

Abstract
Recombinant Norwalk virus-like particles (rNV VLPs) produced in insect cells were evaluated as an oral immunogen in CD1 and BALB/c mice by monitoring rNV-specific serum total and subclass immunoglobulin G (IgG) and intestinal IgA responses. Dose and kinetics of response were evaluated in the presence and absence of the mucosal adjuvant cholera toxin (CT). rNV-specific serum IgG and intestinal IgA were detected in the absence of CT, and the number of responders was not significantly different from that of mice administered VLPs with CT at most doses. The use of CT was associated with induction of higher levels of IgG in serum; this effect was greater at higher doses of VLPs. IgG in serum was detected in the majority of animals by 9 days postimmunization (dpi), and intestinal IgA responses were detected by 24 dpi. In the absence of CT, IgG2b was the dominant IgG subclass response in both mouse strains. Thus, nonreplicating rNV VLPs are immunogenic when administered orally in the absence of any delivery system or mucosal adjuvant. These studies demonstrate that rNV VLPs are an excellent model to study the oral delivery of antigen, and they are a potential mucosal vaccine for NV infections.
Norwalk virus (NV) is classified as a calicivirus based on virion morphology (nonenveloped icosahedral particle with cuplike depressions), biochemical properties (single capsid protein of 58 kDa), and characteristics of the viral genome (single-stranded RNA of positive polarity composed of three open reading frames) (24, 26, 27). NV and NV-related agents are difficult to study because these viruses cannot be cultivated in cell culture and an animal model is not available for virus production or experimentation. In addition, very low concentrations of virus are excreted in stool samples of infected individuals and most excreted antigen is in the form of soluble or proteolytically cleaved capsid protein (17, 21).
Infections with NV and other human caliciviruses (HuCVs) are recognized as the major cause of waterborne or foodborne gastroenteritis not attributable to bacterial pathogens in developed and developing countries (11, 22, 28). In the United States, early estimates indicated that at least 42% of nonbacterial gastroenteritis outbreaks are caused by these viruses (29). More recent estimates with new assays indicate that the incidence of HuCV-associated gastroenteritis is much greater than previously recognized; for example, in 1996 in The Netherlands, almost 90% of reported outbreaks were caused by these viruses (57). Epidemic outbreaks of HuCV infection have occurred in schools, communities, families, recreational facilities, hospitals, nursing homes, day-care centers, and in the military, with illness rates generally exceeding 50% and occasionally exceeding 90% (7, 29). Infections with NV and related viruses occur throughout the year and traditionally were thought to affect school-aged children and adults. However, the enhanced sensitivity of current detection assays has revealed a significant increase in the clinical importance and incidence of NV infections in infants and the elderly (11, 57). A seroprevalence of 85% for Mexican children 2 years of age (25) and 95% for children 0 to 7 years of age in Kuwait (9) indicates that NV infections can occur at an early age. A cost-effective, broadly reactive, efficacious vaccine could be useful.
The symptoms of HuCV infection are self-limited, generally lasting 24 to 48 h, with infected individuals rarely requiring hospitalization or rehydration therapy. However, time away from work, school, or vacation activities can economically impact families and communities. A recent outbreak of NV infection aboard a U.S. aircraft carrier during Operation Desert Storm illustrates the adverse impact of NV or NV-related disease on military operations (51).
Because infection by NV is localized to the intestine, induction of local immunity may be important for protection against infection and disease. Immunoglobulin A (IgA) is the predominant antibody at mucosal surfaces, is locally produced at a level that exceeds that of all of the other immunoglobulins (23, 41), and is important for mucosal immunity. Hence, it is likely that an effective oral NV or NV-related vaccine will need to induce a specific intestinal IgA response. To date, the immune status of NV-infected individuals has not been well defined and constituents of a protective immune response are not known.
The second open reading frame of the NV genome encodes a single viral capsid protein that spontaneously assembles into virus-like particles (VLPs) when expressed in the baculovirus expression system (26). Electron cryomicroscopy studies have shown that these VLPs are composed of 90 dimers of the 58-kDa protein arranged in a T=3 symmetry (49). Several unique properties of NV VLPs are advantageous for a mucosal immunogen. These properties include: (i) recombinant NV (rNV) capsids contain 180 copies of a single protein assembled into particles lacking nucleic acid; (ii) rNV VLPs are easily made and purified in large quantities, frequently more than 20 mg per 9 × 108 cells; (iii) rNV particles are highly immunogenic in experimental animals when administered parenterally with adjuvant (26); (iv) rNV VLPs are stable at low pH (such as the pH of the stomach), to lyophilization, and to long-term storage for up to 1 year in water or phosphate-buffered saline (PBS) at 4°C; (v) rNV VLPs are antigenically similar to native virions (16, 20); (vi) natural NV infections occur by the oral route, so oral delivery of rNV particles could induce similar immune responses; and (vii) the addition of protective epitopes of other pathogens is possible since the VLPs are composed of a single capsid protein and therefore amenable to molecular manipulation. Because of these properties, we tested the potential of rNV VLPs as an oral immunogen by examining the dose response of serum IgG and intestinal IgA and the kinetics of response of serum IgG, serum IgG subclasses, and intestinal IgA in mice after the oral delivery of VLPs in the presence and absence of the mucosal adjuvant cholera toxin (CT). These studies contribute to our understanding of the immune response to NV and serve as a model for the oral delivery of particulate immunogens.
MATERIALS AND METHODS
Recombinant NV VLP preparation and characterization.
The rNV VLPs were prepared in Spodoptera frugiperda (Sf9) insect cells as previously described, with minor modifications (21, 26). Sf9 cells were grown and maintained at 27°C in TNM-FH medium containing 10% fetal bovine serum (FBS) (54). The cells were pelleted (10 min at 55 × g), suspended in Grace’s medium containing 0.5% FBS to a density of 2 × 106 to 3 × 106 cells/ml, and inoculated with rNV-baculovirus (Bac-rNVc8p4) at a multiplicity of infection of between 1 and 3 PFU/cell. After 7 days of culture of the infected cells on a stir bar base (85 rpm; Bellco), intact cells were pelleted (15 min at 490 × g), the supernatant was collected, and debris was removed by centrifugation (30 min at 22,100 × g). The rNV VLPs in the clarified supernatants were pelleted through a 30% sucrose cushion for 3 h at 121,900 × g. The pelleted VLPs were suspended in sterile MilliQ water and banded in a step gradient of sucrose (20 to 65% sucrose; 3.5 ml per step) for 1 h at 112,700 × g. Peak fractions containing the VLPs were pooled, and the VLPs were concentrated by pelleting for 3 h at 121,900 × g and suspended in sterile MilliQ water.
The purified VLPs were characterized by sodium dodecyl sulfate (SDS)-polyacryamide gel electrophoresis (PAGE), followed by staining with Coomassie blue or silver nitrate to confirm the presence of the capsid protein with an apparent molecular weight of 58,000 (58K protein) and to check the purity of the rNV preparation. To ensure that the particles were intact, each preparation was examined by negative-stain electron microscopy (EM), using 1% ammonium molybdate. Protein concentration was determined by the BCA-protein assay according to the manufacturer-specified protocol using bovine serum albumin as the standard (Pierce, Rockford, Ill.). Sterility of each VLP preparation was established by bacteriologic cultures in Lennox L and thioglycolate broth incubated for a minimum of 2 weeks at 37°C, and endotoxin levels were measured by using the Limulus amebocyte lysate assay (Association of Cape Cod, Woods Hole, Mass.) (35, 44). Antigenicity of the preparation was confirmed by Western blot analysis, using mouse hyperimmune antisera prepared against the rNV VLPs (26).
Oral immunization and sample collection.
Adult (24 to 28 g) female CD1 mice (Charles River Laboratories, Portage, Mich.) were immunized by orally administering rNV particles in the presence or absence of 10 μg of CT (Sigma Chemical Co., St. Louis, Mo.) on days 1, 2, 11, and 28. The VLPs (100 μl) were orally delivered by gavage, using a stainless steel intubation needle (Popper and Sons, Inc., New Hyde Park, N.Y.). The concentration of VLPs varied between 5 and 500 μg (5, 25, 50, 75, 100, 200, 300, 400, and 500 μg) in the absence of CT and between 5 and 200 μg (5, 25, 50, 75, 100, and 200 μg) when administered with 10 μg of CT. An alternate immunization schedule was tested in one group of mice that received 500 μg of VLPs on days 1 and 21 in the absence of CT adjuvant. Control mice were given PBS, pH 7.4, or PBS with CT. Individual preserum and stool samples were collected before the first immunization, and individual postimmunization samples were obtained between days 38 and 42. In some experiments, the kinetics of the immune response to rNV VLPs were compared in outbred CD1 and inbred BALB/c mice. For these experiments, 200 μg of rNV particles was orally administered with or without 10 μg of CT. Serum and fecal samples were collected before the first immunization, 1 week following the first two immunizations (day 9), after the third immunization (days 24 to 26), and following the last immunization (day 37 or 45).
Blood samples were collected by tail bleed, and fecal samples were obtained from individual mice by using a fecal collection cage (Baylor College of Medicine). Fecal samples were extracted by making a 1:5 to 1:12 suspension (wt/vol), depending on the weight of the sample, in PBS containing 0.1% Tween 20, 0.1 mg of soybean trypsin inhibitor/ml, and 0.1 mg of merthiolate/ml. Each suspension was thoroughly vortexed, sonicated for 10 min on ice, and centrifuged for 10 min in a microcentrifuge (12,000 × g). The supernatant was collected and centrifuged again for 5 min in a microcentrifuge (12,000 × g). The twice-clarified supernatant was collected and stored at −80°C until tested.
Antibody enzyme-linked immunosorbent assays (ELISAs). (i) Preparation of rNV VLP antigen-coated microtiter plates.
Polyvinyl chloride 96-well plates (Dynatech Lab. Inc., Chantilly, Va.) were coated with rNV antigen by adding 100 μl of rNV particles per well (0.35 μg/ml based on the BCA assay) and incubating for 4 h at room temperature. Nonspecific protein binding was blocked overnight at 4°C with 5% (wt/vol) dry milk in PBS (5% BLOTTO) (serum IgG assay) or 10% (wt/vol) dry milk in PBS (10% BLOTTO) (rNV-specific fecal IgA assay).
(ii) Serum IgG ELISA.
NV-specific IgG titers in serum were determined by testing individual samples on rNV VLP antigen-coated plates using the protocol described previously (15). Test sera were prepared in 5% BLOTTO, serially diluted twofold down the microtiter plate, and incubated for 2 h at 37°C to permit antibody binding. Two control mouse serum samples were added to each plate: (i) a serum with a known NV-specific antibody titer and (ii) a serum sample known to lack NV-specific antibodies. Each test serum sample also was analyzed in a well lacking antigen to determine background binding. The plates were washed six times with 0.05% Tween 20 in PBS (PBS-T) and reacted for 1 h at 37°C with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Hyclone Laboratories, Inc., Logan, Utah) diluted 1:5,000 in 2.5% BLOTTO. The reaction was developed with 100 μl per well of 4% 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase liquid substrate system containing 0.02% hydrogen peroxide (Kirkegaard & Perry Lab., Gaithersburg, Md.) for 7 min, and color development was stopped by the addition of an equal volume of 1 M phosphoric acid. Absorbance measurements were made at 450 nm using a Titertek Multiskan Plus automatic plate reader (ICN Flow, Costa Mesa, Calif.). End point titers are reported as the reciprocal of the highest dilution that had an absorbance value greater than or equal to 0.1 above the background (absorbance of the well lacking antigen).
(iii) Serum IgG subclass ELISA.
Serum samples collected from CD1 and BALB/c mice that were positive for rNV-specific total IgG (above) were evaluated for the presence of NV-specific IgG1, IgG2a, IgG2b, and IgG3 antibodies. These antibodies were evaluated to obtain an indication of the type of response elicited (Th1 versus Th2) by the rNV oral immunogen. Mouse IgG subclass standards and corresponding HRP-conjugated antibodies were obtained from Southern Biotechnology Assoc. (Birmingham, Ala.). The specificity, sensitivity, and optimal dilution of each reagent was confirmed prior to use. Each goat anti-mouse subclass IgG-HRP conjugate was used at a dilution determined to detect an equal quantity of the corresponding IgG subclass. The purified mouse standards were diluted in PBS to a starting concentration of 0.5 μg/ml (IgG1), 0.125 μg/ml (IgG2a and IgG2b), and 0.25 μg/ml (IgG3), serially (twofold) diluted down the microtiter plates (100 μl/well), and adsorbed for 4 h at room temperature. Serial twofold dilutions of the test sera prepared in 5% BLOTTO were added to the rNV-coated wells and incubated for 2 h at 37°C. The standards were incubated in 5% BLOTTO. After six washes in PBS-T, the IgG subclass-specific conjugated antibodies were diluted 1:5,000 (goat anti-mouse IgG1- and IgG2b-HRP), 1:2,500 (goat anti-mouse IgG2a-HRP), or 1:1,000 (goat anti-mouse IgG3-HRP) in 2.5% BLOTTO; 100 μl/well was then added to the appropriate plate and incubated for 1 h at 37°C. The antibody conjugates were diluted to detect between 0.03 and 0.015 ng of the IgG subclass. The reaction was developed with TMB substrate for 7 min, stopped with 1 M phosphoric acid, and evaluated by absorbance measurements at 450 nm. End point titers are reported as the reciprocal of the highest dilution that had an absorbance value greater than or equal to 0.1 above that of the background well lacking antigen.
(iv) Fecal IgA ELISA.
Each stool suspension was assayed for rNV-specific and total IgA in two separate ELISA protocols. To determine the level of rNV-specific fecal IgA, the stool extracts were diluted 1:1 with 2% BLOTTO containing 1% FBS, added to rNV antigen-coated and uncoated wells, serially diluted in 1% BLOTTO containing 0.5% FBS, and reacted for 2 h at 37°C. Purified mouse IgA standard (Sigma Chemical Co.) was diluted in 1% BLOTTO–0.5% FBS and added to each plate at concentrations of 1.0 to 0.002 μg/ml. After six washes in PBS-T, 100 μl of peroxidase-conjugated goat anti-mouse IgA (Sigma) diluted 1:10,000 in 2.5% BLOTTO containing 0.5% FBS was added to each well. The conjugated antibody was incubated for 1 h at 37°C. Both the IgA standard and the anti-mouse IgA conjugate were tested for mouse IgA specificity prior to use and were shown to detect 6.25 ng of IgA with no cross-reactivity with purified IgG at a concentration of 160 ng or IgM at a concentration of 200 ng. The level of rNV-specific fecal IgA was calculated from a standard curve that was determined by the absorbance values of the IgA standard added to each plate. Only the linear portion of the curve was utilized in these calculations.
Total fecal IgA was determined by capturing all IgA molecules contained in the fecal extracts with goat anti-mouse IgA (Southern Biotechnology Assoc.). The capture antibody was diluted in 0.05 M bicarbonate buffer, pH 9.6, and adsorbed to the plates overnight at room temperature. Nonspecific protein binding was blocked with 10% BLOTTO for 3 h at 37°C. The initial dilution of each fecal extract varied according to the results of a preliminary screen of the IgA concentration. In the preliminary assay, the fecal extracts were diluted from 1:400 to 1:5,000 in 1% BLOTTO containing 0.5% FBS and were reacted for 2 h at 37°C. Once the optimal dilution of each stool suspension was determined, at least four dilutions of the fecal extracts were assayed. As positive and negative controls, purified IgA standard (Southern Biotechnology Assoc.) and IgG3, respectively, were diluted serially on each plate (twofold dilutions) beginning at a concentration of 1 μg/ml. Peroxidase-conjugated goat anti-mouse IgA (Sigma) diluted 1:4,500 in 2.5% BLOTTO containing 3% FBS was added to each well, and the reaction was detected with 100 μl per well of TMB peroxidase substrate (Kirkegaard & Perry Lab.).
Each rNV-specific IgA level was expressed in ng/ml, and each corresponding total IgA level was expressed in μg/ml. Fecal IgA responses were expressed as a ratio of the rNV-specific IgA (ng/ml) to total IgA (μg/ml) (nanograms of specific IgA per microgram of total IgA). The intestinal IgA responses were expressed as a ratio because of the daily variation in the IgA concentration in fecal samples.
Antigen ELISA. (i) Detection of NV antigen in stool.
To verify that the antibodies induced by the oral delivery of the rNV VLPs recognize and bind native NV particles in human stool samples, mouse sera collected after the oral delivery of the VLPs were tested in an antigen-capture ELISA (modified from reference 15). Briefly, microtiter plates were coated for 4 h at room temperature with 50 μl/well of rabbit hyperimmune serum prepared against rNV (26) diluted 1:10,000 in PBS. Rabbit preimmune serum and uncoated wells were used to test background binding. The plates were blocked with 5% BLOTTO for 2 h at 37°C, and viral antigen was added from known positive and negative volunteer stool samples (15). The antigen was prepared as 10% fecal suspensions in PBS and extracted with an equal volume of 1,1,2-trichloro-1,1,2-trifluroethane (DuPont, Wilmington, Del.) at 4°C. The antigen was diluted 1:1 in 2% BLOTTO, added to the coated and blocked plates (50 μl/well), and incubated overnight at 4°C. Purified rNV was added at a concentration of 0.5 to 0.25 ng as a positive antigen control. The plates were washed five times with PBS-T and reacted with immune sera from mice orally immunized with VLPs at a dilution of 1:320 to 1:25,600 in 2% BLOTTO for 2 h at 37°C. Three different mouse sera were selected, with total serum IgG titers of 10,240, 51,200, and 204,800. After five washes with PBS-T, HRP-conjugated goat anti-mouse Ig (heavy-chain specific; Kirkegaard & Perry Lab.) diluted 1:7,000 in 1% BLOTTO containing 1% normal rabbit sera was reacted for 1 h at 37°C. The plates again were washed five times, and 100 μl/well of TMB peroxidase substrate was added and reacted for 5 min at room temperature. Color development was stopped by the addition of 100 μl of 1 M phosphoric acid/well, and the absorbance was determined at 450 nm.
Western blot analyses.
The specificity of the reactivity of the mouse antisera to orally delivered rNV VLPs was evaluated by Western blot against Sf9 cell lysates and wild-type baculovirus-infected (Autographa californica nuclear polyhedrosis virus [34, 54]) cell lysates; rNV particles were included as a positive control. Cell lysates were prepared as previously described (34). The rNV particles and cell lysates were mixed with SDS-PAGE sample buffer (1% SDS, 10% 2-mercaptoethanol, 50 mM Tris-HCl [pH 6.8], 0.0025% phenol red, and 10% glycerol), heated at 100°C for 5 min, separated by electrophoresis on SDS–10% polyacrylamide gels, and transferred to nitrocellulose (Hybond-C pure; Amersham Life Science, Arlington Heights, Ill.) in 25 mM Tris, 192 mM glycine, and 20% (vol/vol) methanol. The efficiency of the protein separation and transfer was monitored by Ponceau S staining (Sigma) of the nitrocellulose membranes. A subset of the immune sera from mice that received orally delivered rNV VLPs and with ELISA titers ranging from 10,240 to 204,800 were used as primary antibody at a dilution of 1:500 in 0.1% BLOTTO for the Western blots. Western blot reactivity was developed using chemiluminescence (ECL; Amersham Life Science).
Data and statistical analyses.
Geometric mean titers (GMTs) of IgG in serum were determined for every group of mice. All of the nonresponders were included in the computation of the GMT. Serial, twofold serum dilutions were assayed; the lowest dilution tested was divided by 2 and used as the titer for the negative samples (i.e., negative samples were assigned a titer of 20). Standard errors were calculated on the log-transformed titers. The mean of the ratio of specific to total fecal IgA was determined for each group. Those stool samples in which NV-specific IgA levels were below detection were included in the calculation of the mean and were assigned a value of one-half of the minimal detectable IgA level (3.125 ng). Total IgA levels were determined for 63 of the negative (no detectable NV-specific IgA) fecal samples and for the preimmunization fecal samples. These values were averaged and used as the total IgA for 11 of the negative fecal samples for which the volume was insufficient to complete the total IgA assay.
Statistical analyses were performed using SPSS version 7.0 for Windows (SPSS, Inc., Chicago, Ill.). Nonparametric data were analyzed by χ2, Fisher’s exact, and Wilcoxon’s signed rank tests. GMTs were compared using Student’s t test, the paired t test, or one-way analysis of variance (ANOVA) followed by Tukey’s Honestly Significant Difference (HSD) test. ANOVA was used to compare multiple groups, and when a significant difference was detected, Tukey’s HSD was then applied to determine which group(s) was different. Repeated-measure ANOVA was used to compare GMTs in mice over time, and when a significant difference between groups was detected, paired t tests were used to determine which groups were different. Simple and multiple linear regressions were used to evaluate the relationship of serum antibody level to vaccine dose.
RESULTS
Characterization of the rNV VLPs.
Baculovirus-expressed rNV VLPs were produced as a vaccine candidate and characterized by SDS-PAGE, Western blot, and negative stain EM. SDS-PAGE and Western blot analyses revealed a major band with an apparent molecular weight of 58,000 (58K), and minor bands at approximately 50K and 30K were seen by Western blot (Fig. 1A). EM analysis showed numerous VLPs of high purity which appeared structurally similar to the native virion (Fig. 1B). There was no bacterial growth after bacteriologic culture for 2 weeks at 37°C, and endotoxin levels were below 0.02 endotoxin units per mg of VLPs. These data confirmed the purity and sterility of the VLP preparation prior to oral delivery to mice.
FIG. 1.
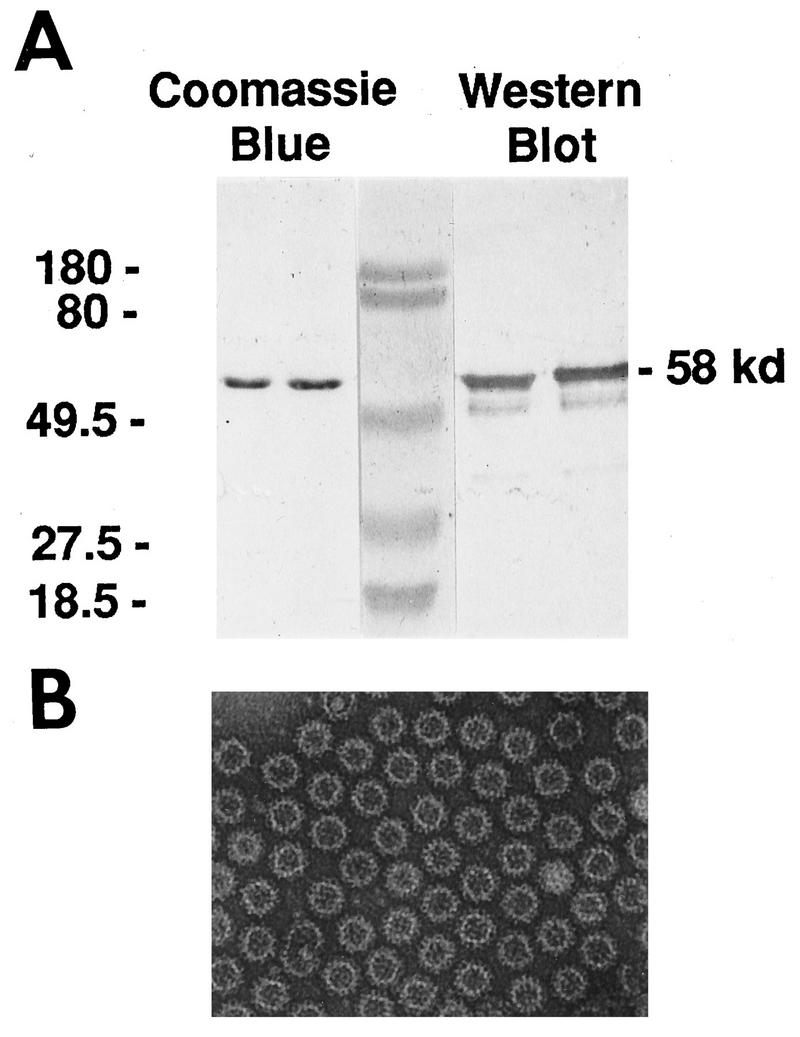
Characterization of the rNV particles. (A) SDS-PAGE (10% polyacrylamide) and Western blot analyses of the baculovirus-expressed rNV capsid protein used in the oral immunization of outbred (CD1) and inbred (BALB/c) mice. The Coomassie blue-stained gel shows a single 58-kDa band corresponding to the NV capsid protein. Western blot analysis using mouse rNV-specific antiserum shows additional bands at 50K and 30K, cleavage products of the full-length capsid protein. The molecular weight markers are shown in the center lane, and the corresponding molecular weights are indicated on the left. (B) EM of rNV particles purified from infected Sf9 insect cells. The particles were purified by centrifugation through a step sucrose gradient (20 to 60%), diluted 1:10 in sterile MilliQ water, and stained with 1% ammonium molybdate, pH 6.0. Homogeneous rNV particles were present in each grid square.
Oral administration of rNV VLPs induces a systemic immune response in mice.
To evaluate the potential of rNV particles as an oral immunogen, various doses of rNV VLPs in the presence or absence of CT were delivered to CD1 mice by oral gavage. Serum antibody responses were measured by ELISA, and the GMT was calculated for each group of mice (Fig. 2). All preimmune serum samples taken prior to the first immunization were negative (titer, <50) for rNV-specific IgG (data not shown). Control mice that received PBS or PBS with CT also lacked serum IgG (titer, <50; lowest dilution tested; data not shown).
FIG. 2.

The IgG responses in serum of CD1 mice to different doses of rNV VLPs orally administered in the absence (A) or presence (B) of CT, as measured by ELISA. The y axis shows the GMT of antibody for each group of animals, and the x axis shows the dose of VLPs orally administered by gavage on days 1, 2, 11, and 28. The open bar in panel A shows the results from a separate group of mice that received an alternate immunization schedule of two oral doses at a 3-week interval. Nonresponders were included in the calculations of the GMT. Above each bar is the number of responders over the total number of mice tested, and the GMT of only the responders is shown in parenthesis. Only one dose of rNV VLPs administered orally in the presence and absence of CT (75 μg) induced significantly different IgG responses when each dose was compared individually (P < 0.001 [∗], Student’s t test). Multiple linear regression showed that CT did influence the magnitude of the response at higher doses of VLPs. The error bars show the standard errors of the mean.
A serum IgG response was obtained with as little as 5 μg of particles in the absence of CT, although vaccine concentrations of 5 and 25 μg induced relatively low antibody titers, ranging from 100 to 400, with a GMT of 73 and 130, respectively. Serum antibody titers induced by 50 to 100 μg of VLPs were variable, ranging from 100 to 204,800, with GMTs of 60 to 622. Doses at or exceeding 200 μg of VLPs without CT consistently induced a serum antibody response in all of the mice, with a GMT ranging from 514 to 1,165. The titers were variable within each group of mice, with maximum titers reaching 409,600 in the absence or presence of adjuvant. Some of the variation in titers may be due to our use of outbred animals for these experiments. Responses to 75 μg of VLPs without CT were significantly lower (P < 0.05, ANOVA and Tukey’s HSD test) than the responses to the 200- and 500-μg doses without CT. These data indicate that rNV VLPs are immunogenic when administered orally to mice and a mucosal adjuvant is not required for the induction of serum IgG.
An alternative immunization schedule of two oral doses at a 3-week interval was tested in one group of mice given a dose of 500 μg of VLPs without CT. All mice (n = 5) in this group developed serum antibody to NV, and when compared to those mice given the four-dose regimen with an equivalent dose of VLPs there were no significant differences (P > 0.05, Student’s t test) in the levels of the response. These data indicate that two oral doses at a 3-week interval are sufficient to elicit a serum IgG response in the absence of CT.
In the presence of 10 μg of CT, a lower concentration of antigen (5 μg) was sufficient to induce a positive serum IgG response in 100% of the mice, with titers ranging from 50 to 200 and a GMT of 94 (Fig. 2). Although CT did not increase the number of responders, when CT was used as a covariant in the multiple regression analyses, CT was found to alter the slope (degree) of the response, with higher responses seen at the higher doses of rNV VLPs. Thus, enhancement of the magnitude of the IgG response by CT was dependent on the dose of the immunogen.
The serum antibody GMT following the oral delivery of 5 μg of VLPs with CT was significantly lower (P < 0.05, ANOVA and Tukey’s HSD test) than the GMTs following the administration of 75, 100, and 200 μg of VLPs with CT. The magnitude of the antibody responses to 25 μg of VLPs administered with CT was significantly lower (P < 0.005) than the antibody titers obtained with 100 and 200 μg of VLPs given with CT; 50 μg of VLPs induced significantly lower antibody titers when compared to the titers elicited by the 200-μg dose in the presence of CT. The serum IgG response to the vaccine delivered with and without CT was dose dependent by simple linear regression (P < 0.001).
Oral administration of rNV VLPs induces an intestinal IgA response in mice.
Stool suspensions were assayed for rNV-specific and total IgA by ELISA to evaluate the induction of mucosal antibody by different doses of rNV VLPs given with and without CT (Fig. 3). Doses of 200 and 500 μg of VLPs resulted in intestinal IgA responses in 60 and 62.5%, respectively, of the mice. The induction of mucosal IgA in the absence of adjuvant occurred more frequently with a higher dose (200 μg) of VLPs than with doses of 100 μg or less (P = 0.015, χ2). These data show that intestinal IgA responses were elicited in the majority of animals when relatively large doses of VLPs were administered without adjuvant.
FIG. 3.

The intestinal IgA responses in CD1 mice to different doses of rNV particles orally administered in the presence or absence of CT. The concentration of VLPs given to each group of mice is shown in the x axis. Each dose was administered by gavage on days 1, 2, 11, and 28. Two independent ELISAs were used to measure the NV-specific and total IgA. The y axis shows the mean of the ratio in nanograms/milliliter of NV-specific IgA to micrograms/milliliter of total IgA or nanograms of specific IgA per microgram of total IgA. The number above each bar depicts the number of responders with detectable specific IgA over the total number of animals tested. The error bars show the standard errors of the mean. When NV-specific IgA was not detected, a value of 3.125 ng (one-half the lowest detectable level of specific IgA) was assigned to that sample, divided by the total IgA concentration, and used in the calculation of the mean and standard error. The number of responders to 100 μg of VLPs was significantly different when administered with or without CT (•) (P = 0.003, Fisher’s exact test). In the 25 μg of rNV dose with CT, one mouse had a ratio of 245 ng of rNV-specific IgA/μg of total IgA which was excluded in calculating the mean (∗). The mean is 26.4 when the 245 ratio is included. In the 200 μg of rNV dose with CT, one mouse had a ratio of 145 ng rNV-specific IgA/μg of total IgA which was excluded in calculating the mean (+). The mean is 29 when the 145 ratio is included.
When the rNV particles were delivered in the presence of CT, lower concentrations of VLPs induced intestinal IgA responses in a larger number of animals. When 100 μg of rNV VLPs was delivered with CT, there was a significant increase (P = 0.003, Fisher’s exact) in the number of animals with a positive IgA response when compared to the number of responders that received antigen alone. Oral administration of VLPs at all doses below 100 μg, and at 200 μg with CT, induced an equivalent level of intestinal IgA (P > 0.05) when compared to the same dose given in the absence of CT (Fig. 3). Intestinal IgA response rates progressively increased when the 5- and 25-, 50- and 75; and 100- and 200-μg groups were compared (P = 0.001, χ2 trend), showing a dose response in the presence of CT adjuvant.
Kinetics of total serum IgG response in CD1 and BALB/c mice.
We next determined if the genetic background of the mice influenced the immune response to the rNV oral immunogen and evaluated the kinetics of the serological response. CD1 outbred and BALB/c inbred mice were orally administered four doses of 200 μg of rNV particles with or without CT and were evaluated for NV-specific serum antibody after each dose (Fig. 4). A dose of 200 μg of VLPs was chosen because 100% of the CD1 mice responded at this dose in the presence and absence of CT in the previous experiment (Fig. 2). After the first two doses (day 9), 90% of the CD1 mice had a positive antibody response in serum, with titers ranging from 160 to 2,560, with a GMT of 724 in the absence of CT (GMT of 452 when the nonrespondes were included), and from 160 to 1,280, with a GMT of 404, in the presence of CT or 242 when the nonresponders were included. The IgG titers in the CD1 mice increased after the third (day 26) and fourth (day 45) dose of VLPs to a final GMT of 3,620 and 2,941 without and with CT, respectively (Fig. 4A; includes nonresponders). There were no significant differences in the IgG responses (P > 0.05, Student’s t test) in the presence or absence of adjuvant. Thus, CT did not augment the serum IgG response to the rNV VLPs in the outbred animals at a dose of 200 μg. When the CD1 mice IgG responses were compared after two, three, or four immunizations, there were significant increases in titer after the third (P = 0.003, paired t test) and fourth (P = 0.004) dose in the absence of CT. With CT, the differences in the IgG titers at 26 and 45 days postimmunization (dpi) were not significant, whereas the titers were significantly increased at 26 dpi (P = 0.013) and 45 dpi (P = 0.004) when compared to the antibody response at 9 dpi.
FIG. 4.

The kinetics of serum IgG responses to the oral delivery of 200 μg of rNV VLPs with and without CT in outbred CD1 (A) and in inbred BALB/c (B) mice as measured by ELISA. The CD1 mice used in this experiment were an independent group of mice from those used for the data shown in Fig. 2 and 3. The y axis shows the GMT of serum IgG in samples taken on days 0, 9, 26, and 45 postimmunization (x axis). The VLPs were administered orally on days 1, 2, 11, and 28 (↑). Data shown are from the second, third, and fourth immunizations. Above each bar are the number of responders over the total number tested. The error bars show the standard errors of the mean. All calculations include the data from the nonresponders. Significant differences in the GMT were seen between the CD1 and BALB/c mice in the absence of CT (P = 0.044 [∗], P = 0.009 [+], P = 0.022 [#]; Student’s t test).
Similar to the CD1 mice, the IgG titers in serum of inbred BALB/c mice increased after each immunization (Fig. 4B). Two immunizations of VLPs (9 dpi) administered to BALB/c mice in the absence of CT resulted in a serum antibody response in 75% of the animals, with a GMT of 190 with nonresponders. In the presence of CT, two doses were sufficient to elicit a serum IgG response in 82% of the inbred mice (GMT = 181). On day 45, the final GMT in the inbred mice was 570 in the absence of CT and 1,646 in the presence of adjuvant (calculations include all mice). The BALB/c mice had similar serum IgG responses in the presence and absence of CT (P > 0.05). There were significant differences between the antibody titers at 9 and 26 dpi, at 9 and 45 dpi, and at 26 and 45 dpi when the VLPs were administered with CT (P = 0.005, <0.001, and 0.002, respectively, paired t test). Antibody responses of the BALB/c mice in the absence of CT were not significantly different at 9 and 26 dpi, but a significant increase in IgG titers was seen at 45 dpi when compared to the titers at 9 dpi (P = 0.006) and 26 dpi (P < 0.001, paired t test).
GMTs were significantly higher in the CD1 mice when compared to the BALB/c mice in the absence of CT at 9 dpi (P = 0.044), at 26 dpi (P = 0.009), and at 45 dpi (P = 0.022). No significant differences in the serological responses to the orally delivered rNV VLPs in the presence of CT were observed between the two mouse strains. In both strains, two doses of VLPs given 1 day apart were sufficient to elicit a serum IgG response in the majority of the animals and there were no differences in the GMT with and without CT. Subsequent doses resulted in an increase in titer. These data indicate that the rNV VLPs are an effective oral immunogen in inbred and outbred mice and tolerance was not induced.
Kinetics of IgG subclass responses in CD1 and BALB/c mice.
Serum IgG subclass responses were monitored in CD1 and BALB/c mice by ELISA to obtain preliminary insights into the nature of the immune response. The serum samples from all mice that were positive for rNV-specific total IgG were included in these assays, i.e., 90% of the CD1 serum samples, and 75 to 100% of the BALB/c serum samples were assayed for subclass IgG responses. In the absence of CT, the predominant subclass induced by the oral delivery of rNV VLPs was IgG2b (Fig. 5A and B), with a GMT of 15,521 in CD1 mice and 5,120 in BALB/c mice. In the presence of CT, IgG2b remained the predominant IgG subclass in CD1 mice (GMT of 11,763), but a large amount of IgG1 was also induced (panel C), reaching a GMT of 5,120. IgG1 (GMT = 3,880) was the predominant IgG subclass produced by BALB/c mice in the presence of CT, followed by an IgG2b response (GMT = 1,689). In addition, IgG2a was induced to a GMT of 485 (panel D). Taken together, a shift to an IgG1 subclass response (Th2 response) was observed when rNV VLPs were administered in the presence of CT in BALB/c mice. While a similar shift did not occur in CD1 mice, there was an increase in the IgG1 response in CD1 mice. The BALB/c mice also produced relatively less rNV-specific IgG2b when compared to the IgG2b secretion in CD1 mice.
FIG. 5.

The kinetics of the serum IgG subclass response in CD1 (A and C) and BALB/c (B and D) mice to the oral administration of 200 μg of rNV particles in the absence (A and B) and the presence (C and D) of 10 μg of CT (data from testing all serum samples positive for rNV-specific total IgG from mice shown in Fig. 4). The x axis shows the days postimmunization, and the y axis shows the GMT of the IgG1, IgG2a, IgG2b, and IgG3 subclasses.
Kinetics of fecal IgA responses in CD1 and BALB/c mice.
The development of rNV-specific intestinal IgA in inbred and outbred animals was evaluated by testing stool extracts in an ELISA (Fig. 6). At 9 dpi, in the absence of CT, NV-specific IgA was not detected in the stools of the CD1 mice, whereas in the presence of CT, 30% of the CD1 mice had detectable intestinal IgA, ranging from 0.36 to 3.32 ng of NV-specific IgA per μg of total IgA (Fig. 6A). In contrast, at 9 dpi, NV-specific intestinal IgA was detected in 42 and 64% of BALB/c mouse stools with and without CT, respectively (Fig. 6B). There were no significant differences (P > 0.05, Fisher’s exact) in the NV-specific IgA responses between the outbred and inbred animals or in the number of mice with detectable NV-specific IgA responses in the presence or absence of CT at all time points.
FIG. 6.

The kinetics of intestinal IgA responses to the oral delivery of 200 μg of rNV VLPs in the presence and absence of CT in CD1 (A) and BALB/c (B) mice. IgA levels were determined by two independent ELISAs which measured rNV-specific and total IgA, respectively. The y axis shows the mean of the ratios of rNV-specific IgA (expressed in nanograms/milliliter) to total IgA (expressed in micrograms/milliliter). The VLPs were administered orally on days 1, 2, 11, and 28 (↑). The numbers above each bar indicate the number of animals with detectable intestinal IgA over the total number of animals tested. The error bars depict the standard errors of the mean. When specific IgA was not detected, a value of 3.125 ng/ml (one-half the detectable level of specific IgA) was used in the calculation of the mean. Because the data are expressed as a ratio, there were instances in which samples that had detectable rNV-specific IgA calculated to a lower ratio than negative samples. This was seen at 9 dpi. The number of CD1 responders significantly increased after the last dose of VLPs (day 37) when compared to the number of responders at 9 dpi in the presence (P = 0.07 [+]) or absence (P = 0.02 [∗]) of CT. Sufficient fecal samples were not available for testing from some animals (day 0, panel B).
After the final immunization (37 dpi), the number of CD1 responders significantly increased to 70% (without CT, P = 0.02) or 80% (with CT, P = 0.07) when compared to those at 9 dpi, but the level of the NV-specific IgA did not significantly increase (P > 0.05). The BALB/c mice did not show a significant difference in the number of responders at any time point.
The antibody induced to orally administered rNV VLPs is specific and reactive with native NV virions.
We also tested the specificity of the antibody response by Western blot analysis. Mouse sera with high ELISA titers after the oral delivery of rNV VLPs were tested with lysates of uninfected Sf9 cells and with wild-type baculovirus-infected cells. Ponceau S staining revealed numerous protein bands in the Sf9 cell lysate lanes and a predominant band with an apparent molecular weight of 71,000 in the baculovirus-infected lysates. The mouse sera tested showed no detectable reactivity with any of the cellular or baculovirus proteins, whereas there was positive reactivity against the rNV capsid protein (data not shown). These data indicate that rNV antibody induced by oral immunization was specific to rNV VLPs and antibody detectable by Western blot was not induced to any potentially minor cellular or baculovirus protein contaminants.
Finally, the mouse antibody induced to the orally delivered rNV was tested for its reactivity with native NV virions in an antigen ELISA. Four volunteer stool samples that contained NV particles as determined by previous testing (positive by reverse transcription-PCR, ELISA, and immunoelectron microscopy) (15, 50) showed high optical density readings (>1.00) in the antigen ELISA when the mouse sera were utilized as the detector antibody, while the same mouse sera had no reactivity to stool samples which lacked NV based on reverse transcription-PCR, ELISA, and immunoelectron microscopy (data not shown). Thus, mouse immune sera generated to rNV VLPs delivered orally recognize and bind native NV virions.
DISCUSSION
This article reports studies of the serum IgG and intestinal IgA responses in mice orally administered rNV VLPs. The results of these studies serve as a model for evaluating responses to an orally administered VLP subunit vaccine and are a first step in evaluating an oral rNV vaccine. We have established a dose response in the absence of adjuvant that will be useful in designing human vaccine trials using an oral subunit NV immunogen.
The rationale to develop an oral vaccine for enteric pathogens is founded in the success of oral, live, attenuated poliovirus vaccines (45–47). Oral immunization offers many practical advantages over parenteral immunization. Oral immunogens are easy to deliver, more acceptable to patients, and the reduction in need for highly trained personnel results in simpler logistics for mass immunization. In addition, oral immunization may induce a vigorous immune response at mucosal surfaces, which are the most common entry sites of infectious agents. However, oral administration of antigen, especially nonreplicating antigen, presents several challenges that must be overcome to achieve an efficacious oral vaccine. The immunogen must maintain its native structure and antigenicity in the acid pH of the stomach, and it must be stable to proteolytic enzyme digestion in the gastrointestinal tract. A variety of oral delivery systems and mucosal adjuvants have been developed to enhance the oral immunogenicity of nonreplicating antigens. Nonreplicating antigens have been incorporated into liposomes (58), gelatin capsules (13, 42), and biodegradable microspheres (30, 33, 43, 48) and delivered by conjugation to anti-major histocompatibility complex class II antibodies (12) in attempts to protect the immunogen from the intralumenal environment, reduce the effective dose, and enhance antigen processing. Although each system has met with some success, liposomes have a reduced uptake and stability in the intestinal environment (58), chemicals used in microencapsulation can degrade key epitopes, only a small portion of the antigen is incorporated into the microspheres, and limited numbers of the microspheres are absorbed from the gastrointestinal tract through the Peyer’s patches (PP; 5- to 10-μm size range) (10, 43).
We have shown that nonreplicating rNV VLPs are immunogenic in the absence of any oral delivery system or mucosal adjuvant. Oral delivery of the particles induced NV-specific serum IgG and mucosal IgA in the majority of animals. There may be several reasons for this success. First, rNV VLPs are stable to acid pH (21) and apparently survive exposure to the enzymes of the gastrointestinal tract, like the native infectious virion. Second, because the rNV VLPs are particulate, they may be taken up by M cells of the PP where they stimulate local precursor IgA plasma cells and activate the common mucosal immune system (38, 40–42). Alternatively, the rNV VLPs could interact specifically with a cellular receptor followed by uptake and presentation to immune cells. Binding and internalization studies using rNV VLPs with cultured human and animal intestinal cells have shown saturable and specific binding to the cell surface followed by low levels of internalization (59). Our present studies in mice have not determined if the VLPs are taken up by PP or bound by a specific cellular receptor, or both. The latter initially seems unlikely because mice are not susceptible to infection with NV. However, the block to NV infection in mice could occur after viral binding or internalization.
The induction of IgA responses after oral delivery of rNV VLPs required higher concentrations of antigen than those required to induce serum IgG responses in the majority of the mice. Additionally, serum IgG responses were observed earlier than intestinal IgA responses. These results are consistent with reports of the oral delivery of soluble protein antigens delivered in the presence of CT (47). The induction of IgG versus IgA with respect to dose and kinetics was similar with rNV VLPs given in the absence or presence of CT. Oral delivery of rNV VLPs alone was sufficient to induce an immune response and may be particularly useful as a mucosal immunogen. The observation that CT was not required for the induction of a systemic or mucosal antibody response is notable, although CT did influence the magnitude of the IgG response at higher doses of VLPs.
The development of an appropriate CD4+ Th-cell subset may be important for disease resolution and for the effectiveness of a vaccine (8, 14). To begin to explore the type of Th or inflammatory cells induced by the rNV VLPs, the IgG subclass response was determined in CD1 and BALB/c mice. Antibodies of the IgG2a isotype (Th1 response) are most often induced by viral infections or viral antigens (4–6, 55), and IgG2a and IgG2b isotypes are the most effective in complement activation as well as antibody-dependent, cell-mediated cytotoxicity (31, 32). We found that in the absence of CT, IgG2b was the dominant antibody isotype induced by the oral delivery of rNV particles in inbred and outbred mice. Although immunological studies have implied that different routes of immunization influence the isotype of the antibody response (8), IgG2b was the predominant subclass in sera from mice intraperitoneally infected with another virus, foot-and-mouth disease virus (FMDV) (14). Thus, with FMDV, the route of immunization did not influence the stimulation of an IgG2b response. It is not known if altering the route of administration of the rNV particles would influence the induction of IgG2b; this will be evaluated in future studies.
Data from other systems have shown that IgG2b secretion is induced by transforming growth factor β which is produced by inflammatory cells such as macrophages (3, 39, 52, 56). The role of macrophages and other inflammatory cells in the induction of an antibody or clinical response to NV is not known, although mononuclear cells have been reported in biopsies from volunteers administered NV (28). Additional studies are needed to determine if the IgG2b response predominates in natural NV infections and if inflammatory cells play a role in NV-induced disease.
In the presence of CT, there was an increase in the level of rNV-specific IgG1 in CD1 mice, although IgG2b remained the dominant IgG subclass response. In BALB/c mice, IgG1 was the predominant subclass response when the VLPs were delivered with CT. A shift to a Th2 response (IgG1 isotype) when antigen is administered in the presence of CT has been previously reported in BALB/c mice (36). In addition, differences in the predominant antibody subclass response in different mouse strains have been reported; in response to bacterial lipopolysaccharide BALB/c mice secreted lower relative amounts of IgG2b compared to IgG3 and IgG1 when directly compared to the IgG subclass response in DBA/2 mice (39). Consistent with these data, the BALB/c mice secreted relatively less rNV-specific IgG2b when compared to the IgG2b secretion in CD1 mice.
The induction of tolerance is a concern with oral immunogens. Immunogens fed daily in small doses or in a single high dose often induce oral tolerance that appears to be mediated by cellular or humoral suppresser factors (2, 37, 53). Our results with oral immunization using the rNV VLPs indicate that tolerance was not induced. After each oral dose of rNV VLPs, the level of IgG and intestinal IgA in serum increased or the number of responders increased. In addition, following a parenteral boost, serum IgG responses increased (data not shown). Soluble proteins are often tolerogenic, whereas particulate antigens are less tolerogenic and can stimulate secretory antibody when multiply administered at a higher dose (1, 18, 19). We hypothesize that the VLPs are ideally folded or have enough structure and/or stability to induce the production of NV-specific secretory IgA without inducing tolerance.
The results from this study are the first step in evaluating an orally delivered rNV VLP immunogen. This study is useful in showing that these VLPs are immunogenic when given orally, and the dose response can be used to design phase I human trials. If this vaccine is shown to be safe and immunogenic in volunteers, the next step will be to test whether protective immunity is induced. Because NV remains refractory to being grown in cell culture and in animal models, we cannot monitor the induction of neutralizing antibody and the immunogenicity of the rNV VLPs shown in mice cannot predict protective efficacy in volunteers. Indeed, currently there are no correlates of protection for any model. However, such correlates may be defined as studies with volunteers proceed and new assays are developed to monitor responses to conserved and variable domains shown recently to be present within the capsid protein (11). Ultimately it should be possible to manipulate the single capsid protein of NV so that chimeric VLPs serve as carriers for protective epitopes of other enteric pathogens. These goals are being facilitated by determination of the structure of this capsid protein, by determination of domains needed for virus assembly, and by the identification of synthetic peptides corresponding to different regions in the capsid that induce human rNV-specific antibody responses following the oral delivery of rNV VLPs.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI 36519 and T32 DK-07664 and by the Advanced Technology Program Grant 004949-003 from the Texas Higher Education Coordinating Board.
We thank Tanya McPherson for technical assistance in the development and performance of the ELISAs and Christopher Barone for assistance with the mice.
REFERENCES
- 1.Bergmann K-C, Waldman R H. Stimulation of secretory antibody following oral administration of antigen. Rev Infect Dis. 1988;10:939–950. doi: 10.1093/clinids/10.5.939. [DOI] [PubMed] [Google Scholar]
- 2.Challacombe S J. Cellular factor in the induction of mucosal immunity by oral immunization. Adv Exp Med Biol. 1987;216b:887–899. [PubMed] [Google Scholar]
- 3.Coffman R L, Lebman D A, Shrader B. Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J Exp Med. 1989;170:1039–1044. doi: 10.1084/jem.170.3.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coutelier J-P, van der Logt J T M, Heessen F W A, Vink A, van Snick J. Virally induced modulation of murine IgG antibody subclasses. J Exp Med. 1988;168:2373–2378. doi: 10.1084/jem.168.6.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coutelier J-P, van der Logt J T M, Heessen F W A, Warnier G, van Snick J. IgG2a restriction of murine antibodies elicited by viral infections. J Exp Med. 1987;165:64–69. doi: 10.1084/jem.165.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coutelier J-P, van Roost E, Lambotte P, van Snick J. The murine antibody response to lactate dehydrogenase-elevating virus. J Gen Virol. 1986;67:1099–1108. doi: 10.1099/0022-1317-67-6-1099. [DOI] [PubMed] [Google Scholar]
- 7.Cukor G, Blacklow N R. Human viral gastroenteritis. Microbiol Rev. 1984;48:157–179. doi: 10.1128/mr.48.2.157-179.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Becker G, Sornasse T, Nabavi N, Bazin H, Tielemans F, Urbain J, Leo O, Moser M. Immunoglobulin isotype regulation by antigen-presenting cells in vivo. Eur J Immunol. 1994;24:1523–1528. doi: 10.1002/eji.1830240710. [DOI] [PubMed] [Google Scholar]
- 9.Dimitrov D H, Dashti S A H, Ball J M, Bishbishi E, Alsaeid K, Jiang X, Estes M K. Prevalence of antibodies to human caliciviruses (HuCVs) in Kuwait established by ELISA using baculovirus-expressed capsid antigens representing two genogroups of HuCVs. J Med Virol. 1997;51:115–118. [PubMed] [Google Scholar]
- 10.Eldridge J H, Gilley R M, Staas J K, Moldoveanu Z, Meulbroek J A, Tice T R. Biodegradable microspheres: vaccine delivery system for oral immunization. Curr Top Microbiol Immunol. 1989;146:59–66. doi: 10.1007/978-3-642-74529-4_6. [DOI] [PubMed] [Google Scholar]
- 11.Estes M K, Atmar R L, Hardy M E. Norwalk and related diarrhea viruses. In: Richman D D, Whitley R J, Hayden F G, editors. Clinical virology. New York, N.Y: Churchill Livinstone; 1997. pp. 1073–1095. [Google Scholar]
- 12.Estrada A, McDermott M R, Underdown B J, Snider D P. Intestinal immunization of mice with antigen conjugated to anti-MHC class II antibodies. Vaccine. 1995;13:901–907. doi: 10.1016/0264-410x(95)00012-p. [DOI] [PubMed] [Google Scholar]
- 13.Farag-Mahmod F I, Wyde P R, Rosborough J P, Six H R. Immunogenicity and efficacy of orally administered inactivated influenza virus vaccine in mice. Vaccine. 1988;6:262–268. doi: 10.1016/0264-410x(88)90222-8. [DOI] [PubMed] [Google Scholar]
- 14.Filgueira D M P, Berinstein A, Smitsaart E, Borca M V, Sadir A M. Isotype profiles induced in Balb/c mice during foot and mouth disease (FMD) virus infection or immunization with different FMD vaccine formulations. Vaccine. 1995;13:953–960. doi: 10.1016/0264-410x(95)00078-f. [DOI] [PubMed] [Google Scholar]
- 15.Graham D Y, Jiang X, Tanaka T, Opekun A R, Madore H P, Estes M K. Norwalk virus infection of volunteers: new insights based on improved assays. J Infect Dis. 1994;170:34–43. doi: 10.1093/infdis/170.1.34. [DOI] [PubMed] [Google Scholar]
- 16.Green K Y, Lew J F, Jiang X, Kapikian A Z, Estes M K. Comparison of the reactivities of baculovirus-expressed recombinant Norwalk virus capsid antigen with those of the native Norwalk virus antigen in serologic assays and some epidemiologic observations. J Clin Microbiol. 1993;31:2185–2191. doi: 10.1128/jcm.31.8.2185-2191.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenberg H B, Valdesuso J R, Kalica A R, Wyatt R G, McAuliffe V J, Kapikian A Z, Chanock R M. Proteins of Norwalk virus. J Virol. 1981;37:994–999. doi: 10.1128/jvi.37.3.994-999.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanson D G. Ontogeny of orally induced tolerance to soluble proteins in mice. I. Priming and tolerance in newborns. J Immunol. 1981;127:1518–1522. [PubMed] [Google Scholar]
- 19.Hanson D G, Miller S D. Inhibition of specific immune responses by feeding protein antigens. V. Induction of the tolerant state in the absence of specific suppressor T cells. J Immunol. 1982;128:2378–2381. [PubMed] [Google Scholar]
- 20.Hardy M E, Tanaka T N, Kitamoto N, White L J, Ball J M, Jiang X, Estes M K. Antigenic mapping of the recombinant Norwalk virus capsid protein using monoclonal antibodies. Virology. 1996;217:252–261. doi: 10.1006/viro.1996.0112. [DOI] [PubMed] [Google Scholar]
- 21.Hardy M E, White L J, Ball J M, Estes M K. Specific proteolytic cleavage of recombinant Norwalk virus capsid protein. J Virol. 1995;69:1693–1698. doi: 10.1128/jvi.69.3.1693-1698.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hedberg C W, Osterholm M T. Outbreaks of food-borne and waterborne viral gastroenteritis. Clin Microbiol Rev. 1993;6:199–210. doi: 10.1128/cmr.6.3.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmgren J, Czerkinsky C, Lycke N, Svennerholm A-M. Mucosal immunity: implications for vaccine development. Immunobiology. 1992;184:157–179. doi: 10.1016/S0171-2985(11)80473-0. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X, Graham D Y, Wang K N, Estes M K. Norwalk virus genome cloning and characterization. Science. 1990;250:1580–1583. doi: 10.1126/science.2177224. [DOI] [PubMed] [Google Scholar]
- 25.Jiang X, Matson D O, Velazquez F R, Zhong W, Hu J, Ruiz-Palacios G, Pickering L K. A study of Norwalk related viruses in Mexican children. J Med Virol. 1995;47:309–316. doi: 10.1002/jmv.1890470404. [DOI] [PubMed] [Google Scholar]
- 26.Jiang X, Wang M, Graham D Y, Estes M K. Expression, self-assembly, and antigenicity of the Norwalk virus capsid protein. J Virol. 1992;66:6527–6532. doi: 10.1128/jvi.66.11.6527-6532.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang X, Wang M, Wang K, Estes M K. Sequence and genomic organization of Norwalk virus. Virology. 1993;195:51–61. doi: 10.1006/viro.1993.1345. [DOI] [PubMed] [Google Scholar]
- 28.Kapikian A Z, Estes M K, Chanock R M. Norwalk group of viruses. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. New York, N.Y: Raven Press; 1996. pp. 783–810. [Google Scholar]
- 29.Kaplan J E, Gary G W, Baron R C, Singh N, Schonberger L B, Feldman R, Greenberg H B. Epidemiology of Norwalk gastroenteritis and the role of Norwalk virus in outbreaks of acute nonbacterial gastroenteritis. Ann Intern Med. 1982;96:756–761. doi: 10.7326/0003-4819-96-6-756. [DOI] [PubMed] [Google Scholar]
- 30.Khoury C A, Moser C A, Speaker T J, Offit P A. Oral inoculation of mice with low doses of microencapsulated, noninfectious rotavirus induces virus-specific antibodies in gut-associated lymphoid tissue. J Infect Dis. 1995;172:870–874. doi: 10.1093/infdis/172.3.870. [DOI] [PubMed] [Google Scholar]
- 31.Kipps T J, Parham P, Punt J, Herzemberg L A. Importance of immunoglobulin isotype in human antibody-dependent, cell-mediated cytotoxicity directed by murine monoclonal antibodies. J Exp Med. 1985;161:1–17. doi: 10.1084/jem.161.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klaus G G B, Pepys M B, Kitajina K, Asconas B A. Activation of mouse complement by different classes of mouse antibody. J Immunol. 1979;38:687–695. [PMC free article] [PubMed] [Google Scholar]
- 33.Klipstein F A, Engert R F, Sherman W T. Peroral immunization of rats with Escherichia coli heat-labile enterotoxin delivered by microspheres. Infect Immun. 1983;39:1000–1003. doi: 10.1128/iai.39.2.1000-1003.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Labbe M, Charpilienne A, Crawford S E, Estes M K, Cohen J. Expression of rotavirus VP2 produces empty corelike particles. J Virol. 1991;65:2946–2952. doi: 10.1128/jvi.65.6.2946-2952.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levin J, Bang F B. Clottable protein in Limulus: its localization and kinetics of its coagulation by endotoxin. Thromb Diath Haemorrh. 1968;19:186–197. [PubMed] [Google Scholar]
- 36.Marinaro M, Staats H F, Hiroi T, Jackson R J, Coste M, Boyaka P N, Okahashi N, Yamamoto M, Kiyono H, Bluethmann H, Fujihashi K, McGhee J R. Mucosal adjuvant effect of cholera toxin in mice results from induction of T helper 2 (Th2) cells and IL-4. J Immunol. 1995;155:4621–4629. [PubMed] [Google Scholar]
- 37.Mattingly J A, Waksman B H. Immunologic suppression after oral administration of antigen. II. Antigen specific helper and suppressor factors produced by spleen cells of rats fed sheep erythrocytes. J Immunol. 1980;125:1044–1047. [PubMed] [Google Scholar]
- 38.McGhee J R, Mestecky J, Elson C O, Kiyono H. Regulation of IgA synthesis and immune response by T cells and interleukins. J Clin Immunol. 1989;9:175–199. doi: 10.1007/BF00916814. [DOI] [PubMed] [Google Scholar]
- 39.McIntyre T M, Klinman D R, Rothman P, Lugo M, Dasch J R, Mond J J, Snapper C M. Transforming growth factor beta-1 selectivity stimulates immunoglobulin G2b secretion by lipopolysaccharide-activated murine B cells. J Exp Med. 1993;177:1031–1037. doi: 10.1084/jem.177.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mestecky J. The common mucosal immune system and current strategies for induction of immune responses in external secretions. J Clin Immunol. 1987;7:265–276. doi: 10.1007/BF00915547. [DOI] [PubMed] [Google Scholar]
- 41.Mestecky J, McGhee J R. Anonymous advances in immunology. New York, N.Y: Academic Press, Inc.; 1987. Immunoglobulin A (IgA): molecular and cellular interactions involved in IgA biosynthesis and immune response; pp. 153–245. [DOI] [PubMed] [Google Scholar]
- 42.Mestecky J, McGhee J R. Prospects for human mucosal vaccines. In: Ciardi J E, et al., editors. Genetically engineered vaccines. New York, N.Y: Plenum Press; 1992. pp. 13–23. [Google Scholar]
- 43.Moldoveanu A, Novak M, Huang W, Gilley R M, Staas J K, Schafer D, Compans R W, Mestecky J. Oral immunization with influenza virus in biodegradable microspheres. J Infect Dis. 1993;167:84–90. doi: 10.1093/infdis/167.1.84. [DOI] [PubMed] [Google Scholar]
- 44.Novitsky T J. Discovery to commercialization: the blood of the horseshoe crab. Oceanus. 1984;27:13–18. [Google Scholar]
- 45.Ogra P L. Mucosal immune response to poliovirus vaccines in childhood. Rev Infect Dis. 1984;6:S361–S368. doi: 10.1093/clinids/6.supplement_2.s361. [DOI] [PubMed] [Google Scholar]
- 46.Ogra P L, Karzon D T. Distribution of poliovirus antibody in serum, nasopharynx, and alimentary tract following segmental immunization of lower alimentary tract with polio vaccine. J Immunol. 1969;102:1423–1430. [PubMed] [Google Scholar]
- 47.Ogra P L, Karzon D T, Righthand F, MacGillivray M. Immunoglobulin response in serum and secretions after immunization with live and inactivated polio vaccine and natural infection. N Engl J Med. 1968;279:893–900. doi: 10.1056/NEJM196810242791701. [DOI] [PubMed] [Google Scholar]
- 48.O’Hagan D T, Jeffery H, Roberts M J J, McGee J P, Davis S S. Controlled release microparticles for vaccine development. Vaccine. 1991;9:768–771. doi: 10.1016/0264-410x(91)90295-h. [DOI] [PubMed] [Google Scholar]
- 49.Prasad B V V, Rothnagel R, Jiang X, Estes M K. Three-dimensional structure of baculovirus-expressed Norwalk virus capsids. J Virol. 1994;68:5117–5125. doi: 10.1128/jvi.68.8.5117-5125.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwab K J, Estes M K, Neill F H, Atmar R L. Use of heat release and an internal RNA standard control in reverse transcription-PCR detection of Norwalk virus from stool samples. J Clin Microbiol. 1997;35:511–514. doi: 10.1128/jcm.35.2.511-514.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharp T W, Hyams K C, Watts D, Trofa A F, Martin G J, Kapikian A Z, Green K Y, Jiang X, Estes M K, Waack M, et al. Epidemiology of Norwalk virus during an outbreak of acute gastroenteritis aboard a U.S. aircraft carrier. J Med Virol. 1995;45:61–67. doi: 10.1002/jmv.1890450112. [DOI] [PubMed] [Google Scholar]
- 52.Snapper C M, Waegell W, Beernink H, Dasch J R. Transforming growth factor-beta 1 is required for secretion of IgG of all subclasses by LPS-activated murine B cells in vitro. J Immunol. 1993;151:4626–4636. [PubMed] [Google Scholar]
- 53.Sosroseno W. A review of the mechanisms of oral tolerance and immunotherapy. J R Soc Med. 1995;88:14–17. [PMC free article] [PubMed] [Google Scholar]
- 54.Summers M D, Smith G E. A manual of methods for baculovirus vectors and insect cell culture procedures, bulletin no. 1555. College Station, Tex: Texas Agricultural Experiment Station; 1987. [Google Scholar]
- 55.Thomsen A R, Volkert M, Marker O. Different isotype profiles of virus-specific antibodies in acute and persistent lymphocytic choriomeningitis virus infection in mice. Immunology. 1985;55:213–223. [PMC free article] [PubMed] [Google Scholar]
- 56.van Ginkel F W, Liu C, Simecka J W, Dong J-Y, Greenway T, Frizzell R A, Kiyono H, McGhee J R, Pascual D W. Intratracheal gene delivery with adenoviral vector induces elevated systemic IgG and mucosal IgA antibodies to adenovirus and beta-galactosidase. Hum Gene Ther. 1995;6:895–903. doi: 10.1089/hum.1995.6.7-895. [DOI] [PubMed] [Google Scholar]
- 57.Vinje J, Altena S A, Koopmans M P G. The incidence and genetic variability of small round structured viruses in outbreaks of gastroenteritis in The Netherlands. J Infect Dis. 1997;176:1374–1378. doi: 10.1086/517325. [DOI] [PubMed] [Google Scholar]
- 58.Wachsmann D, Klein J P, Scholler M, Frank R M. Local and systemic immune responses to orally administered liposome-associated soluble S. mutans cell wall antigens. Immunology. 1985;54:189–193. [PMC free article] [PubMed] [Google Scholar]
- 59.White L J, Ball J M, Hardy M E, Tanaka T N, Kitamoto N, Estes M K. Attachment and entry of recombinant Norwalk virus capsids to cultured human and animal cell lines. J Virol. 1996;70:6589–6597. doi: 10.1128/jvi.70.10.6589-6597.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]