

Abstract
Early stages in vaccinia virus (VV) assembly involve the recruitment of cellular membranes from the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) to virus factories (or virosomes). The key viral factors involved in this process are not yet known. We have previously identified and characterized two viral proteins, of 21 kDa (A17L gene) and 15 kDa (A14L gene), that associate with tubulovesicular elements related to the ERGIC and are localized in viral membranes at all stages of virion assembly. We showed that the 21-kDa protein is not responsible for the recruitment of membranes from the ERGIC to viral factories. However, it appears to be essential for the organization of viral membranes. In this investigation we have generated a VV recombinant, VVindA14L, in which the expression of the A14L gene is inducibly regulated by the Escherichia coli lacI operator-repressor system. Repression of 15-kDa protein synthesis has a dramatic effect on virus yields and severely impairs plaque formation. Compared to wild-type VV, reduced amounts of 15-kDa protein are produced in VVindA14L-infected cells in the presence of IPTG (isopropyl-β-d-thiogalactoside), and this correlates with a small-plaque phenotype and reduced VVindA14L yields under these conditions. In the absence of the 15-kDa protein, early and late viral protein syntheses proceed normally; however, proteolytic cleavage of the major core precursors is inhibited. Electron microscopic examination of cells infected with VVindA14L under nonpermissive conditions reveals the presence of numerous membranous elements that look like unfinished or disassembled crescents interespersed between electron-dense masses. These abnormal membrane elements are usually well separated from the surfaces of the dense structures. These findings show that the 15-kDa protein is essential for VV morphogenesis and indicate that this polypeptide is necessary both for the correct assembly of viral crescents and for their stable attachment to the surfaces of viral factories.
Vaccinia virus (VV), the prototype member of the Poxviridae family, is a large DNA animal virus that replicates exclusively in the cytoplasm of infected cells (reviewed in reference 20). The morphogenesis of VV is a complex multistep process that involves numerous viral elements. Very little is known about the molecular mechanisms that dictate the ordered incorporation of the viral structures during assembly of viral particles. Early stages in virion assembly involve the recruitment of cellular membranes from the endoplasmic reticulum-Golgi intermediate compartment (ERGIC) to specific cytoplasmic areas known as virus factories or virosomes (40) which are the sites of DNA replication. These ERGIC-derived membranes appear to be modified by the gradual incorporation of viral proteins to give rise to the characteristic crescent-shaped membranes that are found on the periphery of the viral factories at early times postinfection. These structures, consisting of two tightly apposed membranes, become spherical while acquiring granular material from the virosomes. The spherical particles, known as immature virions (IVs), evolve to generate the intracellular mature virions (IMVs), where the envelope surrounds a complex core structure (4, 5, 13). This transition from IV to IMV is a poorly understood process that involves proteolytic processing of the major VV core precursors (19, 46). IMVs are the first type of infectious viral particles formed and constitute the majority of the virus particles produced in infected cells. The other major type of infectious particles produced during VV infection are the extracellular enveloped virions (EEVs). These particles are derived from IMVs that become wrapped by cisternae from the trans-Golgi network (12, 38) and exit the cell by fusion with the plasma membrane, a process that results in the loss of the outermost membrane. The remaining Golgi-derived membrane contains several proteins not found in IMVs (24, 25). Four of these proteins, of 37 kDa (F13L gene) (1, 39), 22 to 24 kDa (A34R gene) (2, 7, 17), 42 kDa (B5R gene) (8, 49), and 43 to 50 kDa (A36R gene) (23), have been shown to be required for generation and/or spread of EEVs, but none of them is necessary for IMV formation.
Although a number of proteins are known to be localized on the IMV membrane and the genes coding for some of them have been identified (reviewed in reference 43), very little is known about the key viral components that trigger the recruitment of cellular membranes to the virus factories and modify these membranes to configure them with the characteristics of the mature IMV membrane. In this regard, acquisition of the rigid convex shape of the crescents has been attributed to the 65-kDa protein (D13L gene), which is known to be the target of the VV assembly inhibitor rifampin (18, 44). Since this protein localizes to the inner sides of both crescents and IVs, it has been proposed to act as an internal scaffold (41). The L1R integral membrane protein (9, 26) is also essential for IMV formation, although it appears to be required at a later step of the morphogenetic process, in the transition from IV to IMV (27). Two well-characterized and abundant IMV peripheral proteins are the 14-kDa (A27L gene) (32, 33) and 32-kDa (D8L gene) (16, 22) proteins, but neither of them is critical for IMV assembly (28, 35), although the 14-kDa protein is required for wrapping of IMVs to produce EEVs (35). We have recently identified two membrane proteins, of 21 kDa (A17L gene) and 15 kDa (A14L gene), that form a stable complex with the 14-kDa envelope protein (29–31, 36). By immunoelectron microscopy of infected cells, we detected both the 21- and 15-kDa proteins in membranes of the rough endoplasmic reticulum (RER) and ERGIC and also associated with viral membranes in all stages of virion assembly (36). Moreover, recent studies have shown that both proteins insert into the RER in a cotranslational manner (14, 37). All these results indicate that the 21- and 15-kDa proteins are integral membrane proteins that may participate in the initial sequence of events leading to the formation of VV membranes. Through the generation of recombinant viruses that inducibly express the 21-kDa protein, it has been demonstrated that this protein is indeed essential for VV morphogenesis (30, 50). Repression of 21-kDa protein expression completely abrogates VV assembly at a stage previous to the appearance of viral crescents, although numerous tubulovesicular elements related to the ERGIC can be observed in the peripheries of nascent virus factories (36). These unorganized membranes are intensively labeled with antibodies specific for the 15-kDa protein. Thus, in view of these results, we have speculated that the 21-kDa protein may be involved in organizing the membrane precursors recruited to the locations of VV assembly, while the 15-kDa protein could be one of the viral elements participating in the membrane recruitment process. We have characterized the 15-kDa protein and found that it is myristilated and phosphorylated during infection, and in the virion it appears mostly as disulfide-linked dimers (36). These properties are consistent with its membrane location and potential participation in protein-protein interactions needed for membrane recruitment. Moreover, it has been recently suggested that phosphorylation may be required to initiate the morphogenetic process (45, 47). Phosphorylation of the 15-kDa protein, which has also been reported by Liu et al. (15), may thus be essential for its function.
To study the function of the 15-kDa protein in the VV life cycle and specifically to investigate its role in viral membrane biogenesis, we have generated a conditional mutant virus in which the expression of the A14L gene is regulated by the Escherichia coli lac operator-repressor system. Our results show that the 15-kDa protein is essential for the production of progeny virus and indicate a role for this protein in the assembly of ERGIC-derived membranes into crescents and in the establishment of interactions between these membranes and the granular content of the virosomes.
MATERIALS AND METHODS
Cells, viruses, and antisera.
BSC40 and HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% newborn calf serum (NCS). TK-143 cells were maintained in DMEM containing 10% fetal calf serum. VV strain WR was propagated and titrated in BSC40 cells. Recombinant virus VVindA17L was grown in BSC40 cells in the presence of 2 mM isopropyl-β-d-thiogalactoside (IPTG). The rabbit polyclonal antisera against the VV 15- and 39-kDa proteins have been previously described (6, 36).
Plasmid constructions.
A complete copy of the A14L gene flanked by BamHI and KpnI restriction sites at the 5′ and 3′ ends, respectively, was generated by PCR with VV genomic DNA as the template and oligonucleotide primers A and B, as shown in Fig. 1A. The sequences of the primers are as follows: A, 5′-GCGGGATCCCGATGGACATGATG-CTTATGAT-3′; B, 5′-CGGGTACCCGTTAGTTCATGGAAATAT-3′. The KpnI and BamHI sites are underlined. The 253-bp PCR product was cloned into the plasmid pPR35 (34) previously digested with BamHI and KpnI, generating the insertion plasmid pJR971 (Fig. 1A). The PCR product was sequenced to confirm its identity to the A14L viral sequence. Two DNA fragments homologous to the 5′ and 3′ flanking sequences of the A14L gene were generated by PCR amplification with VV DNA as the template and the following oligonucleotide primers: C, 5′-CCCAAGCTTTATACAGAAGATTTAACT-3′; D, 5′-GCTCTAGAGCTAAATTATTATCGTCCATAT-3′; E, 5′-GCTCTAGAGCTTAA-CTAATAAAAATTTTAA-3′; and F, 5′-CGGAATTCCGATGTTCGTAGACGATAATTC-3′ (Fig. 1B). Primers C and D, containing HindIII and XbaI sites (underlined), respectively, were used to produce a 324-bp fragment corresponding to the A13L open reading frame. Similarly, oligonucleotides E and F, including XbaI and EcoRI sites (underlined), respectively, were used to amplify a 452-bp DNA fragment homologous to the A15L gene, located downstream of the A14L gene (Fig. 1B). The resulting A13L and A15L PCR products were ligated together into HindIII/EcoRI-digested pUC19 to generate pJR972. An XbaI fragment containing the E. coli lacZ gene under the control of the VV p11 promoter (28) was cloned into the XbaI site of pJR972, generating the deletion plasmid pJR973.
FIG. 1.

Strategy for the construction of the VVindA14L recombinant virus. A two-step strategy was followed for the generation of VVindA14L recombinant virus. (A) Introduction of a lacI operator-regulated copy of the A14L gene in the TK locus of the VV genome. A DNA fragment corresponding to the complete A14L gene was produced by PCR amplification from viral DNA with primers A and B (described in Materials and Methods) and was cloned into the pPR35 plasmid, downstream of a hybrid inducible promoter consisting of the VV p4b promoter fused to two lacI operator (op) units. The resulting plasmid, pJR971, which also contains the lacI repressor gene under the control of the VV p7.5 promoter, was used to transfect BSC40 cells infected with wild-type (WR) virus. TK− intermediate viruses, VVTKA14L, were selected after infection of TK-143 cells in the presence of 5-bromodeoxyuridine (BUdR). (B) Deletion from the VVTKA14L genome of the endogenous A14L gene by replacement with the E. coli lacZ gene. Two DNA fragments corresponding to the left and right A14L flanking sequences were amplified by PCR with VV DNA and primers C-D and E-F, respectively, whose sequences are reported in Materials and Methods, and were both cloned into pUC19 to generate the pJR972 plasmid. A DNA fragment containing the E. coli lacZ gene fused to the VV p11 promoter was introduced into the XbaI site of pJR972, between the two A14L flanking sequences. The resulting plasmid, pJR973, was used to transfect BSC40 cells infected with VVTKA14L virus. Recombinant VVindA14L viruses containing only the inducible A14L gene were selected by the blue plaque phenotype in BSC40 cells infected in the presence of IPTG, after addition of X-Gal.
Recombinant virus construction.
An intermediate recombinant virus, VVTKA14L, containing an inducible copy of the A14L gene at the thymidine kinase (TK) locus, was generated by transfecting WR-infected BSC40 cells with the plasmid pJR971. TK− recombinant viruses were selected and plaque purified twice on TK-143 cells infected in the presence of 25 μg of 5-bromodeoxyuridine per ml. Recombinant viruses were distinguished from spontaneous TK− mutants by PCR amplification with oligonucleotide primers 5′-TCGCAGAGTATGCCGGTGTC-3′ and 5′-CTGTCGTGCCAGCTGCATTA-3′, corresponding to the 3′ and 5′ ends of the E. coli lacI gene, respectively. To delete the endogenous A14L gene, BSC40 cells infected with VVTKA14L in the presence of 2 mM IPTG were transfected with the plasmid pJR973. The resulting VVindA14L recombinant viruses were selected by blue plaque phenotype after the addition of 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-Gal) to the infected monolayer.
Metabolic labeling of viral proteins.
Cells were infected (5 PFU/cell) with WR VV or VVindA14L in the presence or absence of 2 mM IPTG, and, where indicated, cells were also maintained in the presence of hydroxyurea (HU) (5 mM). At 6 h postinfection (hpi) cells were washed with methionine-free DMEM and incubated in the same medium for 30 min. Cells were then pulse-labeled with [35S]methionine (100 μCi/ml) for 30 min, chased with a 100-fold excess of unlabeled methionine, and placed on ice or kept in culture for 18 h. Cells were washed three times with ice-cold phosphate-buffered saline, collected, and lysed in 1× sample buffer (62.5 mM Tris [pH 6.8], 2% sodium dodecyl sulfate [SDS], 0.25% bromophenol blue, 5% glycerol, and 5% 2-mercaptoethanol). Samples were boiled for 3 min and resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The gels were dried, and proteins were visualized after autoradiography.
One-step growth of VVindA14L.
Confluent monolayers of BSC40 cells were infected with WR virus or VVindA14L recombinant virus at a multiplicity of infection (MOI) of 2.5 PFU/cell. The inoculum was removed after 1 h, and the cells were washed with DMEM and overlaid with fresh DMEM supplemented with 2% NCS and containing or lacking IPTG (2 mM). Cells were harvested at various times postinfection, and progeny viruses were titrated by plaque assay on monolayers of BSC40 cells in the presence of IPTG.
Electron microscopy.
Monolayers of HeLa cells were infected at an MOI of 5 PFU/cell with the WR strain of VV or with the recombinant virus VVindA14L or VVindA17L in the presence or absence of IPTG. At 24 hpi cells were fixed in situ with a mixture of 2% glutaraldehyde and 2% tannic acid in 0.4 M HEPES buffer (pH 7.5) for 1 h at room temperature. Fixed monolayers were removed from the culture dishes in the fixative and transferred to Eppendorf tubes. After centrifugation and washing with HEPES buffer, the cells were processed for embedding in the resin EML-812 (EML Laboratories, Berks, United Kingdom) as previously described (30), with the following modifications. Postfixation of the cells was done with a mixture of 1% osmium tetroxide and 0.8% potassium ferricyanide in distilled water for 1 h at 4°C. After washing with HEPES buffer, samples were treated with 2% uranyl acetate and dehydrated at 4°C in increasing concentrations of acetone (50, 70, 90, and 100%, 15 min each). Infiltration in EML-812 was done at room temperature. After polymerization, ultrathin (20- to 30-nm) sections of the samples were obtained and stained with saturated uranyl acetate and lead citrate by standard procedures. Samples were studied in a JEOL 1200 EX II electron microscope.
RESULTS
Construction of a recombinant virus with an inducible A14L gene.
In order to investigate the role of the 15-kDa protein, the product of the A14L gene, in VV replication, we used a strategy that is being extensively employed for other VV gene products, consisting of the generation of a recombinant virus in which the expression of the gene of interest is regulated by the E. coli lac operator-repressor system (10, 34). For the production of a recombinant virus with an inducible A14L gene (VVindA14L), we constructed an intermediate virus (VVTKA14L) that contains, in addition to the endogenous A14L gene, a second copy of the gene preceded by two lacI operator sequences and integrated by homologous recombination into the TK region of the VV genome. The strategy used to obtain the recombinant virus VVTKA14L is depicted in Fig. 1A.
The next step was to suppress the wild-type A14L gene from its original locus to obtain recombinant viruses containing only the inducible copy of the gene. This was achieved by replacing the original A14L gene with the sequence coding for E. coli β-galactosidase. To this end, a deletion plasmid containing the lacZ gene regulated by the VV p11 promoter between the A14L left and right flanking sequences was constructed (Fig. 1B). This plasmid, pJR973, was used to transfect BSC40 cells infected with VVTKA14L virus. Progeny viruses generated after 2 days of infection were tested for the blue plaque phenotype by infecting BSC40 cells in the presence of IPTG. After addition of X-Gal to the infected cultures, blue plaques were picked up and used to infect fresh monolayers. At the end of the selection procedure, which was repeated five times, VVindA14L plaque isolates were expanded in BSC40 cells in the presence of 2 mM IPTG.
IPTG-dependent VVindA14L growth.
To characterize the phenotype of the VVindA14L virus, we performed a plaque assay experiment by infecting monolayers of BSC40 cells in the presence or absence of IPTG. As shown in Fig. 2A, plaques produced by VVindA14L in the presence of IPTG were noticeably smaller than those produced by WR. In the absence of IPTG, there was a significant (about 40-fold) reduction in the number of plaques, and the plaques produced under these conditions were larger than the ones formed in the presence of the inducer, suggesting that they most likely represent lacI repression escape mutants, as has been proposed for other conditional lethal mutants (48, 52).
FIG. 2.
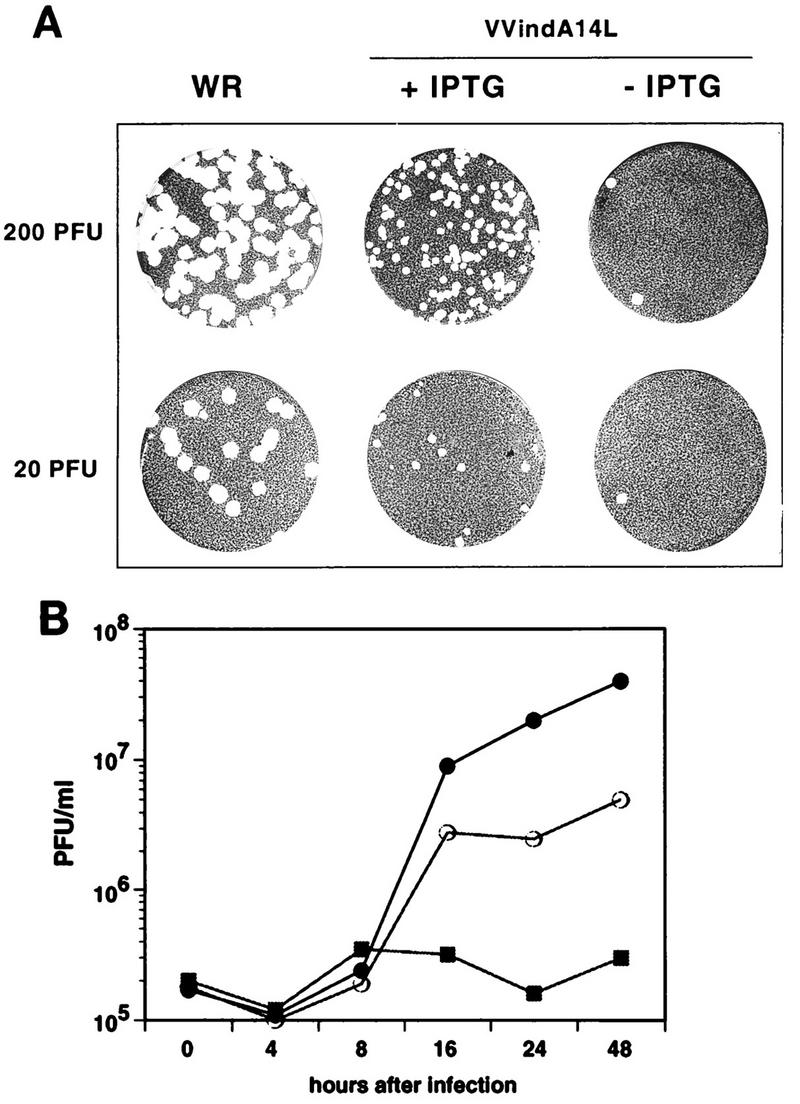
VVindA14L virus growth is dependent on the presence of IPTG. (A) Plaque assay. Confluent monolayers of BSC40 cells were infected with the indicated PFU of either WR or VVindA14L and overlaid with a mixture consisting of DMEM, 0.9% Bacto Agar, and 2% NCS, containing or lacking 2 mM IPTG. After 5 days, the monolayers were stained with 1% crystal violet. (B) One-step growth curves. BSC40 cells were infected at an MOI of 2.5 PFU/cell with WR virus (•) or with VVindA14L in the presence (○) or absence (▪) of 2 mM IPTG. Cells were collected at the indicated times after infection, and virus yields were determined by titration on BSC40 cells in the presence of 2 mM IPTG.
Next, we wished to determine whether the reduction in plaque number in the absence of IPTG was caused by the inability of VVindA14L to replicate under these conditions. For this, a one-step growth analysis was performed (Fig. 2B). In the absence of the inducer, VVindA14L titers remained at background levels during the whole infection period. A significant rise in virus yields was attained upon addition of IPTG to the infected cells. However, maximal yields were lower than those of the parental WR virus. These results show that VVindA14L is indeed an inducer-dependent conditional lethal mutant.
Synthesis of the A14L gene product in cells infected with VVindA14L is dependent on the presence of the inducer.
To confirm that expression of the A14L gene by VVindA14L virus was responsive to IPTG, BSC40 cells infected with WR or VVindA14L in the absence or presence of IPTG were collected at different times postinfection and cell extracts were analyzed by Western blotting with antibodies against the 15-kDa protein. As shown in Fig. 3B (lanes 9 to 12), neither the 15-kDa protein nor its dimer was detectable in VVindA14L-infected cells at any time postinfection in the absence of the inducer. However, in extracts of cells infected under permissive conditions (Fig. 3B, lanes 5 to 8), both forms of the protein (monomer and dimer) were clearly observed at late times postinfection (lanes 7 and 8), although the amount of 15-kDa protein produced in these cells was significantly smaller than that made in WR-infected cells (lanes 1 to 4). To eliminate the possibility that the different degrees of A14L gene expression were related to differences in the efficiency of infection, the blot was also reacted with a polyclonal serum against the VV 39-kDa core protein. As shown in Fig. 3A, the amounts and patterns of expression of this protein were equivalent in the three cases (WR virus and VVindA14L virus with or without IPTG), indicating that cells were equally infected.
FIG. 3.

Western blot analysis of 15-kDa protein synthesis in VVindA14L-infected cells. BSC40 cells were infected (5 PFU/cell) with WR (lanes 1 to 4) or with VVindA14L in the presence (lanes 5 to 8) or absence (lanes 9 to 12) of IPTG (2 mM). Cells were harvested at different times postinfection as indicated and lysed in 1× sample buffer. Proteins were fractionated by SDS-PAGE (12% polyacrylamide gel) and transferred to a nitrocellulose membrane. The membrane was cut in two pieces; the upper part containing the higher-molecular-mass proteins was reacted with anti-39-kDa-protein antibodies (A), and the lower portion of the membrane was incubated with anti-15-kDa-protein serum (B).
Proteolytic maturation of the major VV structural proteins is blocked when synthesis of the A14L gene product is repressed.
We next sought to examine whether inhibition of A14L gene expression would have an impact on the synthesis and/or processing of other VV polypeptides. For this, we carried out a pulse-chase experiment with cells infected with either WR or VVindA14L in the absence or presence of IPTG. At 6 hpi cells were pulse-labeled with [35S]methionine for 30 min and then either harvested immediately or chased with an excess of unlabeled methionine and kept in culture for another 18 h. To examine the pattern of early protein synthesis, cells were infected and pulse-labeled in the presence of HU to inhibit DNA replication and, thereby, late protein synthesis. Labeled proteins from the different infected cultures were analyzed by SDS–10% PAGE. As shown in Fig. 4, the profiles of early proteins obtained from the HU-treated cultures (lanes 1, 4, and 7) were indistinguishable. Similarly, the patterns of late proteins obtained after a 30-min pulse were also identical in VVindA14L-infected cells, treated (lane 5) or not (lane 8) with IPTG, and in WR-infected cells (lane 2), with the only exception being the additional presence of the β-galactosidase marker protein (120 kDa) in cells infected with the recombinant virus. However, the comparison of the 18-h chase samples showed that while in WR-infected cells a conversion of the major p4a and p4b precursors into the 4a and 4b mature products took place (Fig. 4, lane 3), this process was completely inhibited in cells infected with VVindA14L in the absence of the inducer, where both p4a and p4b precursors remained unchanged from the pulse-labeling period (compare lane 9 with lane 8). On the other hand, in IPTG-treated cells proteolytic processing of the precursors did occur, although to a lesser extent than in WR-infected cells as evidenced by the larger amounts of these two polypeptides remaining after the 18-h chase (Fig. 4, lane 6). Thus, these results show that repression of A14L gene expression does not affect VV protein synthesis but results in the inhibition of proteolytic maturation of the major core proteins, and this, in turn, is indicative of a potential blockade in virion morphogenesis.
FIG. 4.

Synthesis and proteolytic processing of viral proteins in VVindA14L-infected cells. BSC40 cells were infected with WR (lanes 1 to 3) or with VVindA14L in the presence (lanes 4 to 6) or absence (lanes 7 to 9) of IPTG (2 mM). One culture of each group of infected cells was treated with HU (5 mM). At 6 hpi untreated and treated cells were pulse-labeled with [35S]methionine for 30 min and then chased with unlabeled methionine. The HU-treated cells (lanes H) and one culture of untreated cells from each group (lanes P) were harvested immediately after the start of the chase period, while the remaining untreated cells (lanes C) were kept in culture for another 18 h. Cells were lysed in sample buffer, and proteins were resolved by SDS-PAGE (12% polyacrylamide gel) and visualized after autoradiography of the dried gel. The positions of the p4a, p4b, 4a, and 4b polypeptides are indicated on the left. Molecular mass markers in kilodaltons are indicated on the right.
VV morphogenesis is arrested in the absence of the A14L gene product.
As mentioned before, the inhibition of proteolytic processing of precursors in cells infected with VVindA14L under nonpermissive conditions suggested that virion assembly was interrupted at some stage previous to IMV formation. To investigate this possibility, HeLa cells infected with VVindA14L were examined by electron microscopy. As shown in Fig. 5, the cytoplasm of cells infected under nonpermissive conditions shows numerous membranous elements that do not organize in crescents, in addition to abnormal crescents that appear to be interrupted or unfinished structures, located between electron-dense masses. Some crescents which are clearly separated from the surfaces of the dense inclusions are also observed (Fig. 5B). Mature virions are totally absent, and a few IV-like particles, whose structure differs from that of control IVs, are occasionally detected (see below). When cells are infected in the presence of IPTG (Fig. 5C), characteristic foci of viroplasmic matrix with viral crescents attached to their surfaces are formed. Mature virions are also visualized, but they are less abundant than in WR VV-infected cells. While WR IVs frequently show condensed DNA bodies inside (Fig. 5A), these are difficult to visualize in IVs from cells infected with VVindA14L under permissive conditions (Fig. 5C).
FIG. 5.
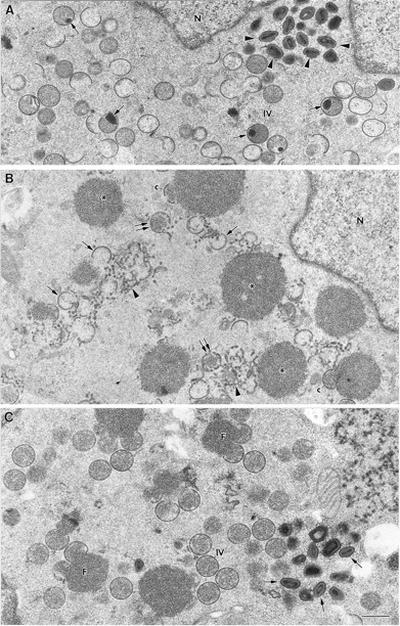
Low-magnification fields of cells infected for 24 h with WR VV or with VVindA14L in the absence or presence of IPTG. (A) IVs, many of them with condensed DNA (arrows), as well as mature virions (arrowheads) accumulate in the cytoplasm of cells infected with WR. (B) The cytoplasm of HeLa cells infected with VVindA14L in the absence of IPTG shows numerous electron-dense masses (asterisks). A few crescent-like structures (c) contact these masses, but most of them do not organize on the surfaces of the masses and accumulate within the cytoplasm (single arrows), as well as membranes that are not organized in crescents (arrowheads). Structures that resemble IVs with interrupted membranes are also seen (double arrows). (C) HeLa cells infected with VVindA14L in the presence of IPTG accumulate characteristic foci of viroplasmic matrix (F) with crescents attached to their surface, IVs, and mature virions (arrows). N, nucleus. Bar, 0.5 μm.
A more detailed analysis of the earliest viral structures formed in HeLa cells infected with the WR, VVindA17L, and VVindA14L viruses is shown in Fig. 6. Numerous IVs accumulate in cells infected with the WR VV. They frequently exhibit condensed DNA inside (Fig. 6A). As has been reported previously (36), in the absence of the 21-kDa protein, vesicular and tubular membranous elements related to the ERGIC are efficiently recruited to the electron-dense masses formed under these conditions (Fig. 6B), but they are not able to organize in viral crescents. However, as shown by the VVindA14L recombinant virus, crescent-like structures are able to assemble in the absence of the 15-kDa protein, although they are not efficiently attached to the surfaces of the dense masses (Fig. 6D). The ends of these crescent-like elements are, in most cases, bent, and although some curvature is observed, they do not acquire a typical spherical shape like the IVs assembled in the presence of the 15-kDa protein (Fig. 6C). Membranous elements that organize not in crescents but with a morphology that differs from that of the characteristic ERGIC elements accumulate around the dense inclusions, in both the presence and absence of the 15-kDa protein (Fig. 5B and 6C and D).
FIG. 6.
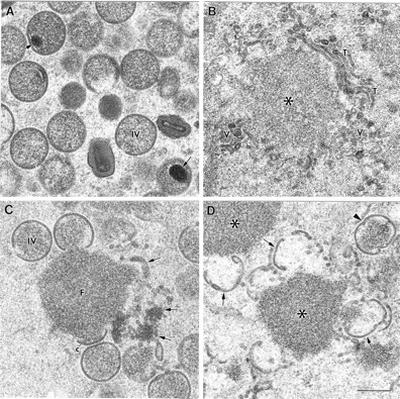
Viral structures distinguished in the cytoplasm of cells after a 24-h infection with WR VV, VVindA17L in the absence of IPTG, or VVindA14L in the presence or absence of IPTG. (A) IVs accumulate in cells infected with WR VV. Arrows point to condensed DNA inside the IVs. (B) Electron-dense masses (asterisk) surrounded by tubular (T) and vesicular (V) membranous elements are assembled in cells infected with VVindA17L in the absence of IPTG (when the 21-kDa protein is not expressed). (C) Cells infected with the VVindA14L virus in the presence of IPTG exhibit characteristic foci of viroplasmic matrix (F) surrounded by viral crescents (c), IVs, and membranous elements (arrows). (D) In the absence of IPTG (when the 15-kDa protein is not present), the VVindA14L virus induces the formation of dense structures similar to viral factories (asterisks) and crescent-like structures (arrows) that do not attach to the surfaces of the masses. IV-like virions (arrowhead) of irregular shape are occasionally seen. Bar, 300 nm.
On the other hand, higher-magnification images of the viral crescents formed by the VVindA14L recombinant virus show that the fine structure of these modified membranes is very similar in both the absence and presence of the 15-kDa protein (Fig. 7). The crescent-like structures that assemble in the absence of the protein look rather fuzzy, but, surprisingly, they exhibit a normal thickness and general organization. However, they frequently open or bend at their edges (Fig. 7B). When the 15-kDa protein is present, the viral crescents attach to the surfaces of the factories, apparently interacting with their dense contents (Fig. 7A). The IVs formed with the 15-kDa protein exhibit a homogeneous distribution of internal dense material, which is in contact with the membrane of the virion (Fig. 7C). The few IV-like particles that assemble in the absence of the protein, however, show a clear gap between the dense content and the viral membrane, suggesting that essential interactions between them are missing (Fig. 7D).
FIG. 7.
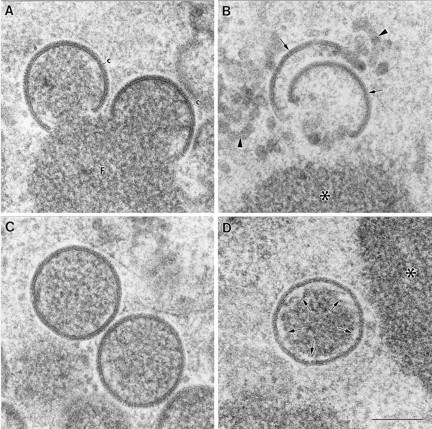
High-magnification fields of the viral crescents and IVs formed in HeLa cells infected for 24 h with VVindA14L in the presence or absence of IPTG. (A) Viral crescents (c) formed on the surfaces of the viroplasmic foci (F) in the presence of IPTG exhibit a thickness and organization indistinguishable from those of the structures generated by the WR VV. (B) The crescent-like structures (arrows) assembled in the absence of IPTG acquire the characteristic curvature of the crescent, but they exhibit a more diffuse appearance. They are usually separated from the surfaces of the dense masses (asterisk), and there are also some membranous elements of irregular shape (arrowheads). (C) IVs from cells infected with the VVindA14L in the presence of IPTG show the same size and apparent organization as the WR VV virions. (D) HeLa cells infected with VVindA14L in the absence of IPTG assemble a few IV-like virions, whose dense internal contents are separated from the membrane of the viral particle (arrows). Bar, 200 nm.
Taken together, these data indicate that the 15-kDa protein is required for the correct assembly of immature viral membranes around, and enclosing, the dense content of the factories, to produce first the characteristic crescents and then the IVs.
DISCUSSION
During VV assembly, about 100 viral polypeptides are incorporated, together with the genomic DNA, into macromolecular structures to form the complex viral particles. Many aspects of this elaborate process remain unknown. From conventional electron microscopy studies it has been traditionally believed that virus morphogenesis commences with de novo formation of the viral membranes (5, 42). However, it has been recently proposed that viral membranes are derived from cisternal membranes of the ERGIC and thus consist of two lipid bilayers that are difficult to visualize in the virion because they become tightly apposed (40). This latest model, which was first based on structural and immunocytochemical data, is being strengthened by the identification of viral membrane proteins that are targeted to this cellular compartment. We have identified three abundant viral proteins, i.e., the 21-kDa protein (A17L gene) (29), the 15-kDa protein (A14L gene) (36), and p8 (A13L gene) (37), that are found associated with the RER and ERGIC and are localized in the viral membrane at all stages of virion assembly (14, 31, 36, 37). Moreover, we have established that the 21-kDa protein is essential for virion assembly (30). In its absence, numerous tubulovesicular elements, related to the ERGIC, appear in the boundaries of dense structures that resemble viral factories (36). These membranous structures do not acquire the characteristic curved morphology of the crescent. The 65-kDa protein, the product of the D13L gene, is responsible for conferring the rigid convex shape to the membrane, and both in the absence of this protein (52) and in the presence of rifampin (11, 21) ruffled membranes are observed around electron-dense masses which are referred to as rifampin bodies. These irregularly shaped membranes show the same thickness as the crescents, indicating that the two membrane bilayers are already tightly bound at this stage. As opposed to these ruffled membranes, the tubulovesicular elements do not show a continuity around the dense masses and are clearly wider and less compact. These data indicate a function for the 21-kDa protein in the organization of the ERGIC-derived membranes, in which the participation of the 65-kDa protein and, probably, other membrane proteins gives rise to the compact and rigid structure of the crescents.
Since these tubulovesicular membrane precursors have already incorporated the 15-kDa protein, we suggested that this protein could be involved in the membrane recruitment process from the ERGIC to the virus factories (36). The generation of a recombinant VV in which the expression of the A14L gene is inducibly regulated by IPTG has enabled us to investigate in depth the function of the 15-kDa protein. In cells infected in the presence of IPTG, the levels of expression of the 15-kDa protein were considerably lower than those obtained in cells infected with wild-type WR virus. This reduced expression of the 15-kDa protein could be anticipated, since it was previously reported that the strong repression of transcription achieved by the presence of two lacI operators next to the target gene is difficult to overcome completely upon addition of the inducer (34).
Our results show that the 15-kDa protein is essential for virus replication. Compared with that of WR virus, at least a 2-log-unit reduction in VVindA14L titers was observed when IPTG was omitted. Even in the presence of the inducer, there was a decrease in VVindA14L yields, and this is likely due to reduced expression of the 15-kDa protein under these conditions. The lower levels of 15-kDa protein in the presence of IPTG can also account for the small-plaque phenotype of VVindA14L. On the other hand, in the absence of the inducer, plaque formation was essentially abolished.
Early and late protein synthesis proceeds normally in the absence of the 15-kDa protein; however, proteolytic cleavage of the core precursors is inhibited. This defect has been found to be associated with conditions that result in a blockade of VV morphogenesis (19, 21, 27, 30, 45–47, 50–52).
Electron microscopic analysis of cells infected with VVindA14L revealed that VV assembly is arrested when the 15-kDa protein is not expressed. In the absence of the 15-kDa protein, electron-dense masses that resemble those produced by VVindA17L in the absence of the 21-kDa protein accumulated in the cytoplasm of infected cells. Aberrant membranous elements, some of which look like unfinished or disassembled crescents, appeared interspersed between the electron-dense masses, from which they were clearly separated. Thus, contrary to our previous hypothesis, membrane recruitment to the sites of virion assembly appears to be independent of the 15-kDa protein. Mature IMVs were completely absent, and only a few abnormal IVs could be observed. These anomalous IVs are distinguishible from IVs formed in the presence of the 15-kDa protein by the apparent lack of contact between the internal material and the surrounding membrane.
Although all virion forms were present in the cytoplasm of cells infected under permissive conditions, unassembled membranous elements were also abundant in these cells, which again indicates that the amount of 15-kDa protein produced under these conditions is not enough to completely rescue the wild-type phenotype.
Taken together, these data indicate a role for the 15-kDa protein in the correct organization of the crescent and its binding to the content of the viral factory. Given that crescent-like structures are formed under nonpermissive conditions, it seems that the 65-kDa scaffolding protein is able to curve the membranes in the absence of the 15-kDa protein, although the process appears to be incomplete since the structures are not perfectly spherical. On the other hand, it is also possible that a minimal amount of the 15-kDa protein is produced under nonpermissive conditions due to some leakiness of the system, perhaps enough to allow for the assembly of the crescents and the few IV-like particles observed. Similar to the case for the IVs formed in the absence of the 15-kDa protein, formation of IVs with a gap between the internal content and the membrane has been reported to occur when synthesis of VP8 core protein (L4R gene) is repressed (48). This anomalous phenotype is likely caused by a defect in viroplasm-membrane interactions. Thus, it is tempting to speculate that essential interactions between the VP8 present in the viroplasm and the 15-kDa protein located in the viral membrane may occur during virion assembly. However, even if this is the case, other protein-protein interactions may be required to establish or maintain the contact between the two structures, e.g., the suggested interaction between the 39-kDa core protein (A4L gene) and the 21-kDa membrane protein (3).
Our studies provide genetic evidence for an essential role of the 15-kDa protein in VV morphogenesis. The 15-kDa protein is necessary both for the correct assembly of the viral crescent and for its stable attachment to the surface of the viral factory, as a first step in the formation of the immature virus.
ACKNOWLEDGMENTS
We thank Paco Rodríguez and José Sánchez-Serrano for critical readings of the manuscript, Angel Sanz and Inés Poveda for excellent photography work, and Victoria Jiménez for skilled technical assistance.
This work was supported by a grant from Comisión Interministerial de Ciencia y Tecnología (CICYT) (BIO95-0022) to M.E. and by grant PB91-0109 from the Dirección General de Investigación Científica y Técnica of Spain to J.L.C. D.R. and C.R. were recipients of contracts from the C.S.I.C.-Fundación Ramón Areces, and J.R.R. was the recipient of a contract from the M.E.C. of Spain.
REFERENCES
- 1.Blasco R, Moss B. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-dalton outer envelope protein. J Virol. 1991;65:5910–5920. doi: 10.1128/jvi.65.11.5910-5920.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blasco R, Sisler J R, Moss B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J Virol. 1993;67:3319–3325. doi: 10.1128/jvi.67.6.3319-3325.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cudmore S, Blasco R, Vincentelli R, Esteban M, Sodeik B, Griffiths G, Krijnse Locker J. A vaccinia virus core protein, p39, is membrane associated. J Virol. 1996;70:6909–6921. doi: 10.1128/jvi.70.10.6909-6921.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dales S, Siminovitch L. The development of vaccinia virus in Earles L strain cells as examined by electron microscopy. J Biophys Biochem Cytol. 1961;10:475–503. doi: 10.1083/jcb.10.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dales S, Pogo B G T. Biology of poxviruses. Virology monographs. Vol. 18. New York, N.Y: Springer-Verlag; 1981. [DOI] [PubMed] [Google Scholar]
- 6.Demkowicz W E, Maa J S, Esteban M. Identification and characterization of vaccinia virus genes encoding proteins that are highly antigenic in animals and are immunodominant in vaccinated humans. J Virol. 1992;66:386–398. doi: 10.1128/jvi.66.1.386-398.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duncan S A, Smith G L. Identification and characterization of an extracellular envelope glycoprotein affecting vaccinia virus egress. J Virol. 1992;66:1610–1621. doi: 10.1128/jvi.66.3.1610-1621.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelstad M, Smith G L. The vaccinia virus 42-KDa envelope protein is required for the envelopment and egress of extracellular virus and for virus virulence. Virology. 1993;194:627–637. doi: 10.1006/viro.1993.1302. [DOI] [PubMed] [Google Scholar]
- 9.Franke C A, Wilson E M, Hruby D E. Use of a cell-free system to identify the vaccinia virus L1R gene product as the major late myristylated virion protein M25. J Virol. 1990;64:5988–5996. doi: 10.1128/jvi.64.12.5988-5996.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuerst J R, Fernández M P, Moss B. Transfer of the inducible lac repressor/operator system from Escherichia coli to a vaccinia virus expression vector. Proc Natl Acad Sci USA. 1989;86:2549–2553. doi: 10.1073/pnas.86.8.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grimley P M, Rosemblum E N, Mims S J, Moss B. Interruption by rifampicin of an early stage in vaccinia virus morphogenesis: accumulation of membranes which are precursors of virus envelopes. J Virol. 1970;6:519–533. doi: 10.1128/jvi.6.4.519-533.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiller G, Weber K. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J Virol. 1985;55:651–659. doi: 10.1128/jvi.55.3.651-659.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichihashi Y, Matsumoto S, Dales S. Biogenesis of poxviruses: role of A-type inclusions and host cell membranes in virus dissemination. Virology. 1971;46:507–532. doi: 10.1016/0042-6822(71)90056-0. [DOI] [PubMed] [Google Scholar]
- 14.Krijnse-Locker J, Schleich S, Rodríguez D, Goud B, Snijder E J, Griffiths G. The role of a 21kDa viral membrane protein in the assembly of vaccinia virus from the intermediate compartment. J Biol Chem. 1996;271:14950–14958. doi: 10.1074/jbc.271.25.14950. [DOI] [PubMed] [Google Scholar]
- 15.Liu, K., R. Rollins, and P. Traktman. Submitted for publication.
- 16.Maa J-S, Rodríguez J F, Esteban M. Structural and functional characterization of a cell surface binding protein of vaccinia virus. J Biol Chem. 1990;265:1569–1577. [PubMed] [Google Scholar]
- 17.McIntosh A A, Smith G L. Vaccinia virus glycoprotein A34R is required for infectivity of extracellular enveloped virus. J Virol. 1996;70:272–281. doi: 10.1128/jvi.70.1.272-281.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McNulty-Kowalczyk A, Paoletti E. Mutations in ORF D13L and other genetic loci alter the rifampicin phenotype of vaccinia virus. Virology. 1993;194:638–646. doi: 10.1006/viro.1993.1303. [DOI] [PubMed] [Google Scholar]
- 19.Moss B, Rosenblum E N. Protein cleavage and poxvirus morphogenesis: tryptic peptide analysis of core precursors accumulated by blocking assembly with rifampicin. J Mol Biol. 1973;81:267–269. doi: 10.1016/0022-2836(73)90195-2. [DOI] [PubMed] [Google Scholar]
- 20.Moss B. Poxviridae and their replication. In: Fields B N, Knipe D M, editors. Virology. 2nd ed. New York, N.Y: Raven Press; 1990. pp. 2079–2111. [Google Scholar]
- 21.Nagayama A, Pogo B G T, Dales S. Biogenesis of vaccinia virus: separation of early stages from maturation by means of rifampicin. Virology. 1970;4:1039–1051. doi: 10.1016/0042-6822(70)90150-9. [DOI] [PubMed] [Google Scholar]
- 22.Niles E G, Seto J. Vaccinia virus gene D8 encodes a virion transmembrane protein. J Virol. 1988;62:3772–3778. doi: 10.1128/jvi.62.10.3772-3778.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkinson J E, Smith G L. Vaccinia virus gene A36R encodes a Mr 43-50 K protein on the surface of extracellular enveloped virus. Virology. 1994;204:376–390. doi: 10.1006/viro.1994.1542. [DOI] [PubMed] [Google Scholar]
- 24.Payne L G. Polypeptide composition of extracellular enveloped vaccinia virus. J Virol. 1978;27:28–37. doi: 10.1128/jvi.27.1.28-37.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Payne L G. Characterization of vaccinia virus glycoproteins by monoclonal antibody precipitation. Virology. 1992;187:251–260. doi: 10.1016/0042-6822(92)90313-e. [DOI] [PubMed] [Google Scholar]
- 26.Ravanello M P, Hruby D. Characterization of the vaccinia virus L1R myristyl protein as a component of the intracellular virion envelope. J Gen Virol. 1994;75:1479–1483. doi: 10.1099/0022-1317-75-6-1479. [DOI] [PubMed] [Google Scholar]
- 27.Ravanello M P, Hruby D. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virion assembly. J Virol. 1994;68:6401–6410. doi: 10.1128/jvi.68.10.6401-6410.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodríguez D, Rodríguez J R, Esteban M. Insertional inactivation of the vaccinia virus 32-kDa gene is associated with attenuation in mice and reduction of viral gene expression in polarized epithelial cells. J Virol. 1992;66:183–189. doi: 10.1128/jvi.66.1.183-189.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodríguez D, Rodríguez J R, Esteban M. The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L gene. J Virol. 1993;67:3435–3440. doi: 10.1128/jvi.67.6.3435-3440.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez D, Esteban M, Rodriguez J R. Vaccinia virus A17L gene product is essential for an early step in virion morphogenesis. J Virol. 1995;69:4640–4648. doi: 10.1128/jvi.69.8.4640-4648.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodríguez D, Risco C, Rodríguez J R, Carrascosa J L, Esteban M. Inducible expression of vaccinia virus A17L gene provides a synchronized system to follow sorting of viral proteins during morphogenesis. J Virol. 1996;70:7641–7653. doi: 10.1128/jvi.70.11.7641-7653.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodríguez J F, Janezko R, Esteban M. Isolation and characterization of neutralizing monoclonal antibodies to vaccinia virus. J Virol. 1985;56:482–488. doi: 10.1128/jvi.56.2.482-488.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodríguez J F, Esteban M. Mapping and nucleotide sequence of the vaccinia virus gene that encodes a 14-kilodalton fusion protein. J Virol. 1987;61:3550–3554. doi: 10.1128/jvi.61.11.3550-3554.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodríguez J F, Smith G L. Inducible gene expression from vaccinia virus vectors. Virology. 1990;177:239–250. doi: 10.1016/0042-6822(90)90477-9. [DOI] [PubMed] [Google Scholar]
- 35.Rodríguez J F, Smith G L. IPTG-dependent vaccinia virus: identification of a virus protein enabling virion envelopment by Golgi membrane and egress. Nucleic Acids Res. 1990;18:5347–5351. doi: 10.1093/nar/18.18.5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodríguez J R, Risco C, Carrascosa J L, Esteban M, Rodríguez D. Characterization of early stages in vaccinia virus membrane biogenesis: implication of the 21-kDa and a newly identified 15-kDa envelope protein. J Virol. 1997;71:1821–1833. doi: 10.1128/jvi.71.3.1821-1833.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salmons T, Kuhn A, Schleich S, Rodríguez J R, Rodríguez D, Esteban M, Griffiths G, Krijnse Locker J. Vaccinia virus membrane proteins P8 and P16 are cotranslationally inserted into the rough endoplasmic reticulum and retained in the intermediate compartment. J Virol. 1997;71:7404–7420. doi: 10.1128/jvi.71.10.7404-7420.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmelz M, Sodeik B, Ericsson M, Wolffe E J, Shida H, Hiller G, Griffiths G. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans-Golgi network. J Virol. 1994;68:130–147. doi: 10.1128/jvi.68.1.130-147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmutz C, Payne L G, Gubser J, Wittek R. A mutation in the gene encoding the vaccinia virus 37,000-Mr protein confers resistance to an inhibitor of virus envelopment and release. J Virol. 1991;65:3435–3442. doi: 10.1128/jvi.65.7.3435-3442.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sodeik B, Doms R W, Ericsson M, Hiller G, Machamer C E, van’t Hof W, van Meer G, Moss B, Griffiths G. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J Cell Biol. 1993;121:521–541. doi: 10.1083/jcb.121.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sodeik B, Griffiths G, Ericsson M, Moss B, Doms R W. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J Virol. 1994;68:1103–1114. doi: 10.1128/jvi.68.2.1103-1114.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stern W, Dales S. Biogenesis of vaccinia: concerning the origin of the envelope phospholipids. Virology. 1974;62:293–306. doi: 10.1016/0042-6822(74)90393-6. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi T, Oie M, Ichihashi Y. N-terminal amino acid sequences of vaccinia virus structural proteins. Virology. 1994;202:844–852. doi: 10.1006/viro.1994.1406. [DOI] [PubMed] [Google Scholar]
- 44.Tartaglia J, Paoletti E. Physical mapping and DNA sequence analysis of the rifampin resistance locus in vaccinia virus. Virology. 1985;147:394–404. doi: 10.1016/0042-6822(85)90141-2. [DOI] [PubMed] [Google Scholar]
- 45.Traktman P, Caligiuri A, Jesty S A, Sankar U. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of virion morphogenesis. J Virol. 1995;69:6581–6587. doi: 10.1128/jvi.69.10.6581-6587.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.VanSlyke J K, Lee P, Wilson E M, Hruby D E. Isolation and analysis of vaccinia virus previrions. Virus Genes. 1993;7:311–324. doi: 10.1007/BF01703388. [DOI] [PubMed] [Google Scholar]
- 47.Wang S, Shuman S. Vaccinia virus morphogenesis is blocked by temperature-sensitive mutations in the F10 gene, which encodes protein kinase 2. J Virol. 1995;69:6376–6388. doi: 10.1128/jvi.69.10.6376-6388.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilcock D, Smith G L. Vaccinia virus core protein VP8 is required for virus infectivity, but not for core protein processing or for INV and EEV formation. Virology. 1994;202:294–304. doi: 10.1006/viro.1994.1346. [DOI] [PubMed] [Google Scholar]
- 49.Wolffe E J, Isaacs S N, Moss B. Deletion of the vaccinia virus B5R gene encoding a 42-kilodalton membrane glycoprotein inhibits extracellular virus envelope formation and dissemination. J Virol. 1993;67:4732–4741. doi: 10.1128/jvi.67.8.4732-4741.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolffe E J, Moore D M, Peters P J, Moss B. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J Virol. 1996;70:2797–2808. doi: 10.1128/jvi.70.5.2797-2808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Moss B. Vaccinia virus morphogenesis is interrupted when expression of the gene encoding an 11-kilodalton phosphorylated protein is prevented by the Escherichia coli lac repressor. J Virol. 1991;65:6101–6110. doi: 10.1128/jvi.65.11.6101-6110.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Moss B. Immature viral envelope formation is interrupted at the same stage by lac operator-mediated repression of the vaccinia virus D13L gene and by the drug rifampicin. Virology. 1992;187:643–653. doi: 10.1016/0042-6822(92)90467-4. [DOI] [PubMed] [Google Scholar]