

Abstract
Reovirus-induced acute myocarditis in mice serves as a model to investigate non-immune-mediated mechanisms of viral myocarditis. We have used primary cardiac myocyte cultures infected with a large panel of myocarditic and nonmyocarditic reassortant reoviruses to identify determinants of viral myocarditic potential. Here, we report that while both myocarditic and nonmyocarditic reoviruses kill cardiac myocytes, viral myocarditic potential correlates with viral spread through cardiac myocyte cultures and with cumulative cell death. To address the role of secreted interferon (IFN), we added anti-IFN-α/β antibody to infected cardiac myocyte cultures. Antibody benefited nonmyocarditic more than myocarditic virus spread (P < 0.001), and this benefit was associated with the reovirus M1 and L2 genes. There was no benefit for a differentiated skeletal muscle cell line culture (C2C12 cells), suggesting cell type specificity. IFN-β induction in reovirus-infected cardiac myocyte cultures correlated with viral myocarditic potential (P = 0.006) and was associated with the reovirus M1, S2, and L2 genes. Sensitivity to the antiviral effects of IFN-α/β added to cardiac myocyte cultures also correlated with viral myocarditic potential (P = 0.004) and was associated with the same reovirus genes. Several reoviruses induced IFN-β levels discordant with their myocarditic phenotypes, and for those tested, sensitivity to IFN-α/β compensated for the anomalous induction levels. Thus, the combination of induction of and sensitivity to IFN-α/β is a determinant of reovirus myocarditic potential. Finally, a nonmyocarditic reovirus induced cardiac lesions in mice depleted of IFN-α/β, demonstrating that IFN-α/β is a determinant of reovirus-induced myocarditis. This provides the first identification of reovirus genes associated with IFN induction and sensitivity and provides the first evidence that IFN-β can be a determinant of viral myocarditis and reovirus disease.
Acute myocarditis (1) has most likely occurred in more than 5% of the human population (69). It is often fatal in infants, while in older individuals it can progress to dilated cardiomyopathy (36). A wide variety of viruses have been implicated (35, 65), but most research has focused on enteroviruses, responsible for 20 to 50% or more of human myocarditis (65). While much evidence suggests that enterovirus-induced cardiac damage in mice is mediated predominantly by the immune response (10, 51, 69), enteroviruses are cytopathic to murine cardiac myocytes (23) and they can induce myocarditis in mice lacking immune cells (8). Moreover, in a large clinical trial immunosuppressive agents were not therapeutic (37), indicating that the role of immune cells is complex. How do other viruses, responsible for 50% or more of human myocarditis, induce the disease? Little is known from human studies; however, in adenovirus- and human immunodeficiency virus-associated myocarditis, the extent of myocardial inflammation and damage does not correlate with the severity of cardiac dysfunction (13, 35), suggesting that inflammatory cells may play a minimal or indirect role. Importantly, non-immune-mediated mechanisms of cardiac tissue damage remain largely unexplored.
Reovirus-induced acute viral myocarditis in mice is characterized by a mild inflammatory infiltrate with marked necrosis (22, 61, 62), in contrast to the massive cellular infiltrate characteristic of the enterovirus-induced disease (49). Indeed, reovirus induces myocarditis in SCID mice (60) and macrophage inflammatory protein-1α (MIP-1α) knockout mice (61a), demonstrating that reovirus-induced myocarditis is not immune cell mediated. Reovirus therefore presents an ideal model for studying non-immune-mediated viral myocarditis.
The reovirus genome is composed of 10 segments of double-stranded RNA (dsRNA), and with one exception, each gene segment encodes one protein (reviewed in reference 44). “Reassortant” viruses can be generated that contain mixtures of gene segments from two virus parents, and genetic analyses with reassortant viruses have been invaluable tools in identifying gene product functions. We have used genetic analyses to identify the determinants of reovirus-induced acute myocarditis and found that the M1 gene was implicated in every analysis while the L1 and L2 gene associations with disease varied among viruses (58, 59). These three genes encode viral core proteins likely to form a structural unit involved in viral RNA synthesis in the core. Specifically, the L1-encoded λ3 protein has a polymerase function (15, 63) and lies at the base of a pentameric channel formed by the L2-encoded λ2 protein (16, 19, 40), which is a guanylyl transferase (9, 34). The M1-encoded μ2 protein, which lies adjacent to λ3 (19), has been implicated in RNA synthesis in genetic analyses (11, 57, 70) and is an RNA-binding protein (7) associated with nucleoside triphosphatase activity (47). Thus, the genetic analyses suggested that viral RNA synthesis in cardiac myocytes was likely to be involved in reovirus-induced myocarditis.
Accordingly, we used our large panel of myocarditic and nonmyocarditic reassortant reoviruses to identify parameters of reovirus replication in primary cardiac myocyte cultures that correlate with viral myocarditic potential (57). While RNA synthesis in cardiac myocytes correlated with viral myocarditic potential, the yield of infectious virus did not (57). This suggested that some other consequence of viral RNA synthesis, such as cytokine response, was likely to be a determinant of the disease. One mechanism for this would be induction of interferon (IFN).
IFNs are divided into three classes (α, β, and γ) and are synthesized by specific types of cells as one of the first responses to viruses (reviewed in reference 68). IFN-α or -β is secreted, binds to cell receptors (the type I IFN-α/β receptor), and, through a phosphorylation cascade, rapidly upregulates transcription of a large family of genes (IFN-responsive genes) that contain IFN-stimulated regulatory elements in their promoter regulatory regions (reviewed in reference 30). While IFN-α/β induces transcription of multiple genes that have antiviral activities, three of these gene products remain latent until activated (directly or indirectly) by dsRNA (reviewed in reference 27). Specifically, the dsRNA-activated protein kinase (PKR [reviewed in reference 52]), 2′,5′-oligo(A) synthetase, and RNase L (activated by the 2′,5′-oligo(A) synthetase-generated oligomers, and thus indirectly activated by dsRNA) each remain latent until dsRNA is suitably presented. Thus, our previous demonstration that reovirus RNA synthesis in cardiac myocyte cultures correlates with viral myocarditic potential could reflect differential induction of IFN and/or differential activation of the IFN-induced antiviral proteins (i.e., differential sensitivity to IFN).
In the studies presented here, we have investigated whether reovirus-induced IFN is a determinant of myocarditic potential. Our results demonstrate that in cardiac myocyte cultures, nonmyocarditic viruses induce more IFN-β and are more sensitive to IFN-α/β than myocarditic viruses and that both functions are associated with viral core proteins. Furthermore, a nonmyocarditic virus induces cardiac lesions in mice depleted of IFN-α/β. Thus, reovirus induction of and sensitivity to IFN-β are determinants of reovirus-induced myocarditis.
MATERIALS AND METHODS
Viruses and cells.
All reovirus stocks (triply plaqued and passaged twice in mouse L cells) were characterized previously for their myocarditic phenotypes (58, 59). Virus 8B is a reassortant virus derived from a mouse infected with the strains serotype 1 Lang (T1L) and serotype 3 Dearing (T3D) (59). All other reassortant viruses (Fig. 1) were derived from mouse L cells infected with the indicated viruses (59). Viruses EB121 and E3 are reassortant viruses derived from T1L and T3D. All EW-series reassortant viruses were derived from 8B and EB121, while all DB-series reassortant viruses were derived from EW60 and E3. Viral myocarditic potentials were determined by injecting 2 × 105 to 2 × 106 (58) and 4 × 106 to 5 × 107 (59) PFU into 2-day-old Cr:NIH(S) mice and examining their hearts for macroscopic lesions (gross myocarditis) at death or at 14 days postinjection (58, 59).
FIG. 1.

Panel of reassortant reoviruses used for analyses. Gene segment derivations are indicated as follows: 1, T1L origin; 3, T3D origin; black box, myocarditic (8B) origin; white box, nonmyocarditic (EB121) origin. Viral myocarditic potentials (expressed as the percent of mice with gross myocarditis) were determined previously (58, 59). NON-STRUC, nonstructural.
Primary cardiac myocyte cultures were prepared as described previously (3). Briefly, Cr:NIH(S) term fetuses or 1-day-old neonates (National Cancer Institute) were sacrificed and the apical two-thirds of their hearts were minced and trypsinized. The cell suspension was plated on 6-well clusters and incubated at 37°C in 5% CO2 to allow fibroblasts to adhere, at which time the nonadherent cells were removed to fresh 96-well clusters (myocyte cultures containing 5 to 15% fibroblasts as previously reported [3]). The cultures were incubated in Dulbecco modified Eagle medium (Gibco BRL) supplemented with 7% fetal bovine serum (HyClone) (completed DMEM) and 0.06% thymidine.
C2C12 cells are a skeletal muscle cell line (ATCC CRL-1771 [6]) maintained in completed DMEM (10% fetal bovine serum, 0.1% gentamycin). For differentiation, the cells were split 1:4 and plated at 104 per well of 96-well clusters. After 2 days the serum was lowered to 4%, and after 2 or 3 more days, more than 90% of the cells had differentiated into myotubes (by microscopic examination), which were used for infections, as for the cardiac myocyte cultures.
Infections and harvests.
Two days after preparation of primary cardiac myocyte cultures, or when C2C12 cells were differentiated (see above), viable cells were counted (typically 2 × 105 to 4 × 105 per well in each case) and infected at a multiplicity of infection (MOI) of 0.1 or 5 PFU per cell in completed DMEM with 0.06% thymidine (myocytes only). Duplicate wells were infected with each virus, except when assayed by MTT (catalog no. M-5655; Sigma) (see below), in which case triplicate wells were infected. Cultures were incubated at 37°C in 5% CO2. If indicated, cultures (300 μl before infection, 150 μl after infection) were treated with one of the following reagents: 900 international reference units (3 μl) of IFN-α/β (catalog no. I-1258; Sigma) or control buffer (100 mM NaCl, 10 mM Tris [pH 7.4], 1 mM EDTA); for additional daily additions (see Fig. 6 and 7), 90 international reference units (2 μl) of IFN-α/β or diluted control buffer (15 mM NaCl, 1.5 mM Tris [pH 7.4], 0.15 mM EDTA); or 3 μl of antibody containing 165 National Institutes of Health neutralizing units of rabbit anti-mouse IFN-α/β (catalog no. 21032; Lee Biomolecular Research, Inc., San Diego, Calif.) or control rabbit antibody.
FIG. 6.
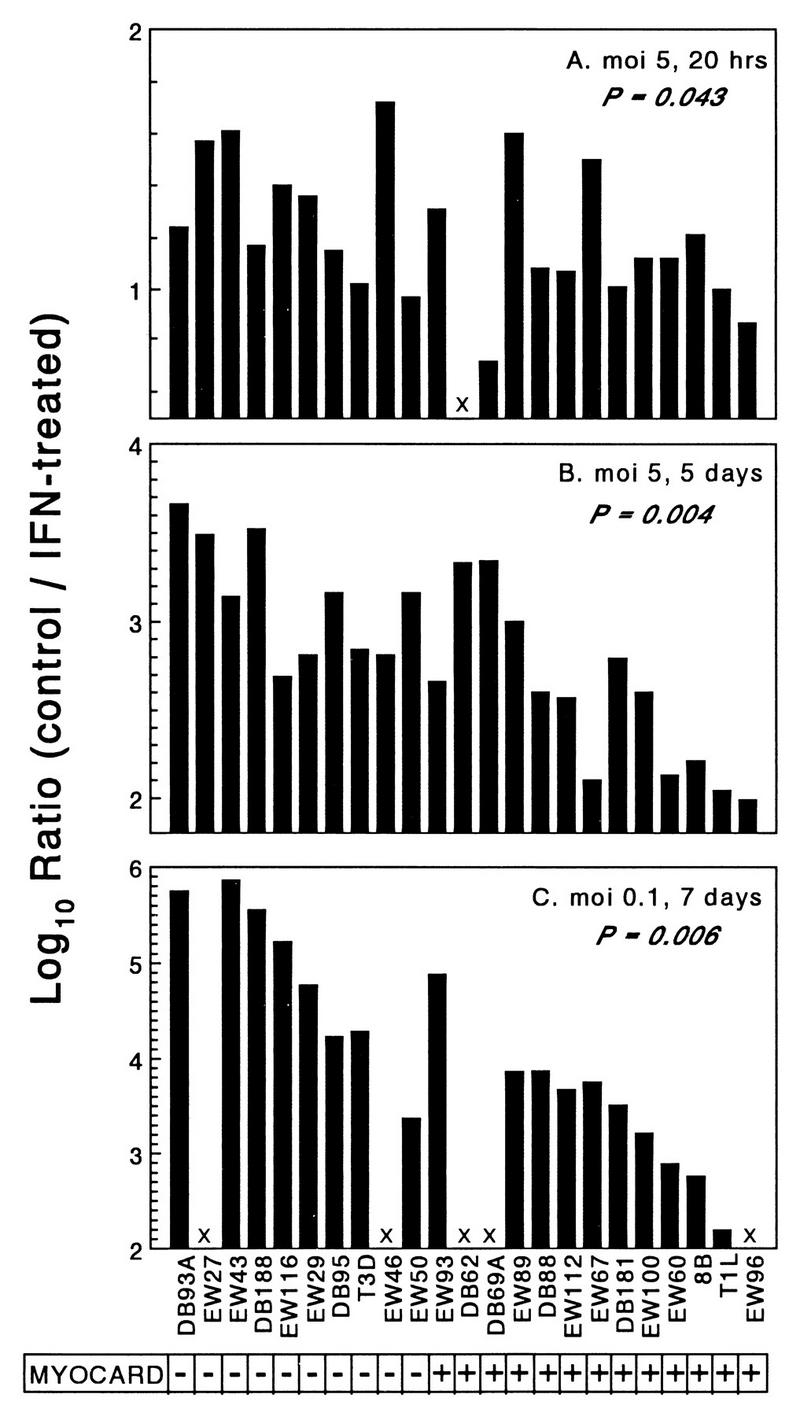
Nonmyocarditic viruses are more sensitive to IFN-α/β in cardiac myocytes than myocarditic viruses are. Primary cardiac myocyte cultures were infected under three different conditions (duplicate wells for each data point) and lysed at the indicated times, and viral titers were determined by plaque assay. Results are expressed as the ratio of viral yield in control-treated cultures relative to that in IFN-treated cultures. (A) Infected at an MOI of 5, control cultures received control buffer, IFN-treated cultures received IFN, and all cultures were harvested at 20 h postinfection. (B) Infected at an MOI of 5, control cultures received anti-IFN-α/β antibody and control buffer, IFN-treated cultures received control antibody and IFN, and all cultures were harvested at 5 days postinfection. (C) Infected at an MOI of 0.1, cultures were treated as for panel B and harvested at 7 days postinfection. MYOCARD, myocarditic potential. X, not determined.
FIG. 7.
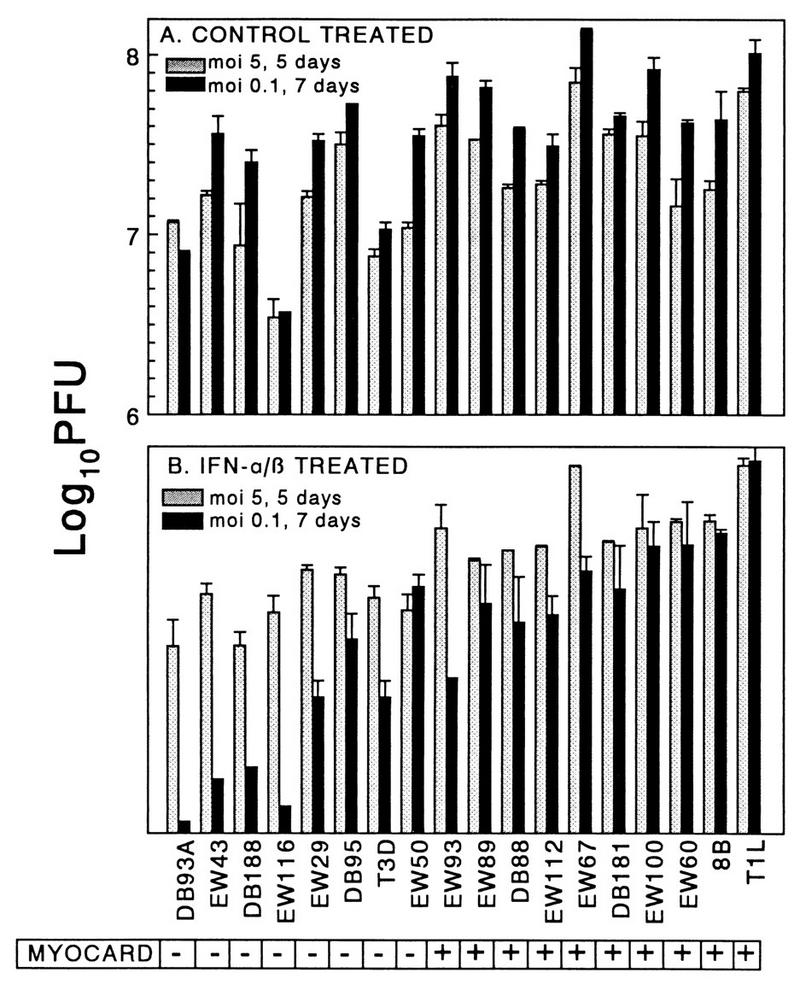
IFN-α/β inhibition of reovirus replication is amplified over time. The results from Fig. 6B and C are expressed as actual viral yields rather than ratio of viral yields. The results are expressed as the means of duplicate samples plus standard deviations. MYOCARD, myocarditic potential.
Cell viability assay.
At 20 h or 5 days postinfection, 15 μl of a solution of 6 mg of MTT/ml in completed DMEM was added to each 150-μl culture well and incubated for 4 h at 37°C in 5% CO2. The culture plates were centrifuged (750 × g) for 8 min, supernatants were aspirated, and 100 μl of 0.04 N HCl in isopropanol was added to each well. After 15 min at room temperature, 100 μl of H2O was added to each well and the optical density at 550 nm was determined on an automated microplate reader.
Plaque assays.
Culture wells were frozen at −70°C, subjected to two additional freeze-thaw cycles, and then lysed in 0.5% Nonidet P-40. Virus titers were determined by plating serial dilutions on mouse L-cell monolayers, overlaying with agar, and staining with neutral red as previously described (57).
IFN bioassay.
Supernatants were removed from the culture wells and stored at −70°C until they were ready for assay. One-third of each culture well supernatant was assayed as follows, with commercial IFN-α/β treated in parallel to generate standard curves for each assay. After diluting with MEM, reactions were acidified to pH 2 to 3 with 2 N HCl, incubated for 24 h at 4°C (to inactivate reovirus), and then neutralized with 2 N NaOH. Duplicate aliquots (equivalent to one-sixth culture well each) or control medium was added to 96-well clusters containing 100 μl of completed MEM and seeded 24 h earlier with 4 × 104 mouse L cells, and each sample was then serially twofold diluted five times. After 24 h of incubation at 37°C in 5% CO2, 4 × 105 PFU of encephalomyocarditis virus in 50 μl of completed MEM was added to each well. After 24 h of incubation at 37°C in 5% CO2, L-cell viability was determined by MTT assay as described above. IFN concentrations were calculated based on standard curves generated with commercial IFN-α/β.
IFN RT-PCR.
RNA from infected cardiac myocyte cultures was harvested 20 h postinfection as previously described (57) with TriReagent (Molecular Research Center, Cincinnati, Ohio). Pooled RNA samples from duplicate culture wells were treated with RNase-free DNase, and then half of each sample was treated with DNase-free RNase (negative controls). Mouse L-cell DNA was used as a positive control. Samples were denatured in H2O at 70°C for 5 min, snap cooled on ice, and then incubated for 1 h at 42°C in a reverse transcription (RT) reaction mixture [10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris (pH 8.8), 2 mM MgSO4, 0.1% Triton X-100, 1 mM dithiothreitol, 2.5 μM oligo(dT), 1 mM each deoxynucleoside triphosphate, 0.5 U of RNA inhibitor/μl, and 15 U of avian myoblastosis virus reverse transcriptase]. Each reaction mixture was divided into three aliquots for PCR. Primers for PCR were as follows. For IFN-α, the known mouse IFN-α gene sequences (murine IFN-α 1 through 8) were aligned and nucleotide regions conserved among all sequences were identified to design an upstream primer (TCTCTCCTGCCTGAAGGAC; bases 147 to 165) and a downstream primer (TCCTCACAGCCAGCAGGG; bases 416 to 433) predicted to generate a 286-bp product. For IFN-β, sequences were identified that had no homology with any of the IFN-α genes to design an upstream primer (TTCGGAAATGTCAGGAGCTC; bases 104 to 123) and a downstream primer (CTGCAACCACCACTCATTCT; bases 633 to 654) predicted to generate a 550-bp product. For glyceraldehyde-3-phosphate dehydrogenase (G3PDH), the upstream primer (TCACCACCATGGAGAAGGC; bases 345 to 363) and downstream primer (CAAAGTTGTCATGGATGACC; bases 526 to 545) were predicted to generate a 200-bp product. In each case, the upstream primer was between 700 and 900 bases from the 3′ end of the mRNA. PCR was conducted in 100 μl of reaction mixture (1× Taq DNA polymerase buffer, 1.5 mM MgCl2, 0.2 mM each deoxynucleoside triphosphate, 1 μM each primer) overlaid with mineral oil and hot started with 1 μl of Taq DNA polymerase. The reactions were incubated at 99°C for 2 min, 52°C for 1 min, and 72°C for 1 min and then were incubated for 35 cycles at 94°C for 1 min, 52°C for 1 min, and 72°C for 1 min. Reactions were completed with a 5-min incubation at 72°C, and then 10% was electrophoresed on a 3% NuSieve GTG agarose–1% agarose gel and visualized by ethidium bromide staining. RNase-treated samples (negative controls) generated no detectable products (data not shown), indicating that RT-PCR products were generated from RNA, not DNA, templates.
Mouse depletion of IFN-α/β, injection, and analysis.
Two-day-old randomized litters of Cr:NIH(S) mice (National Cancer Institute) were injected intraperitoneally with 1,100 National Institutes of Health neutralizing units (20 μl) of rabbit anti-mouse IFN-α/β (see above) or control rabbit antibody. Six hours later, the mice were injected intramuscularly in the hindlimb with 20 μl of MEM containing 2 × 105 PFU of the nonmyocarditic reassortant reovirus DB188 (Fig. 1). At 2 and 4 days postinjection, the mice were injected with anti-IFN-α/β or control antibody as described above. At 7 days postinjection, the mice were sacrificed and their hearts were removed to 10% buffered formalin for sectioning and hematoxylin-eosin staining. Slides containing 27 to 30 sections from each heart were scored (by an observer with no knowledge of the heart source) for independent cardiac lesions (i.e., cardiac lesions found in consecutive sections were scored only once).
Statistical analysis.
The nonparametric Kruskal-Wallis analysis, provided in Systat software (SPSS Federal Systems, Chicago, Ill.), was used. A P value less than or equal to 0.05 was considered significant.
RESULTS
Cumulative viral CPE in primary cardiac myocyte cultures correlates with viral myocarditic potential.
When a panel of myocarditic and nonmyocarditic reassortant viruses was used to infect primary cardiac myocyte cultures at an MOI of 5 PFU per cell, cytopathic effect (CPE) at 3 days postinfection correlated with viral myocarditic potential (3). Later experiments revealed that despite our using an MOI statistically sufficient to infect all of the cells, only a fraction of the cells were initially infected and additional cells were then infected by released progeny virus (57). Thus, measurements made several days postinfection actually reflected both primary and secondary infections (secondary infections made possible perhaps by the high local concentration of virus generated from primary infections). To determine whether viral cytopathogenicity in primary infections alone is also a determinant of viral myocarditic potential, a panel of myocarditic and nonmyocarditic viruses was used to compare CPE in primary infections (at 20 h postinfection, sufficient for one cycle of replication [57]) with CPE following spread to secondary infections (at 5 days postinfection). The results (Fig. 2) confirmed our initial observations that CPE following viral spread through the culture correlates with viral myocarditic potential (P = 0.004). This CPE was associated with the viral M1 (P = 0.004) and S1 (P = 0.002) genes. In contrast, CPE in primary infections did not correlate with viral myocarditic potential (P > 0.05), despite evidence of varying CPE (killing 4 to 33% of the cells). This CPE was associated only with the S1 gene (P = 0.001). Thus, both myocarditic and nonmyocarditic reoviruses kill cardiac myocytes but myocarditic reoviruses induce greater cumulative cell death, perhaps reflecting more efficient spread between myocytes.
FIG. 2.
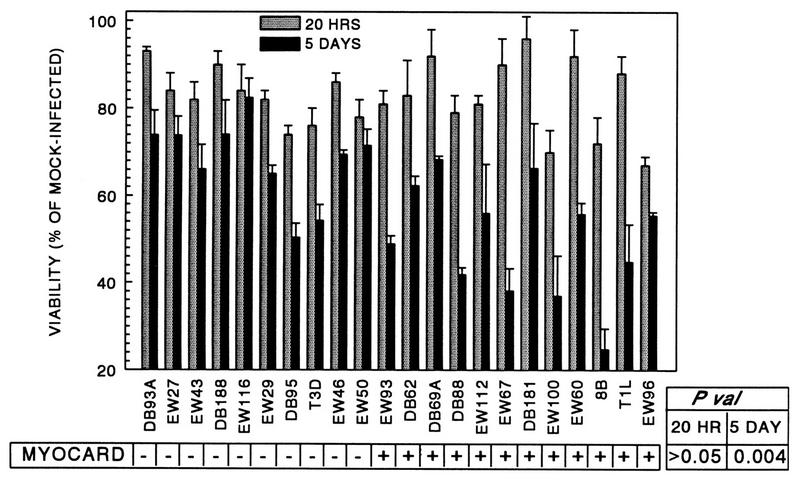
Cumulative cell death in primary cardiac myocyte cultures correlates with viral myocarditic potential. Primary cardiac myocyte cultures were infected with reoviruses at an MOI of 5 PFU per cell. At 20 h or 5 days postinfection, cultures were harvested and cell viability was determined by MTT metabolic assay. Results are expressed as the mean percent viability of triplicate wells relative to that of mock-infected controls plus the standard deviation. P val, P value; MYOCARD, myocarditic potential.
IFN-α/β is a determinant of viral spread between cardiac myocytes and correlates with viral myocarditic potential.
In primary cardiac myocyte cultures, viral RNA synthesis during initial (primary) infections and viral spread and cumulative cell death correlate with viral myocarditic potential, irrespective of viral yield from the primary infections (57) and of cell death during the primary infections (above). One mechanism by which viral RNA synthesis can determine viral spread is by induction of IFN during primary infections, which would subsequently affect the efficiency of secondary infections. To test whether IFN was a determinant of viral spread, cardiac myocyte cultures were infected with a panel of viruses at an MOI of 0.1 PFU per cell, anti-IFN-α/β antibody or control antibody was added, and viral yields were determined at 7 days postinfection (Fig. 3). Viral replication in cultures receiving control antibody varied 3 logs between viruses (Fig. 3A) and correlated with the potential to induce myocarditis (P < 0.001), confirming our earlier observations (38, 58). This replication was associated with the viral M1 (P < 0.001) and L2 (P = 0.049) genes. Anti-IFN-α/β antibody enhanced replication of every virus tested (Fig. 3A), demonstrating that induced IFN was a determinant of replication and spread for every virus. The benefit from anti-IFN-α/β antibody, however, varied 1,000-fold between viruses (Fig. 3B) and correlated with induction of myocarditis (P < 0.001) and the M1 (P < 0.001) and L2 (P = 0.012) genes. To determine whether induced IFN inhibits reovirus spread in other types of cells, the experiment was repeated in a differentiated skeletal muscle cell line (C2C12 cells). In these differentiated muscle cells, viral yield in control cultures varied only 1.5 logs between viruses (6.4 to 8.1 log10 PFU per well) and did not correlate with viral myocarditic potential (data not shown). The benefit from anti-IFN-α/β antibody was maximally only threefold (Fig. 3B), and the benefit did not correlate with viral myocarditic potential. Therefore, the role of IFN-α/β in controlling reovirus spread is cell type specific, and in cardiac myocyte cultures, nonmyocarditic viruses induce more IFN-α/β and/or are more sensitive to the antiviral effects of IFN-α/β than myocarditic viruses are.
FIG. 3.
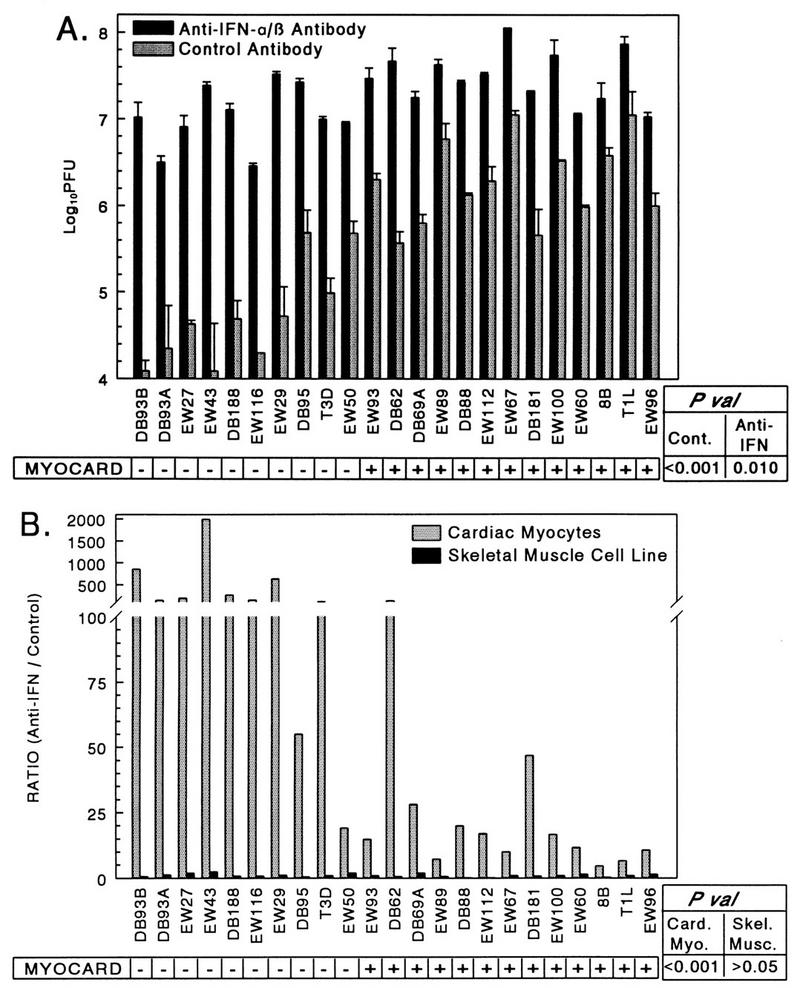
IFN-α/β is a determinant of viral spread between cardiac myocytes but not between differentiated skeletal muscle cells. Replicate wells of primary cardiac myocyte cultures or a differentiated skeletal muscle cell line (C2C12 cells) were infected at an MOI of 0.1 PFU per cell and overlaid with anti-IFN-α/β antibody or control antibody. At 7 days postinfection, the cultures were lysed and viral titers were determined by plaque assay. (A) Results from cardiac myocyte cultures, expressed as mean viral yield of duplicate wells plus standard deviation (skeletal muscle cell culture data is not shown). (B) Results from cardiac myocyte cultures and skeletal muscle cell cultures, expressed as the ratio of viral yield in anti-IFN-α/β antibody-treated wells relative to that in control-treated wells. P val, P values; Card. Myo., cardiac myocytes; Skel. Musc., skeletal muscle cells; Cont., control antibody; MYOCARD, myocarditic potential.
Nonmyocarditic viruses induce more IFN-β in infected cardiac myocyte cultures than myocarditic viruses do.
To determine whether the anti-IFN-α/β antibody benefit to viral spread reflected induction of IFN, cardiac myocyte cultures were infected with a panel of viruses at an MOI of 5 PFU per cell and culture supernatants were titered by bioassay at 10 and 20 h postinfection for IFN levels (Fig. 4). At 10 h postinfection, 8 of 12 nonmyocarditic viruses had induced detectable levels of IFN while only 2 of 15 myocarditic viruses had done so, and the magnitude of IFN induction correlated with viral myocarditic potential (P = 0.006). This IFN induction was associated with the M1 (P = 0.040), S2 (P = 0.004), and L2 (P = 0.037) genes. At 20 h postinfection, 22 of the 27 viruses induced detectable IFN, and again, the magnitude of IFN induction correlated with viral myocarditic potential (P = 0.007). This IFN induction was again associated with the M1 (P = 0.038), S2 (P = 0.012), and L2 (P = 0.044) genes. Thus, nonmyocarditic viruses induced more IFN than myocarditic viruses did in cardiac myocyte cultures.
FIG. 4.

Nonmyocarditic viruses induce more IFN-α/β in cardiac myocytes than myocarditic viruses do. Primary cardiac myocyte cultures were infected at an MOI of 5 PFU per cell, and supernatants were removed at 10 and 20 h postinfection. After acidification (to inactivate reovirus) and neutralization of samples and commercial IFN-α/β standards, IFN units were determined by bioassay. P val, P value; MYOCARD, myocarditic potential.
To distinguish between IFN-α and IFN-β, cardiac myocyte cultures were mock infected or infected with each of three nonmyocarditic viruses that induced high (T3D and EW29) or low (DB93A) levels of IFN (Fig. 4). RNA was harvested at 20 h postinfection for RT-PCR (Fig. 5). IFN-β mRNA was amplified from each of the three infected cultures but not from the mock infection. In contrast, IFN-α mRNA was not amplified from any culture, despite the strong positive signal from a DNA control. The G3PDH mRNA, a control constitutively expressed gene, was amplified from all cultures, verifying the quality of the RNA harvests. Therefore, the IFN induced by nonmyocarditic viruses (Fig. 4) is IFN-β.
FIG. 5.

Reovirus-induced IFN in cardiac myocytes is IFN-β. Primary cardiac myocyte cultures were infected with three nonmyocarditic reoviruses or mock infected, and at 20 h postinfection RNA was harvested. Following DNase treatment, cDNAs were generated with oligo(dT) and PCR amplification was performed with primer pairs specific for IFN-α, IFN-β, and G3PDH genes. One RT-PCR sample contained no RNA template (negative control), and one sample contained mouse DNA (positive control). Samples (10%) were electrophoresed and ethidium bromide stained. Control samples that were RNase treated before RT-PCR generated no bands (data not shown). MW, molecular weight standards.
Nonmyocarditic viruses are more sensitive to IFN-α/β in infected cardiac myocyte cultures than myocarditic viruses are.
In addition to the observed varying induction of IFN-β, the anti-IFN-α/β benefit to viral spread could reflect varying viral sensitivity to IFN-β’s antiviral effects. To measure viral sensitivity to IFN-α/β’s antiviral effects, cardiac myocyte cultures were treated with IFN-α/β or control buffer, infected 24 h later with a panel of viruses at an MOI of 5 PFU per cell, and titered for virus at 20 h postinfection. For every virus tested, pretreatment with IFN-α/β decreased viral yield compared to control treatment, indicating that all viruses were sensitive to IFN-α/β’s antiviral effects. However, the IFN-α/β inhibition of replication varied only 1 log between viruses (Fig. 6A). Despite this narrow range, the degree of inhibition correlated with viral myocarditic potential (P = 0.043) and was associated with the reovirus M1 gene (P = 0.043). The experiment was repeated, but in order to amplify the effects of IFN-α/β, the infections were not harvested until 5 days postinfection to allow multiple rounds of infection through the culture. Because some viruses induced more IFN-β than others (Fig. 4), the control-treated cultures received anti-IFN-α/β antibody instead of buffer alone, so that IFN-α/β-treated cultures could be compared to cultures devoid of IFN. Again, IFN-α/β treatment decreased viral yield and, as expected, did so to a greater extent than when measured at 20 h postinfection, although the inhibition varied only 1.7 logs between viruses. Again the degree of IFN-α/β inhibition correlated with viral myocarditic potential (Fig. 6B) (P = 0.004), and was associated with the reovirus M1 and S2 genes (P = 0.004 and 0.020, respectively). To further amplify the effects of IFN-α/β, an MOI of 0.1 PFU per cell was used, IFN-α/β or anti-IFN-α/β was added every day, and the cultures were not harvested until 7 days postinfection. Under these conditions, the degree of inhibition varied 4.5 logs between viruses (Fig. 6C) and again correlated with the potential to induce myocarditis (P = 0.006) and was associated with the reovirus M1 and L2 genes (P = 0.006 and 0.013, respectively). The dramatic difference in IFN-α/β effects at an MOI of 0.1 and harvested at 7 days (Fig. 6C) compared to an MOI of 5 harvested at 5 days (Fig. 6B) did not reflect differences in replication in the presence of anti-IFN-α/β antibody (Fig. 7A) but instead reflected differences in the effects of IFN-α/β (Fig. 7B). Replication following IFN-α/β treatment was reduced as much as 6 logs under conditions of low MOI and longer incubation time (Fig. 7B), presumably reflecting the cumulative effects of IFN inhibition of viral replication through multiple rounds of infection. Thus, in addition to inducing more IFN-β in cardiac myocyte cultures, nonmyocarditic viruses were also more sensitive to IFN-α/β’s antiviral effects in those cells than myocarditic viruses were.
A nonmyocarditic reovirus induces cardiac lesions in mice depleted of IFN-α/β.
In order to determine directly whether IFN-α/β is a determinant of reovirus-induced myocarditis in mice, neonates were injected with anti-IFN-α/β or control antibody and challenged with a nonmyocarditic reassortant reovirus, DB188 (Fig. 1). At 7 days postinjection, slides containing 27 to 30 cardiac sections per mouse were examined for independent lesions (i.e., cardiac lesions found in consecutive sections were scored only once). While none of the 86 sections from the three mice injected with control antibody contained a single cardiac lesion, each of the four mice injected with anti-IFN-α/β antibody contained six to nine independent cardiac lesions (Table 1 and Fig. 8). Thus, IFN-α/β is a determinant of reovirus myocarditic potential. Since the antibody neutralized an unknown fraction of IFN-α/β, and it is uncertain whether neutralization was uniform throughout the tissues (including the heart), it is likely that complete and uniform depletion would magnify the effects seen here.
TABLE 1.
Frequency of necrotic lesions in cardiac sections from DB188-infected mice
| Mouse | Treatment antibody | No. cardiac sections examined | No. of independent lesionsa |
|---|---|---|---|
| 1 | Control | 29 | 0 |
| 2 | Control | 29 | 0 |
| 3 | Control | 28 | 0 |
| 4 | Anti-IFN-α/β | 25 | 6 |
| 5 | Anti-IFN-α/β | 30 | 6 |
| 6 | Anti-IFN-α/β | 28 | 6 |
| 7 | Anti-IFN-α/β | 27 | 9 |
A lesion was scored only once if found on consecutive cardiac sections.
FIG. 8.
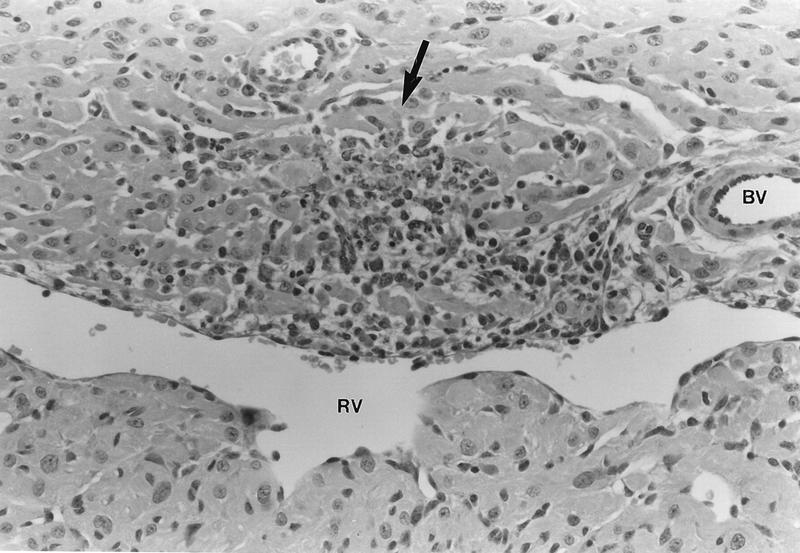
A nonmyocarditic reovirus induces cardiac lesions in mice depleted of IFN-α/β. Neonatal mice were injected with anti-IFN-α/β antibody or control antibody and then challenged with the nonmyocarditic reassortant reovirus DB188 (Fig. 1). Following two more antibody injections, mice were sacrificed at 7 days postinjection with virus and cardiac sections were hematoxylin and eosin stained (results are summarized in Table 1). The photograph represents a typical lesion observed in mice injected with anti-IFN-α/β antibody (magnification, ×80). While an inflammatory infiltrate is evident in the necrotic region (arrow), our previous investigations (60) suggest that this mediates protection rather than damage. RV, right ventricular chamber; BV, blood vessel.
DISCUSSION
Reovirus-induced acute myocarditis in mice serves as a model to investigate non-immune-mediated mechanisms of myocarditis. Previously, we demonstrated that reovirus RNA synthesis in cardiac myocytes correlates with viral myocarditic potential but that generation of infectious virus does not, suggesting that some other consequence of viral RNA synthesis determines disease. We demonstrate here that while both nonmyocarditic and myocarditic reoviruses can kill cardiac myocytes, viral spread through primary cardiac myocyte cultures and the resulting cumulative cell death correlate with viral myocarditic potential. Anti-IFN-α/β antibody has a greater effect on the spread of nonmyocarditic than myocarditic reoviruses, and this is due to both increased induction of IFN-β and increased sensitivity to the antiviral effects evoked by IFN-α/β. Finally, a nonmyocarditic reovirus induces cardiac lesions in mice depleted of IFN-α/β, demonstrating directly that IFN is a determinant of reovirus-induced myocarditis. These results provide the first evidence that viruses induce IFN in cardiac myocyte cultures and that this IFN is a determinant of viral myocarditis.
We have shown here that both myocarditic and nonmyocarditic reoviruses can kill cardiac myocytes in culture (Fig. 2). While we have not directly assayed for apoptosis, our previous electron microscopic studies of infected cardiac sections were consistent with necrosis alone (61). Reovirus pathogenesis in the mouse has been well studied (66, 67), and recent evidence has demonstrated that reoviruses can induce apoptosis in central nervous system cells (48) and in other cell types (50); we will directly address apoptosis in future investigations. This is a particularly intriguing possibility, given our previous evidence that viral RNA synthesis in cardiac myocytes correlates with viral myocarditic potential and the recent report that dsRNA in vaccinia virus-infected cells induces apoptosis (28).
While both nonmyocarditic and myocarditic reoviruses kill cardiac myocytes, viral spread and the resulting cumulative cell death correlate with viral myocarditic potential (Fig. 2). While this could have merely reflected efficiency of infection during the initial infections and/or yield of virus from the initial infections, we have shown that neither of those parameters correlates with viral myocarditic potential (57). Instead, our previous evidence suggested that viral RNA synthesis in the initial infections determines spread and viral myocarditic potential (57), consistent with a model where reovirus-induced IFN in initial infections controlled the spread of the virus to neighboring cells. Indeed, in the present study we found that in cardiac myocyte cultures, anti-IFN-α/β antibody had a greater effect on the spread of nonmyocarditic reoviruses than myocarditic reoviruses (Fig. 3A). Interestingly, anti-IFN-α/β had little effect on virus spread in the C2C12 differentiated skeletal muscle cell culture (Fig. 3B). The range of viral yields from control treated C2C12 cells was similar to that in anti-IFN-α/β antibody-treated cardiac myocyte cultures, suggesting that virus spread through C2C12 cell cultures occurred as if in the absence of IFN. We are currently investigating whether this reovirus behavior in C2C12 cells reflects failure to induce IFN, resistance to IFN, or both.
Using a bioassay (Fig. 4) and RT-PCR (Fig. 5), we demonstrated that nonmyocarditic reoviruses induced more IFN-β than myocarditic reoviruses did in cardiac myocyte cultures. Several nonmyocarditic viruses induced less IFN than would be expected (DB93A, DB93B, DB188, EW27, EW46, and EW116) given the overall correlation between high IFN induction and low myocarditic potential. Five of those six viruses were tested for sensitivity to the antiviral effects of IFN (Fig. 6) (DB93B was not tested). Of those five, four were among the most IFN sensitive of the 27 viruses tested. The remaining virus (EW46) was the most sensitive under one set of conditions (Fig. 6A), was intermediate under another set (Fig. 6B), and was not tested in the third set (Fig. 6C). Thus, while some nonmyocarditic reoviruses may induce only moderate levels of IFN, their marked sensitivity to IFN apparently compensates to control virus spread and myocarditis. Two myocarditic reoviruses (EW25 and 8B) induced more IFN than would be expected; the IFN sensitivity of one (8B) was tested (Fig. 6), and it was among the least IFN sensitive of the 27 viruses tested. Thus, resistance to IFN can also compensate for IFN induction to determine virus spread and myocarditis. Reoviruses have been shown to induce IFN in several cell lines (18, 29, 33), but reovirus induction of IFN in differentiated cells or primary cell cultures has not previously been investigated. Indeed, IFN induction in cardiac myocyte cultures has not been investigated previously for any viruses. IFN-β transcription is stimulated by a number of viruses and by dsRNA and is controlled by a complex panel of positive and negative regulatory factors (reviewed in reference 24). We are currently investigating the role of several regulatory factors in reovirus induction of IFN in cardiac myocyte cultures.
Using three different infection conditions, we found that nonmyocarditic reoviruses were more sensitive to IFN-α/β added to cardiac myocyte cultures than myocarditic reoviruses were (Fig. 6). Reoviruses are sensitive to IFN treatment in many cell types. As described in the introduction, PKR, 2′,5′-oligo(A) synthetase, and RNase L are all induced by IFN but are only activated (directly or indirectly) in the presence of dsRNA (27), which is stimulatory even when provided only as a local secondary structure in single-stranded RNA. Reoviruses activate PKR in fibroblast cell lines, resulting in phosphorylation of the α subunit of eukaryotic initiation factor 2 and inhibition of host protein synthesis (21, 25, 32). While many studies have demonstrated that PKR activation can inhibit viral replication, including reovirus replication (12, 41, 45, 54), 2′,5′-oligo(A) synthetase can be critical to the IFN-induced antiviral state and the anti-reoviral state as well (2, 46). The relative importance of each of these effectors to the antiviral effect is virus strain specific and cell specific. For example, IFN-β treatment of mouse L929 cells inhibits reovirus strain Dearing replication more than it does reovirus strain Lang replication (26). While PKR mediates the antiviral effect in mouse L929 cells (5, 53), IFN-β can inhibit reovirus replication in other mouse cell lines without concomitant elevation of PKR activity (31) and IFN treatment of HeLa cells results in RNase L-mediated cleavage of reovirus RNA (2, 46). We are currently investigating the mediators of reovirus sensitivity to IFN in cardiac myocyte cultures.
Both induction of and sensitivity to IFN-β in cardiac myocyte cultures were associated with the viral M1, S2, and L2 genes (Fig. 4 and 6). The M1-encoded μ2 protein is an RNA-binding protein, the L2-encoded λ2 protein is a guanylyl transferase, and both proteins are found at the icosahedral vertices of the core in a complex that likely functions in core viral RNA synthesis (see the introduction). The S2-encoded ς2 protein is an abundant core protein (reviewed in reference 44) with no known function, but which has limited homology with an Escherichia coli RNA polymerase (14). Together, the data are consistent with a mechanism where reovirus RNA synthesis determines both induction of IFN-β and sensitivity to IFN-α/β. In our studies of viral RNA synthesis in cardiac myocyte cultures (57), however, we found that myocarditic reoviruses generate single-stranded RNA and dsRNA faster than nonmyocarditic viruses. Those results are inconsistent with a mechanism where the rate of reovirus dsRNA synthesis directly determines induction of and sensitivity to IFN. An alternative explanation is that the rate of reovirus synthesis determines the availability of reovirus mRNA for translation and that the latter determines reovirus induction of and sensitivity to IFN. Indeed, the reovirus S4-encoded ς3 protein inhibits PKR, a mediator of the antiviral effects of IFN. Specifically, genetic analyses have identified the reovirus S4 gene as determining the level of inhibition of host protein synthesis (56), and multiple studies have demonstrated that ς3 can bind dsRNA to abrogate activation of PKR (4, 25, 32) and therefore abrogate phosphorylation of the α subunit of eukaryotic initiation factor 2 and inhibition of protein synthesis. The association of ς3 with the M2-encoded μ1c protein inhibits ς3 binding to dsRNA, and consequently ς3-μ1c association is also a determinant of PKR activation and inhibition of protein synthesis (55, 71). Given these ς3 and μ1c functions, why weren’t the S4 and M2 genes identified as determinants of reovirus induction of and sensitivity to IFN in our genetic analyses? One likely explanation is that the ς3 proteins encoded by the T1L- and T3D-derived S4 genes function quite similarly to each other in inhibition of protein synthesis compared to the ς3 proteins from other reovirus strains (55), and thus a genetic analysis of these two alleles would not identify them as determinants of the degree of inhibition of protein synthesis. Thus, while previous studies have implicated particular reovirus proteins (ς3 and μ1c) in regulating cell functions that are activated by dsRNA, our results suggest that the kinetics of reovirus protein synthesis (as determined by viral RNA synthesis kinetics) are also important determinants of these cell functions. Indeed, our results are completely consistent with previous evidence that the degree of inhibition of host protein synthesis correlates with the kinetics of reovirus protein synthesis but not with the final yield of virus (43).
Finally, a nonmyocarditic reovirus induced multiple cardiac lesions in each of four mice depleted of IFN-α/β (Table 1 and Fig. 8), demonstrating directly that IFN-α/β is a determinant of reovirus-induced myocarditis. In many virus systems, IFN-α/β plays a critical role in determining survival of the infected host, as demonstrated with anti-IFN-α/β antiserum (20) and, more recently, knockout mice that eliminate IFN activity (17, 39, 42, 64). Currently, we are using IFN-α/β receptor knockout mice to continue our investigations of the role of IFN-α/β in reovirus-induced myocarditis.
ACKNOWLEDGMENTS
We are grateful to Bob Johnston, Nancy Davis, William Klimstra, Fred Fuller, and Diana Noah for many invaluable discussions.
This research was supported by Public Health Service grant AI-31250 from the NIAID and grant 204743 from the North Carolina State University College of Veterinary Medicine.
Footnotes
This article is dedicated to Bernie Fields, who knew long ago that IFN would prove to be critical in reovirus disease.
REFERENCES
- 1.Aretz H T, Billingham M E, Edwards W D, Factor S M, Fallon J T, Fenoglio J J, Olsen E G J, Schoen F J. Myocarditis: a histopathologic definition and classification. Am J Cardiovasc Pathol. 1986;1:3–14. [PubMed] [Google Scholar]
- 2.Baglioni C, De Benedetti A, Williams G J. Cleavage of nascent reovirus mRNA by localized activation of the 2′-5′-oligoadenylate-dependent endoribonuclease. J Virol. 1984;52:865–871. doi: 10.1128/jvi.52.3.865-871.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baty C J, Sherry B. Cytopathogenic effect in cardiac myocytes but not in cardiac fibroblasts is correlated with reovirus-induced acute myocarditis. J Virol. 1993;67:6295–6298. doi: 10.1128/jvi.67.10.6295-6298.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beattie E, Denzler K L, Tartaglia J, Perkus M E, Paolettie E, Jacobs B L. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J Virol. 1995;69:499–505. doi: 10.1128/jvi.69.1.499-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bischoff J R, Samuel C E. Mechanism of interferon action. Activation of the human P1/eIF-2 alpha protein kinase by individual reovirus s-class mRNAs: s1 mRNA is a potent activator relative to s4 mRNA. Virology. 1989;172:106–115. doi: 10.1016/0042-6822(89)90112-8. [DOI] [PubMed] [Google Scholar]
- 6.Blau H M, Choy-Pik C, Webster C. Cytoplasmic activation of human nuclear genes in stable heterokaryons. Cell. 1983;32:1171–1180. doi: 10.1016/0092-8674(83)90300-8. [DOI] [PubMed] [Google Scholar]
- 7.Brentano, L., D. Noah, E. G. Brown, and B. Sherry. The reovirus protein mu 2, encoded by the M1 gene, is an RNA-binding protein. Submitted for publication. [DOI] [PMC free article] [PubMed]
- 8.Chow L H, Beisel K W, McManus B M. Enteroviral infection of mice with severe combined immunodeficiency. Evidence for direct viral pathogenesis of myocardial injury. Lab Invest. 1992;66:24–31. [PubMed] [Google Scholar]
- 9.Cleveland D R, Zarbl H, Millward S. Reovirus guanylyltransferase is L2 gene product lambda 2 protein. J Virol. 1986;60:307–311. doi: 10.1128/jvi.60.1.307-311.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook D N, Beck M A, Coffman T M, Kirby S L, Sheridan J F, Pragnell I B, Smithies O. Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science. 1995;269:1583–1585. doi: 10.1126/science.7667639. [DOI] [PubMed] [Google Scholar]
- 11.Coombs K. Identification and characterization of a double-stranded RNA reovirus temperature-sensitive mutant defective in minor core protein mu2. J Virol. 1996;70:4237–4245. doi: 10.1128/jvi.70.7.4237-4245.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Benedetti A, Williams G J, Comeau L, Baglioni C. Inhibition of viral mRNA translation in interferon-treated L cells infected with reovirus. J Virol. 1985;55:588–593. doi: 10.1128/jvi.55.3.588-593.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Castro S, D’Amati G, Gallo P, Cartoni D, Santopadre P, Vullo V, Cirelli A, Migliau G. Frequency of development of acute global left ventricular dysfunction in human immunodeficiency virus infection. J Am Coll Cardiol. 1994;24:1018–1024. doi: 10.1016/0735-1097(94)90864-8. [DOI] [PubMed] [Google Scholar]
- 14.Dermody T S, Schiff L A, Nibert M L, Coombs K M, Fields B N. The S2 gene nucleotide sequences of prototype strains of the three reovirus serotypes: characterization of reovirus core protein ς2. J Virol. 1991;65:5721–5731. doi: 10.1128/jvi.65.11.5721-5731.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drayna D, Fields B N. Activation and characterization of the reovirus transcriptase: genetic analysis. J Virol. 1982;41:110–118. doi: 10.1128/jvi.41.1.110-118.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dryden J A, Wang G, Yeager M, Nibert M L, Coombs K M, Furlong D B, Fields B N, Baker T S. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol. 1993;122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durbin J E, Hackenmiller R, Simon M C, Levy D E. Targeted disruption of the mouse STAT1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- 18.Ellis M N, Eidson C S, Brown J, Kleven S H. Studies on the interferon induction and interferon sensitivity of avian reoviruses. Avian Dis. 1983;27:927–936. [PubMed] [Google Scholar]
- 19.Farsetta D L, Dryden K A, Nibert M L. Scientific program and abstracts. American Society for Virology annual meeting. 1996. Identification of three proteins in reovirus top component particles that contribute to an internal structure seen by cryo-electron microscopy and image reconstruction, abstr. W10-9. [Google Scholar]
- 20.Gresser I, Tovey M G, Maury C, Bandu M-T. Role of interferon in the pathogenesis of virus diseases in mice as demonstrated by the use of anti-interferon serum. II. Studies with herpes simplex, Moloney sarcoma, vesicular stomatitis, Newcastle disease, and influenza viruses. J Exp Med. 1976;144:1316–1322. doi: 10.1084/jem.144.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta S L, Holmes S L, Mehra L L. Interferon action against reovirus: activation of interferon-induced protein kinase in mouse L929 cells upon reovirus infection. Virology. 1982;120:495–499. doi: 10.1016/0042-6822(82)90051-4. [DOI] [PubMed] [Google Scholar]
- 22.Hassan S A, Rabin E R, Melnick J L. Reovirus myocarditis in mice: an electron microscopic, immunofluorescent, and virus assay study. Exp Mol Pathol. 1965;4:66–80. doi: 10.1016/0014-4800(65)90024-9. [DOI] [PubMed] [Google Scholar]
- 23.Herzum M, Ruppert V, Kuytz B, Jomaa H, Nakamura I, Maisch B. Coxsackievirus B3 infection leads to cell death of cardiac myocytes. J Mol Cell Cardiol. 1994;26:907–913. doi: 10.1006/jmcc.1994.1108. [DOI] [PubMed] [Google Scholar]
- 24.Hiscott J, Nguyen H, Lin R. Molecular mechanisms of interferon beta gene induction. Semin Virol. 1995;6:161–174. [Google Scholar]
- 25.Imani F, Jacobs B. Inhibitory activity for the interferon-induced protein kinase is associated with the reovirus serotype 1 sigma 3 protein. Proc Natl Acad Sci USA. 1988;85:7887–7891. doi: 10.1073/pnas.85.21.7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs B L, Ferguson R E. The Lang strain of reovirus serotype 1 and the Dearing strain of reovirus serotype 3 differ in their sensitivities to beta interferon. J Virol. 1991;65:5102–5104. doi: 10.1128/jvi.65.9.5102-5104.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacobs B L, Langland J O. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219:339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- 28.Kibler K V, Shors T, Perkins K B, Zeman C C, Banaszak M P, Biesterfeldt J, Langland J O, Jacobs B L. Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J Virol. 1997;71:1992–2003. doi: 10.1128/jvi.71.3.1992-2003.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai M-H T, Joklik W K. The induction of interferon by temperature-sensitive mutants of reovirus. UV-irradiated reovirus, and subviral reovirus particles. Virology. 1973;51:191–204. doi: 10.1016/0042-6822(73)90379-6. [DOI] [PubMed] [Google Scholar]
- 30.Levy D E. Interferon induction of gene expression through the JAK-STAT pathway. Semin Virol. 1995;6:181–190. [Google Scholar]
- 31.Lewis J A. Induction of an antiviral state by interferon in the absence of elevated levels of 2,5-oligo(A) synthetase and eIF-2 kinase. Virology. 1988;162:118–127. doi: 10.1016/0042-6822(88)90400-x. [DOI] [PubMed] [Google Scholar]
- 32.Lloyd R M, Shatkin A J. Translational stimulation by reovirus polypeptide ς3: substitution for VAI RNA and inhibition of phosphorylation of the α subunit of eukaryotic initiation factor 2. J Virol. 1992;66:6878–6884. doi: 10.1128/jvi.66.12.6878-6884.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long W F, Burke D C. Interferon production by double-stranded RNA: a comparison of interferon induction by reovirus RNA to that by a synthetic double-stranded polynucleotide. J Gen Virol. 1971;12:1–11. doi: 10.1099/0022-1317-12-1-1. [DOI] [PubMed] [Google Scholar]
- 34.Mao Z X, Joklik W K. Isolation and enzymatic characterization of protein lambda 2, the reovirus guanylyltransferase. Virology. 1991;185:377–386. doi: 10.1016/0042-6822(91)90785-a. [DOI] [PubMed] [Google Scholar]
- 35.Martin A, Webber S, Fricker F, Jaffe R, Demmler G, Kearney D, Zhang Y-H, Bodurtha J, Gelb B, Ni J, Bricker T, Towbin J A. Acute myocarditis: rapid diagnosis by PCR in children. Circulation. 1994;90:330–339. doi: 10.1161/01.cir.90.1.330. [DOI] [PubMed] [Google Scholar]
- 36.Martino T A, Liu P, Sole M J. Viral infection and pathogenesis of dilated cardiomyopathy. Circ Res. 1994;74:182–188. doi: 10.1161/01.res.74.2.182. [DOI] [PubMed] [Google Scholar]
- 37.Mason J W, O’Connell J B, Herskowitz A, Rose N R, McManus B M, Billingham M E, Moon T E Myocarditis Treatment Trial Investigators. A clinical trial of immunosuppressive therapy for myocarditis. N Engl J Med. 1995;333:269–275. doi: 10.1056/NEJM199508033330501. [DOI] [PubMed] [Google Scholar]
- 38.Matoba Y, Sherry B, Fields B N, Smith T W. Identification of the viral genes responsible for growth of strains of reovirus in cultured mouse heart cells. J Clin Invest. 1991;87:1628–1633. doi: 10.1172/JCI115177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meraz M A, White J M, Sheehan K C F, Bach E A, Rodig S C, Dighe A S, Kaplan D H, Riley J K, Greenlund A C, Campbell D, Carver-Moore K, DuBois R N, Clark R, Aguet M, Schreiber R D. Targeted disruption of the STAT1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signalling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 40.Metcalf P, Cyrklaff M, Adrian M. The three-dimensional structure of reovirus obtained by cryo-electron microscopy. EMBO J. 1991;10:3129–3136. doi: 10.1002/j.1460-2075.1991.tb04874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyamoto N G, Jacobs B L, Samuel C E. Mechanism of interferon action. Effect of double-stranded RNA and the 5′-O-monophosphate form of 2′,5′-oligoadenylate on the inhibition of reovirus mRNA translation in vitro. J Biol Chem. 1983;258:15232–15237. [PubMed] [Google Scholar]
- 42.Muller U, Steinhoff U, Reis L F L, Hemmi S, Pavlovic J, Zinkernagel R M, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 43.Munemitsu S M, Samuel C E. Biosynthesis of reovirus-specified polypeptides. Multiplication rate but not yield of reovirus serotypes 1 and 3 correlates with the level of virus-mediated inhibition of cellular protein synthesis. Virology. 1984;136:133–143. doi: 10.1016/0042-6822(84)90254-x. [DOI] [PubMed] [Google Scholar]
- 44.Nibert M L, Schiff L A, Fields B N. Reoviruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Vol. 2. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 1557–1596. [Google Scholar]
- 45.Nilsen T W, Maroney P A, Baglioni C. Inhibition of protein synthesis in reovirus-infected HeLa cells with elevated levels of interferon-induced protein kinase activity. J Biol Chem. 1982;257:14593–14596. [PubMed] [Google Scholar]
- 46.Nilsen T W, Maroney P A, Baglioni C. Synthesis of (2′-5′)oligoadenylate and activation of an endoribonuclease in interferon-treated HeLa cells infected with reovirus. J Virol. 1982;42:1039–1045. doi: 10.1128/jvi.42.3.1039-1045.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noble S, Nibert M L. Core protein μ2 is a second determinant of nucleoside triphosphatase activities by reovirus cores. J Virol. 1997;71:7728–7735. doi: 10.1128/jvi.71.10.7728-7735.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oberhaus S M, Smith R L, Clayton G H, Dermody T S, Tyler K L. Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol. 1997;71:2100–2106. doi: 10.1128/jvi.71.3.2100-2106.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabin E R, Hassan S A, Jenson A B, Melnick J L. Coxsackievirus B3 myocarditis in mice. An electron microscopic, immunofluorescent and virus-assay study. Am J Pathol. 1964;44:775–797. [PMC free article] [PubMed] [Google Scholar]
- 50.Rodgers S E, Barton E S, Oberhaus S M, Pike B, Gibson C A, Tyler K L, Dermody T S. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol. 1997;71:2540–2546. doi: 10.1128/jvi.71.3.2540-2546.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose N R, Hill S L. The pathogenesis of postinfectious myocarditis. Clin Immunol Immunopathol. 1996;80:S92–S99. doi: 10.1006/clin.1996.0146. [DOI] [PubMed] [Google Scholar]
- 52.Samuel C E. The eIF-2alpha protein kinases, regulators of translation in eukaryotes from yeast to humans. J Biol Chem. 1993;268:7603–7606. [PubMed] [Google Scholar]
- 53.Samuel C E, Duncan R, Knutson G S, Hershey J W. Mechanism of interferon action. Increased phosphorylation of protein synthesis initiation factor eIF-2 alpha in interferon-treated reovirus-infected mouse L929 fibroblasts in vitro and in vivo. J Biol Chem. 1984;259:13451–13457. [PubMed] [Google Scholar]
- 54.Samuel C E, Knutson G S. Mechanism of interferon action. Kinetics of decay of the antiviral state and protein phosphorylation in mouse fibroblasts treated with natural and cloned interferons. J Biol Chem. 1982;257:11796–11801. [PubMed] [Google Scholar]
- 55.Schmechel S, Chute M, Skinner P, Anderson R, Schiff L. Preferential translation of reovirus mRNA by a sigma 3-dependent mechanism. Virology. 1997;232:62–73. doi: 10.1006/viro.1997.8531. [DOI] [PubMed] [Google Scholar]
- 56.Sharpe A H, Fields B N. Reovirus inhibition of cellular RNA and protein synthesis: role of the S4 gene. Virology. 1982;122:381–391. doi: 10.1016/0042-6822(82)90237-9. [DOI] [PubMed] [Google Scholar]
- 57.Sherry B, Baty C J, Blum M A. Reovirus-induced acute myocarditis in mice correlates with viral RNA synthesis rather than generation of infectious virus in cardiac myocytes. J Virol. 1996;70:6709–6715. doi: 10.1128/jvi.70.10.6709-6715.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sherry B, Blum M A. Multiple viral core proteins are determinants of reovirus-induced acute myocarditis. J Virol. 1994;68:8461–8465. doi: 10.1128/jvi.68.12.8461-8465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sherry B, Fields B N. The reovirus M1 gene, encoding a viral core protein, is associated with the myocarditic phenotype of a reovirus variant. J Virol. 1989;63:4850–4856. doi: 10.1128/jvi.63.11.4850-4856.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sherry B, Li X-Y, Tyler K L, Cullen J M, Virgin H W., IV Lymphocytes protect against and are not required for reovirus-induced myocarditis. J Virol. 1993;67:6119–6124. doi: 10.1128/jvi.67.10.6119-6124.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sherry B, Schoen F J, Wenske E, Fields B N. Derivation and characterization of an efficiently myocarditic reovirus variant. J Virol. 1989;63:4840–4849. doi: 10.1128/jvi.63.11.4840-4849.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61a.Sherry, B., and D. Cook. Unpublished data.
- 62.Stangl, E., W. Aschauer, J. Zahringer, and G. Hubner. 1987. Reovirus myocarditis. Eur. Heart J. 8(Suppl. J):407–409.
- 63.Starnes M C, Joklik W K. Reovirus protein lambda 3 is a poly(C)-dependent poly(G) polymerase. Virology. 1993;193:356–366. doi: 10.1006/viro.1993.1132. [DOI] [PubMed] [Google Scholar]
- 64.Steinhoff U, Müller U, Schertler A, Hengartner H, Aguet M, Zinkernagel R M. Antiviral protection by vesicular stomatitis virus-specific antibodies in alpha/beta interferon receptor-deficient mice. J Virol. 1995;69:2153–2158. doi: 10.1128/jvi.69.4.2153-2158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tracy S, Chapman N M, McManus B M, Pallansch M A, Beck M A, Carstens J. A molecular and serologic evaluation of enteroviral involvement in human myocarditis. J Mol Cell Cardiol. 1990;22:403–414. doi: 10.1016/0022-2828(90)91476-n. [DOI] [PubMed] [Google Scholar]
- 66.Tyler K L, Fields B N. Reoviruses. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. 3rd ed. Vol. 2. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 1597–1624. [Google Scholar]
- 67.Virgin H W, Tyler K L, Dermody T S. Reovirus. In: Nathanson N, editor. Viral pathogenesis. New York, N.Y: Lippincott-Raven; 1997. pp. 669–702. [Google Scholar]
- 68.Welsh R M, Sen G. Non-specific host responses to viral infection. In: Nathanson N, editor. Viral pathogenesis. New York, N.Y: Lippincott-Raven; 1997. pp. 109–142. [Google Scholar]
- 69.Woodruff J F. Viral myocarditis, a review. Am J Pathol. 1980;101:427–479. [PMC free article] [PubMed] [Google Scholar]
- 70.Yin P, Cheang M, Coombs K M. The M1 gene is associated with differences in the temperature optimum of the transcriptase activity in reovirus core particles. J Virol. 1996;70:1223–1227. doi: 10.1128/jvi.70.2.1223-1227.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yue Z, Shatkin A J. Double-stranded RNA-dependent protein kinase (PKR) is regulated by reovirus structural proteins. Virology. 1997;234:364–371. doi: 10.1006/viro.1997.8664. [DOI] [PubMed] [Google Scholar]