as estimated on a molar basis (data are the mean and standard deviation of three experiments). The cRNA synthesized from the wild-type templates used in these experiments may be a competent template for vRNA synthesis. However, previous results with model vRNA templates similar to the 49-mer template studies here gave no evidence of vRNA synthesis (25). If it occurs at all, vRNA synthesis is likely to be at very low levels due to the small amount of cRNA synthesized.
FIG. 2.

Development of the [α-32P]ATP incorporation assay with the 49-mer template. (A) Autoradiograph of 16% polyacrylamide–7 M urea gel. Lane 1, 49-nt vRNA marker; lane 2, complete reaction (see Materials and Methods), showing high-molecular-weight products (X) and low-molecular-weight cRNA products, with one near the 49-nt vRNA marker and the other nearby (Y); lane 3, rerun product Y (eluted product); lanes 4 and 5, transcription in the absence of RNP and template, respectively. The weak signal at the origin in lane 5 is thought to be due to transcription of residual endogenous RNP and/or nonspecific binding of radiolabel. WT, wild type. (B) Polyacrylamide gel electrophoresis analysis as described for panel A, showing products of transcription before (lane 1) and after (+; lane 2) RNase A digestion. The mRNA, cRNA, and poly(A) products are indicated. A trace of the cRNA product and a partial digestion product are seen at the bottom of lane 2, and most of the poly(A) signal remains. (C) Analysis on a 10% polyacrylamide–7 M urea gel of unbound (poly(A)− [lane 1]) and bound (poly(A)+ [lane 2]) RNA isolated by oligo(dT)-cellulose separation; lane 3, RNA markers (in nucleotides).
The cRNA product typically runs as two bands, an upper band (band Y) (Fig. 2A, lane 2) of slightly slower mobility than that of a 49-nt marker and a lower band of approximately the same mobility as that of the marker. When the upper band was eluted and rerun under similar gel conditions, it migrated as the faster form, demonstrating that the two bands contain the same-sized species (Fig. 2A, lane 3). The fast band of correct mobility was observed exclusively when smaller proportions of the reaction were loaded, whereas the upper band was favored with larger loadings (data not shown). Thus, we believe band Y to be an incompletely denatured form of cRNA that retains some secondary structure. Attempts to improve the RNA denaturing conditions by increasing the formamide concentration in the sample loading buffer and heating for longer or increasing the urea concentration in the gel to 8 M failed to shift the upper band to the expected position. The authenticity of the cRNA product synthesized from short templates of this type under similar transcription conditions has been rigorously demonstrated before (25).
The sensitivities of cRNA bands and the putative mRNA smear to RNase A were assessed, since poly(A) is known to be resistant to digestion with this enzyme. Figure 2B demonstrates that under conditions where cRNA was degraded, the high-molecular-weight smear remained, albeit with a reduced molecular weight due to degradation of non-poly(A) sequences. This is consistent with the presence of poly(A) tails. Further experiments demonstrated that a chemically synthesized A30 was stable to RNase A digestion under conditions where cRNA, but not mRNA, was degraded (data not shown).
Labelled transcription products were investigated for their ability to bind oligo(dT)-cellulose to provide further evidence for polyadenylation. The high-molecular-weight smear was shown to bind oligo(dT)-cellulose, whereas cRNA bands did not, demonstrating that the putative mRNA smear contains polyadenylated molecules (Fig. 2C, lanes 1 and 2). There was evidence of discrete bands in the poly(A)+ fraction (Fig. 2C, lane 2), which may reflect traces of nonspecific binding of nonpolyadenylated transcripts, since they were not present after RNase treatment (Fig. 2B, lane 2).
Formal proof of the presence of poly(A) tails was gained by sequencing cloned cDNAs obtained by using the 5′-GC-clamped T20 primer protocol (see below).
RT-PCR assay for poly(A) with 5′-GC-clamped T20.
The second polyadenylation assay investigated influenza virus transcription reaction mixtures containing a 717-nt influenza virus-like vRNA synthesized by T7 RNA polymerase (Fig. 1A) (see Materials and Methods for template details). A 5′-GC-clamped T20 primer (5′GCCCCGGGATCCT20) was employed to generate cDNA molecules containing more A residues than would have been present if mispriming at the run of six A residues present in cRNA had occurred. During reverse transcription, the T20 part of the primer can prime throughout the poly(A) tail, thus producing a population of first-strand cDNA molecules of various lengths. This length heterogeneity is preserved during amplification by the 5′ GC clamp; providing the PCR annealing temperature is high enough to prevent priming due to the T20 part of the primer alone, this ensures that the length of each cDNA is preserved during amplification. The resultant population of molecules appears as a broad band on agarose gels (Fig. 3, lane 2). The broad band was present only when the influenza reaction mixtures contained both RNP preparations and the added wild-type influenza virus-like 717-nt RNA (Fig. 3, lanes 2 through 4). The reverse transcription step requires the presence of both the 5′-GC-clamped primer and RT (Fig. 3, lanes 5 and 6). The internal CAT-specific primer used (see Materials and Methods) should give a minimum fragment size of 312 nt when priming occurs exactly at the poly(A) junction. The broad band observed in all experiments started at this expected length and typically extended approximately 150 nt. Cloned cDNAs derived from the broad band were isolated and sequenced to confirm the presence of poly(A) tails (see below).
FIG. 3.

Development of the RT-PCR assay with a 5′-GC-clamped T20 primer and a 717-nt vRNA template. Lane 1, DNA markers; lane 2, complete reaction, showing a broad poly(A)+ band (see Materials and Methods); lanes 3 through 6, reactions without (−) template, RNP, Moloney murine leukemia virus RT, and 5′-GC-clamped T20 primer, respectively. WT 717 nt, wild-type influenza virus-like 717-nt vRNA.
Estimates of poly(A) tail length.
The broad band observed when products from the 717-nt template were assayed (Fig. 3) is approximately 150 nt wide (see above), providing an estimate of poly(A) tail length. Fifteen clones, eight from the broad band and seven from similar material derived from transcription of the 49-mer template, were sequenced and found to contain poly(A) sequences of up to 120 and 150 nt, respectively. Because the 5′-GC-clamped T20 primer can anneal anywhere on the poly(A) tail during reverse transcription, the resultant cDNA clones are likely to underestimate poly(A) tail lengths. In all of the clones examined, the poly(A) tail was found at the correct junction opposite the template U6 track.
The length of the poly(A) tail was estimated directly by using RNA size markers generated by T7 RNA polymerase runoff transcription of restriction enzyme-cleaved plasmids. This analysis (Fig. 2C) suggested that the poly(A) lengths range from 10 to >350 residues (see Materials and Methods). By using phosphorimage analysis (two independent experiments) and correcting for ATP incorporation, the most abundant species was estimated to have poly(A) tail lengths of about 70 nt, with the bulk (90%) ranging from 30 to 180 nt. These values may be an underestimate because of possible radiolytic or RNase degradation. Previous literature estimates for poly(A) tail length of 60 to 350 nt (20) would decrease and become closer to our estimates if a similar correction were made to account for the incorporation of labelled ATP.
Effect of mutations in 5′ vRNA conserved sequences on polyadenylation.
The [α-32P]ATP incorporation assay was used to study a 5′ deletion mutant that was 45 nt long, based on the 49-mer vRNA template (Fig. 1B), and lacked the first 4 5′ residues. Figure 4 shows that this mutant retained the ability to transcribe cRNA at a reduced efficiency but that mRNA production was below the detection level (estimated quantitatively as <3% of that of the wild type) (Fig. 4, lane 2). We then investigated two point mutations within this region by both polyadenylation assays. Position 3′ (U→A) and 4′ (A→U) mutants of the 49-mer template were synthesized and assayed by the [α-32P]ATP incorporation assay. The mRNA signal was below detection levels (<3% of that of the wild type) for the position 3′ U→A mutant (Fig. 5A, lane 2), whereas the position 4′ A→U mutant produced an mRNA similar in size and yield (estimated by phosphorimage analysis) to that of the wild type (compare lanes 1 and 3). Both mutant RNAs, 3′ U→A and 4′ A→U, were competent templates for cRNA production (Fig. 5A, lanes 2 and 3). The same mutations were also made as 717-nt templates, and these were assayed for mRNA and cRNA production. The 5′-GC-clamped T20 primer assay gave a broad band in reactions derived from the wild type and position 4′ A→U mutant (Fig. 5B, lanes 2 and 4, respectively), indicating the presence of polyadenylated mRNA, whereas no signal was seen for the position 3′ U→A mutant (lane 3). The synthesis of cRNA from 717-nt templates was investigated by [α-32P]CTP incorporation and polyacrylamide gel electrophoresis to visualize the full-length cRNA band. A signal that was of the correct size and similar in yield (estimated by phosphorimage analysis) was seen from each of the mutants and the wild-type template (Fig. 5C, lanes 2 through 4), demonstrating that all three templates initiated transcription.
FIG. 4.
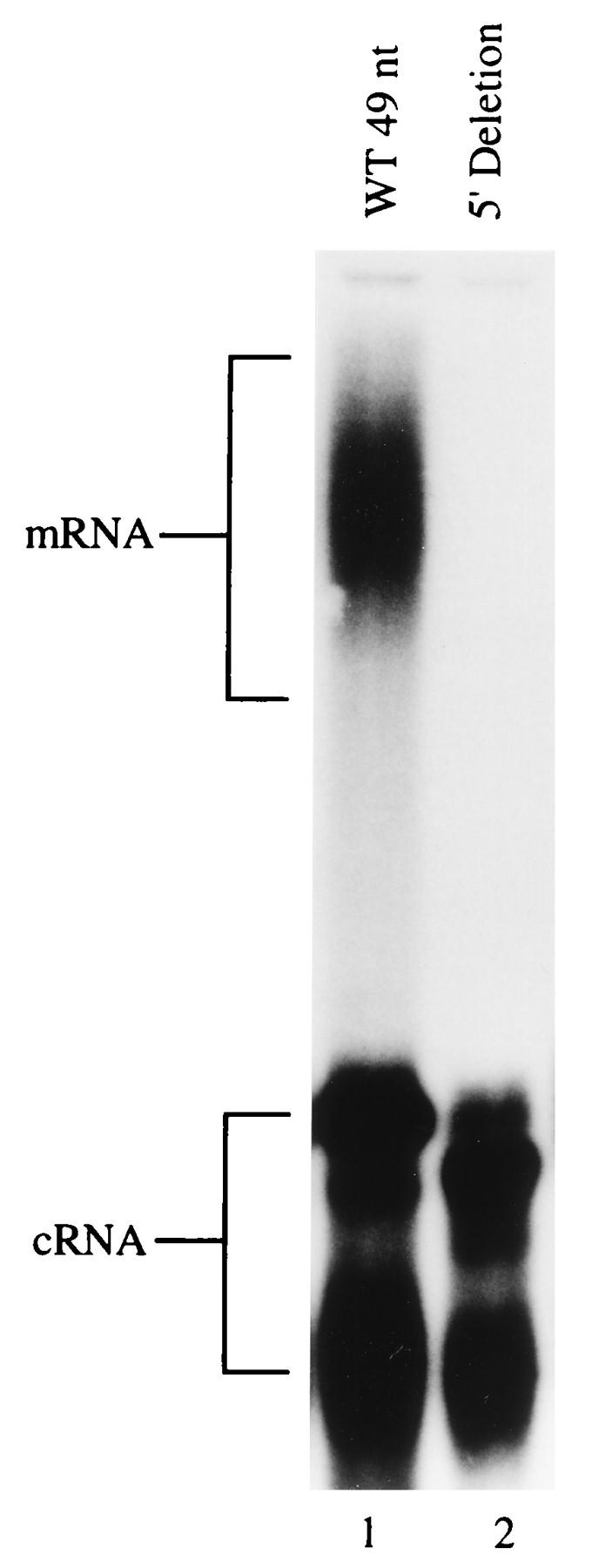
Effect of deleting the first 4 residues of the 5′ strand on polyadenylation activity. The [α-32P]ATP incorporation assay was used to examine transcription products from the wild-type (WT) 49-mer vRNA template (lane 1) and the 5′ deletion mutant (lane 2).
FIG. 5.

Effect of point mutations at positions 3′ and 4′ in the 5′ strand of the RNA fork on polyadenylation activity. (A) [α-32P]ATP incorporation assay of the wild-type (WT) 49-mer vRNA (lane 1) and point mutants (lanes 2 and 3). (B) RT-PCR assay with the 5′-GC-clamped T20 primer and 717-nt vRNA. Lane 1, DNA markers; lane 2, wild-type (WT) 717-nt vRNA; lanes 3 and 4, point mutants; lane 5, no template. (C) In vitro transcription reactions of wild-type (WT) 717-nt vRNA and point mutants examined by [α-32P]CTP assay for cRNA. Lane 1, 717-nt RNA marker; lane 2, WT 717-nt vRNA; lanes 3 and 4, point mutants; lane 5, no template.
DISCUSSION
In this work, we investigated whether transcripts synthesized in reconstituted influenza virus in vitro transcription assays containing added vRNA-like templates were polyadenylated. Since there was no evidence of polyadenylated mRNA in any of the previously reported reconstituted in vitro transcription assays (14, 18, 25), we investigated more sensitive methods for the detection of mRNA. We developed two independent assays which demonstrated that polyadenylation occurred on about 5% of transcripts. This amount was below the level of detection in the previously reported assays (18, 26). One method was an RT-PCR assay, and the other was a direct method where [α-32P]ATP was incorporated into high-molecular-weight poly(A)-containing mRNA. Applying these assays to products transcribed from mutated vRNA-like templates demonstrated that 5′ conserved sequences, previously identified as being crucial for polymerase binding, are specifically required for polyadenylation in vitro.
ApG-primed transcription was used throughout this study. During viral infection, polyadenylation is linked to capped-primer initiation by the endonuclease function, which has also previously been shown to require 5′ sequences (5). Since some of the ApG-primed transcripts reported here are polyadenylated, capped-primer synthesis is not an absolute requirement for polyadenylation in this system. Conversely, when globin mRNA is used as a source of capped primer in the in vitro transcription assay, the main product produced is not polyadenylated (25). Furthermore, extensive analysis performed with many templates has previously shown that ApG-primed synthesis essentially mimics capped-primer initiation (4). We conclude that ApG is a valid primer for the study of mRNA polyadenylation.
Previous works from this laboratory and another laboratory have shown that the polymerase complex binds to 5′-terminal sequences of vRNA (3, 27). The linkage of 5′ binding to transcription initiation (3) and the observation that 5′ binding is required for primer utilization and therefore mRNA production (5) suggested a model to account for mRNA/cRNA switching, depending on whether the polymerase complex remained bound to the 5′ end of the template RNA which it was transcribing. If the polymerase remained attached to the 5′ sequences, it would be unable to copy to the end of the template; instead, it would reiteratively copy the nearby U5-7 track. Since in the in vitro transcription assay, initiation is independent of influenza virus-like sequences at the 5′ end of the added template, presumably because of complementation by 5′ sequences endogenous to the polymerase preparation (3), we were interested to see whether the 5′ sequences of the vRNA-like template may play a role specifically in mRNA production, as predicted by the model. Both wild-type 49-mer and 717-nt templates have influenza virus vRNA-like sequences at their 3′ and 5′ termini, and both are competent templates for mRNA and cRNA synthesis (Fig. 2A, 3, and 5C). Using the [α-32P]ATP incorporation assay, we initially examined the products transcribed from a 45-nt template which retains the U6 track and adjacent base-paired region but has the first 4 5′ residues deleted. The experiment clearly showed that cRNA was produced but that mRNA production was reduced to below detection levels (Fig. 4). We then analyzed templates carrying point mutations at positions 3′ and 4′ in the deleted region. These mutations were chosen because the same mutants had been previously characterized in a polymerase cross-linking assay with short model 5′ sequences (3). In the cross-linking assay, the two mutants behaved very differently from one another. The 4′ A→U mutant retained the pattern and intensity of cross-linking of the three polymerase proteins similar to those of the wild type, whereas the 3′ U→A mutant failed to bind the polymerase proteins. When they were tested in the [α-32P]ATP incorporation assay, both mutants were templates for cRNA synthesis, which is consistent with the wild-type status of their 3′ ends. The 4′ A→U mutant gave rise to mRNA at levels similar to those of the wild-type 49-mer, whereas the 3′ U→A mutant failed to synthesize detectable mRNA (Fig. 5A).
The same point mutants were synthesized in the 717-nt form and tested for polyadenylation by the 5′-GC-clamped T20 RT-PCR assay and for full-length cRNA synthesis (Fig. 5B and C). The results exactly mirrored those discussed above. cRNAs were made by both mutants, but mRNA was made only by the 4′ A→U mutant. Together, these findings validate the methodology described here and further add to our understanding of how regulated expression of mRNA occurs for influenza virus. Clearly, a functional polymerase binding site at the 5′ end of the template molecule is required for polyadenylation. This finding extends previous in vivo studies, which demonstrated that the U6 track and an adjacent double-stranded region were required for polyadenylation (13, 14). The 5′ deletion mutant and position 3′ U→A mutant retain both of these key features but fail to polyadenylate.
For cRNA synthesis, as opposed to mRNA synthesis, read-through may be achieved by detachment of the polymerase from the 5′ end after transcription initiation. Alternatively, and as must occur in the in vitro assay where added templates lacking 5′ vRNA sequences are competent transcription templates, a trans-acting polymerase complex which initiates transcription without ever being bound to the 5′ end of the template molecule being transcribed may exist. In the in vitro assay, cRNA synthesis is probably dependent on the presence of 5′ ends which remain after nuclease digestion (3, 4). Whether a trans-acting polymerase complex which is associated with a 5′ terminus but is not covalently linked to the template being transcribed is used for cRNA synthesis in vivo is unknown. Whatever the mechanism for cRNA production in vivo, we have demonstrated that mutated template molecules differ in their ability to polyadenylate transcripts in the in vitro transcription assay, depending on their ability to bind polymerase. These findings suggest that for polyadenylation to occur, the polymerase has to be bound to the 5′ end of the template molecule being transcribed. Approximately 5% of transcripts made in this system are polyadenylated. This is a much lower proportion, relative to cRNA, than that observed in virus-infected cells. We believe that the inefficient polyadenylation in our system is due to the limited rate at which the polymerase complex can dissociate from endogenous 5′ ends and subsequently associate with the 5′ ends of the added template RNA. The [α-32P]ATP incorporation assay is ideally suited to study the switching between cRNA and mRNA synthesis since both types of RNA are assayed in the same reaction.
In summary, we have shown for the first time by two independent assays that polyadenylation occurs in reconstituted influenza virus in vitro transcription reactions containing appropriate influenza virus-like model vRNA templates. Our results indicate that 5′ conserved sequences of vRNA are required for polyadenylation and support the hypothesis that polymerase binding to the 5′ end of the template being transcribed is required for polyadenylation. Studies of mutations at other positions, both in the 5′ strand and elsewhere, are needed to fully determine the cis-acting requirements for poly(A) formation.
ACKNOWLEDGMENTS
D.C.P. was supported by the MRC (project grant no. G9427296PB to G.G.B.), E.F. was supported by the Welcome Trust (grant 047079), and L.L.M.P. was supported by the Croucher Foundation.
REFERENCES
- 1.Cianci C, Tiley L, Krystal M. Differential activation of the influenza virus polymerase via template RNA binding. J Virol. 1995;69:3995–3999. doi: 10.1128/jvi.69.7.3995-3999.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desselberger U, Racaniello V R, Zazra J J, Palese P. The 3′ and 5′ terminal sequences of influenza A, B and C virus RNA segments are highly conserved and show partial inverted complementarity. Gene. 1980;8:315–328. doi: 10.1016/0378-1119(80)90007-4. [DOI] [PubMed] [Google Scholar]
- 3.Fodor E, Pritlove D C, Brownlee G G. The influenza virus panhandle is involved in the initiation of transcription. J Virol. 1994;68:4092–4096. doi: 10.1128/jvi.68.6.4092-4096.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fodor E, Pritlove D C, Brownlee G G. Characterization of the RNA-fork model of the virion RNA in the initiation of transcription in influenza A virus. J Virol. 1995;69:4012–4019. doi: 10.1128/jvi.69.7.4012-4019.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hagen M, Chung T D Y, Butcher J A, Krystal M. Recombinant influenza virus polymerase: requirement of both 5′ and 3′ viral ends for endonuclease activity. J Virol. 1994;68:1509–1515. doi: 10.1128/jvi.68.3.1509-1515.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hay A J, Skehel J J. Characterisation of influenza virus RNA transcripts synthesized in vitro. J Gen Virol. 1979;44:599–608. doi: 10.1099/0022-1317-44-3-599. [DOI] [PubMed] [Google Scholar]
- 7.Honda A, Ueda K, Nagata K, Ishihama A. RNA polymerase of influenza virus: role of NP on RNA chain elongation. J Biochem. 1988;104:1021–1026. doi: 10.1093/oxfordjournals.jbchem.a122569. [DOI] [PubMed] [Google Scholar]
- 8.Hsu M, Parvin J D, Gupta S, Krystal M, Palese P. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc Natl Acad Sci USA. 1987;84:8140–8144. doi: 10.1073/pnas.84.22.8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang T S, Palese P, Krystal M. Determination of influenza virus proteins required for genome replication. J Virol. 1990;64:5669–5673. doi: 10.1128/jvi.64.11.5669-5673.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Z, Szostak J W. A simple method for 3′-labelling of RNA. Nucleic Acids Res. 1996;24:4360–4361. doi: 10.1093/nar/24.21.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura N, Nishida M, Nagata K, Ishihama A, Oda K, Nakada S. Transcription of a recombinant influenza virus RNA in cells that can express the influenza virus RNA polymerase and nucleoprotein genes. J Gen Virol. 1992;73:1321–1328. doi: 10.1099/0022-1317-73-6-1321. [DOI] [PubMed] [Google Scholar]
- 12.Lamb R F, Krug R M. Orthomyxoviridae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven Publishers; 1996. pp. 1353–1395. [Google Scholar]
- 13.Li X, Palese P. Characterization of the polyadenylation signal of influenza virus RNA. J Virol. 1994;68:1245–1249. doi: 10.1128/jvi.68.2.1245-1249.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo G, Luytjes W, Enami M, Palese P. The polyadenylation signal of influenza virus RNA involves a stretch of uridines followed by the RNA duplex of the panhandle structure. J Virol. 1991;65:2861–2867. doi: 10.1128/jvi.65.6.2861-2867.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luytjes W, Krystal M, Enami M, Parvin J D, Palese P. Amplification, expression, and packaging of a foreign gene by influenza virus. Cell. 1989;59:1107–1113. doi: 10.1016/0092-8674(89)90766-6. [DOI] [PubMed] [Google Scholar]
- 16.Martin J, Albo C, Ortin J, Melero J A, Portela A. In vitro reconstitution of active influenza virus ribonucleoprotein complexes using viral proteins purified from infected cells. J Gen Virol. 1992;73:1855–1859. doi: 10.1099/0022-1317-73-7-1855. [DOI] [PubMed] [Google Scholar]
- 17.Nagata K, Takeuchi K, Ishihama A. In vitro synthesis of influenza virus RNA: biochemical complementation assay of factors required for influenza virus replication. J Biochem. 1989;106:205–208. doi: 10.1093/oxfordjournals.jbchem.a122833. [DOI] [PubMed] [Google Scholar]
- 18.Parvin J D, Palese P, Honda A, Ishihama A, Krystal M. Promoter analysis of the influenza virus RNA polymerase. J Virol. 1989;63:5142–5152. doi: 10.1128/jvi.63.12.5142-5152.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perales B, De La Luna S, Palacios I, Ortin J. Mutational analysis identifies functional domains in the influenza A virus PB2 polymerase subunit. J Virol. 1996;70:1678–1686. doi: 10.1128/jvi.70.3.1678-1686.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plotch S J, Krug R M. Influenza virion transcriptase: synthesis in vitro of large, polyadenylic acid-containing complementary RNA. J Virol. 1977;21:24–34. doi: 10.1128/jvi.21.1.24-34.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pritlove, D. C. Unpublished data.
- 22.Pritlove D C, Fodor E, Seong B L, Brownlee G G. In vitro transcription and polymerase binding studies of the termini of influenza virus cRNA: evidence for a cRNA panhandle. J Gen Virol. 1995;76:2205–2213. doi: 10.1099/0022-1317-76-9-2205. [DOI] [PubMed] [Google Scholar]
- 23.Robertson J S. 5′ and 3′ terminal nucleotide sequences of the RNA genome segments of influenza virus. Nucleic Acids Res. 1979;6:3745–3757. doi: 10.1093/nar/6.12.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robertson J S, Schubert M, Lazzarini R A. Polyadenylation sites for influenza virus mRNA. J Virol. 1981;38:157–163. doi: 10.1128/jvi.38.1.157-163.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seong B L, Brownlee G G. A new method for reconstituting influenza polymerase and RNA in vitro: a study of the promoter elements for cRNA and vRNA synthesis in vitro and viral rescue in vivo. Virology. 1992;186:247–260. doi: 10.1016/0042-6822(92)90079-5. [DOI] [PubMed] [Google Scholar]
- 26.Seong B L, Kobayashi M, Nagata K, Brownlee G G, Ishihama A. Comparison of two reconstitution systems for in vitro transcription and replication of influenza virus. J Biochem. 1992;111:496–499. doi: 10.1093/oxfordjournals.jbchem.a123786. [DOI] [PubMed] [Google Scholar]
- 27.Tiley L S, Hagen M, Matthews J T, Krystal M. Sequence-specific binding of the influenza virus RNA polymerase to sequences located at the 5′ ends of the viral RNAs. J Virol. 1994;68:5108–5116. doi: 10.1128/jvi.68.8.5108-5116.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]