

Abstract
Triple-negative breast cancer (TNBC) is a highly aggressive subtype of breast cancer, lacking the expression of estrogen receptor, progesterone receptor, and HER2, which leads to poor prognosis and limited treatment options. Despite advances in targeted therapies, TNBC patients often fail to benefit due to its heterogeneity, drug resistance, and immune evasion. Gene editing technologies such as CRISPR/Cas9 and RNA interference offer promising strategies for precise gene modulation, yet their clinical translation is challenged by delivery efficiency, off-target effects, and safety concerns. This review systematically summarizes key regulatory genes implicated in TNBC progression, including those involved in invasion, proliferation, and chemoresistance, such as BRCA1/2, TP53, MUC1, EGFR, MYC, and others. We further discuss advances in gene editing tools and their combination with targeted delivery materialsranging from viral vectors (AAV, lentivirus) to nonviral platforms (lipid nanoparticles, polymer-based systems, and bioderived vesicles)highlighting their potential to enhance editing specificity, minimize immune response, and overcome tumor microenvironment barriers. Finally, we address biosafety, ethical concerns, and mitigation strategies such as high-fidelity Cas9 variants and AI-assisted off-target prediction. Collectively, this review provides a comprehensive framework for future research and clinical application of targeted gene editing in TNBC, aiming to develop safer, more effective, and personalized treatment strategies.


1. Introduction
Breast cancer is the most common malignancy in women and the second leading cause of cancer-related deaths worldwide, after lung cancer. TNBC accounts for approximately 15% to 20% of all breast cancers, and it is characterized by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) after immunohistochemical staining (IHC). TNBC is a heterogeneous group of diseases characterized by high aggressiveness, poor prognosis, and high recurrence rates. Current treatment options are limited to surgery, adjuvant chemotherapy, and radiation. To date, treatments targeting specific molecular targets have rarely resulted in clinically significant improvements in the prognosis of patients with TNBC, and nonspecific chemotherapy remains the standard treatment method. Furthermore, the majority of patients who respond to standard treatments are confined to the nonmetastatic stage; however, these standard treatment approaches have not significantly improved overall survival rates. Therefore, there is an urgent need to develop more precise treatment regimens and implement them in clinical practice. Targeted gene editing may emerge as an effective approach for treating TNBC. While numerous studies have yielded promising results, there are still many challenges in translating these findings into clinical applications. Currently, there have been numerous scientific investigations into TNBC; however, due to the diverse nature of the research, there is a lack of systematic reviews, resulting in a limited comprehensive understanding of this field. Therefore, this review aims to systematically analyze the key genes influencing the progression of TNBC and explore the potential mechanisms of targeted gene editing therapy for these genes. We will delve into the mechanisms of action of each key gene in TNBC, including their roles in cell proliferation, metastasis, drug resistance, and other aspects of tumor development. Additionally, we will focus on the expression patterns of these genes, particularly those related to clinical pathological features and prognosis. In addition to reviewing key gene studies, we will also examine and analyze the application of various targeted gene editing techniques in TNBC treatment. These techniques include but are not limited to CRISPR/Cas9, RNA interference, etc. We will evaluate their advantages and limitations in treating TNBC and propose possible improvement strategies. Through this review, we hope to provide comprehensive references for further research, promote the application of targeted gene editing technology in TNBC treatment, and offer more effective treatment options for TNBC patients.
2. Key Regulatory Genes Influencing TNBC Progression
2.1. Regulatory Genes of Tumor Cells
2.1.1. Regulation of Invasion and Metastasis
2.1.1.1. Nuclear Protein-Encoding Genes
BRCA1/2: BRCA1 and BRCA2 mutations are common genetic factors in TNBC. BRCA1 mutations are found in 70% of breast cancer patients, while BRCA2 mutations are seen in 16–23% of TNBC cases. These genes play critical roles in maintaining genomic integrity and DNA repair. Dysfunctional BRCA1 or BRCA2 in TNBC results in homologous recombination repair defects, causing reliance on error-prone DNA repair mechanisms, which leads to genomic instability. This instability increases the frequency of microhomology-mediated insertions and deletions, affecting genomic stability and promoting tumor invasion and metastasis.
TP53: TP53 is a tumor suppressor gene encoding the p53 protein, which is involved in DNA repair, apoptosis, and cell cycle regulation. Approximately 80% of TNBC cases harbor TP53 mutations, which are critical for early tumorigenesis. Loss of p53 is associated with chemotherapy resistance. p53 inhibits tumor cell invasion and metastasis by suppressing epithelial-mesenchymal transition (EMT), thereby inhibiting TNBC progression.
MCTS1: MCTS1 (MCTS1 Re-Initiation And Release Factor) encodes a protein that is overexpressed in aggressive TNBC, correlating with poor prognosis. MCTS1 induces EMT and matrix metalloproteinase activation, enhancing the invasiveness of TNBC cells. Additionally, MCTS1 promotes IL-6 secretion, driving macrophage polarization to the M2 phenotype, and increasing TNBC invasiveness. The MCTS1/IL-6/Nrf2/MnSOD/mROS axis is a key mechanism that enhances TNBC stemness and invasiveness.
YTHDC1: YTHDC1(YTH N6-methyladenosine RNA binding protein C1) is an m6A RNA binding protein that regulates mRNA splicing and stability. It plays a critical role in TNBC progression by enhancing cell survival and promoting TGF-β-mediated EMT, which facilitates distant metastasis. TGF-β, known for inhibiting cell proliferation, also promotes invasion, angiogenesis, and metastasis, and is associated with chemotherapy resistance and CSC generation in TNBC.
MALAT1: Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1), a nuclear speckle-localized lncRNA, interacts with pre-mRNA splicing factors to regulate gene expression and alternative splicing. In TNBC, MALAT1 is significantly overexpressed compared to normal tissue and promotes tumor progression by enhancing cell migration, invasion, and angiogenesis. Importantly, CRISPR/Cas9-mediated deletion of the MALAT1 promoter increases TNBC sensitivity to neoadjuvant chemotherapy, suggesting that MALAT1-targeted gene editing could inhibit TNBC proliferation and metastasis while improving chemosensitivity.
BRD4: Bromodomain-containing protein 4 (BRD4), an epigenetic reader, regulates oncogene transcription by mediating superenhancer assembly. In TNBC, BRD4 isoforms exhibit opposing functions: the short isoform (BRD4-S) promotes tumor growth and metastasis, while the long isoform (BRD4-L) shows tumor-suppressive effects. Recent studies reveal that BRD4-S restricts the LOXL2 promoter (a transcriptional activator of cell cycle genes), and dual inhibition of BRD4/LOXL2 synergistically suppresses TNBC proliferation both in vitro and in vivo. Additionally, BRD4 mediates upregulation of mutant p53.
CTCF: CTCF, a highly conserved eukaryotic protein with 11 zinc finger motifs, recognizes specific genomic sequences. In breast cancer, CTCF regulates multiple genes through interactions with DNA methylation, estrogen receptor (ER), and miRNAs. Strikingly, CTCF shows 100% deactivation frequency in metastatic cancer patients. The expression of CTCF can suppress the migration of TNBC cells, while its low expression is closely associated with poor prognosis in TNBC. , Inserting exogenous CTCF genes may help treat metastatic TNBC by integrating them into the genomes of cancer cells, compensating for the low CTCF levels.
CDK5: CDK5 plays multifaceted roles in cancer progression through its distinct regulatory mechanisms. As an atypical cyclin-dependent kinase activated by p35 (CDK5R1) and p39 (CDK5R2), CDK5 demonstrates oncogenic properties across multiple pathological processes. In breast cancer, elevated CDK5/p35 expression correlates with advanced tumor stage and poor prognosis, with TGFβ1 stimulation further enhancing their transcription. Mechanistically, CDK5 promotes tumorigenesis by (1) facilitating cell migration and EMT through actin cytoskeleton remodeling, (2) suppressing apoptotic pathways to sustain tumor survival, and (3) modulating tumor immunity via the IRF2-IRF2BP2-PD-L1 regulatory axis. , Particularly noteworthy is CDK5′s dual role in metastasis promotion and immune evasion, where its activity maintains PD-L1 expression through transcriptional regulation of interferon response factors.
SEPT9: SEPT9, particularly its oncogenic isoform SEPT9_i1, plays a significant role in breast cancer progression. Generated from transcript variant SEPT9_v1, SEPT9_i1 is frequently amplified and overexpressed in breast tumors, correlating with advanced disease stages and poor clinical outcomes. Functionally, SEPT9_i1 promotes tumor aggressiveness by enhancing cancer cell migration and invasion through both 2D and 3D microenvironments, while simultaneously facilitating extracellular matrix (ECM) degradation to support metastatic dissemination.
2.1.1.2. Genes Encoding Extranuclear Proteins
UBR5: The ubiquitin ligase UBR5 plays a multifaceted role in TNBC progression. It promotes tumor invasiveness by degrading the tumor suppressor CDC73 and is significantly upregulated in TNBC tissues. CRISPR/Cas9-mediated UBR5 knockout impairs tumor angiogenesis, induces apoptosis/necrosis, and causes growth arrest. Additionally, UBR5 upregulates PKR-STAT1-IRF1 signaling to enhance PD-L1 expression, facilitating immune evasion.
Notch1: Notch1 signaling is aberrantly activated in triple-negative breast cancer (TNBC), where it drives tumor aggressiveness through multiple mechanisms. Compared to other breast cancer subtypes, TNBC exhibits significantly higher Notch1 signaling activity, which correlates with more invasive features and poorer patient survival. At the molecular level, Notch1 promotes TNBC progression by inducing epithelial-mesenchymal transition (EMT) to facilitate metastasis and dysregulating cell cycle control to sustain proliferation. This pathway also maintains cancer stem cell populations and contributes to chemotherapy resistance. The therapeutic potential of targeting Notch1 is highlighted by miR-3178’s ability to suppress TNBC growth and metastasis through Notch1 inhibition while the frequent occurrence of NOTCH1 alterations across cancers further underscores its importance in TNBC pathogenesis.
NAA20: The EGFR signaling pathway is typically highly active in TNBC, and it is closely associated with poor survival outcomes and prognosis in TNBC patients. NAA20 can promote the progression of TNBC by modulating the EGFR signaling pathway mediated by Rab5A. Knocking out NAA20 significantly inhibits the survival, migration, and invasion capabilities of TNBC cells, while reducing the expression level of EGFR.
NRG1: Neuregulin 1 (NRG1), a member of the epidermal growth factor family, promotes TNBC progression through a unique signaling cascade. In TNBC, NRG1 activates an ERK1/2-Fbxw7-c-Myc pathway to upregulate Fra-1 expression, which subsequently drives epithelial-mesenchymal transition (EMT) and facilitates metastasis. This NRG1-Fra-1 axis represents a promising therapeutic target, as its inhibition could potentially suppress TNBC’s aggressive metastatic behavior.
SEAS1: SEAS1 (also known as SEMA3B-AS1) is a long noncoding RNA that is downregulated in TNBC and is associated with poor prognosis. Knockout of SEAS1 significantly enhances the proliferation, migration, and invasion of TNBC cells, while its overexpression reverses these effects. Mechanistically, SEAS1 suppresses tumor progression by targeting the miR-3940/KLLN axis, highlighting its potential as a diagnostic biomarker and therapeutic target in TNBC.
MUC1: MUC1 is aberrantly amplified in approximately 90% of TNBC cases, where its C-terminal subunit (MUC1-C) drives oncogenic progression through multifaceted mechanisms. , MUC1-C activates diverse signaling pathways, induces chromatin remodeling, and confers aggressive phenotypes including DNA damage resistance, drug tolerance, stem-like properties, and immune evasion. Notably, MUC1 contributes to an immunosuppressive tumor microenvironment by promoting CD8+ T cell depletion and dysfunction, highlighting its dual role in tumor cell-intrinsic progression and extrinsic immune modulation.
Additionally, research suggests that compared to primary TNBC, there is a more pronounced enrichment of genes such as PIK3CA, GATA3, CDH1, MAP3K1, PTEN, and PIK3R1 in metastatic TNBC. Metastatic TNBC typically exhibits mutation patterns similar to those of primary TNBC. In some rare gene mutations, a higher mutation frequency has been observed, such as PTPRD, TSC2, PLCG1, ARID1B, CREBBP, and FAM47C, among others. These may all serve as potential targets for targeted gene therapy in TNBC.
2.1.2. Genes Regulating Proliferation
PIK3CA: In TNBC, PIK3CA mutations (frequently coinciding with AKT1 activation and PTEN loss) cause constitutive PI3K/AKT/mTOR pathway activation. Approximately 25–30% of advanced TNBC cases harbor coactivating PIK3CA and AKT1 mutations. This pathway reprograms tumor metabolism by enhancing nutrient transporter and metabolic enzyme activities, with moderately elevated ROS levels promoting tumor growth through modulation of multiple metabolic processes. Notably, PIK3CA mutations lead to hyperactivation of mTORC1 downstream signaling, representing a key mechanism driving TNBC progression.
EGFR: EGFR (ErbB1/HER1), a key member of the ErbB/HER receptor tyrosine kinase family, is frequently overexpressed in TNBC and correlates with poor patient prognosis. This overexpression drives TNBC progression through multiple signaling pathways, including PI3K/Akt, Ras/ERK, and STAT3, enhancing tumor cell proliferation, migration, and invasion. Notably, EGFR monomers have been identified as primary activators of STAT3 signaling in TNBC, particularly under nutrient-limited conditions in the tumor microenvironment, suggesting potential therapeutic vulnerabilities.
USP19: The endoplasmic reticulum (ER)-resident deubiquitinase USP19 demonstrates tumor-suppressive functions in TNBC through ER stress regulation. Downregulated in TNBC tissues, USP19 maintains ER homeostasis by stabilizing BAG6 and BCL2, thereby controlling protein folding and calcium flux. Loss of USP19 activity disrupts these processes, leading to enhanced ER stress that ultimately suppresses TNBC proliferation and triggers apoptosis.
BIRC6: BIRC6 is highly expressed in TNBC cells and tissues, and its overexpression is associated with decreased patient survival rates. The signaling cascade of the EGFR serves as the major upstream regulatory factor driving the high expression of BIRC6. BIRC6 can inhibit SMAC-mediated apoptosis, thereby promoting the survival of TNBC cells and the development of tumors.
YTHDF2: The m6A reader protein YTHDF2 plays an essential role in TNBC progression by coordinating mRNA metabolism under cellular stress conditions. Particularly critical in MYC-driven TNBC cells, YTHDF2 maintains cell viability while regulating migration and proliferation. Mechanistically, YTHDF2 deficiency upregulates EMT pathway genes and, when combined with PRSS23 deletion, partially restores BiP expression, reduces ROS levels by 50%, and attenuates apoptotic signaling, revealing its multifaceted role in TNBC pathogenesis.
RSRC2: RSRC2 is a recently discovered tumor suppressor gene with lower expression levels in TNBC tissues. The low expression of RSRC2 is associated with poorer prognosis in breast cancer patients. RSRC2 inhibits tumor cell proliferation, adhesion, migration, and invasion in TNBC by negatively regulating SCIN-mediated cellular functions, providing a potential target for TNBC treatment.
CASP9: Caspase-9 (CASP9), a key executioner of intrinsic apoptosis, functions as a tumor suppressor in TNBC by responding to cellular damage signals including genomic instability, oxidative stress, and aberrant proliferation. Its activation triggers the caspase cascade that leads to programmed cell death, counteracting the apoptosis evasion that characterizes TNBC progression suggesting its potential as a therapeutic target for restoring apoptotic sensitivity.
PKN3: Protein kinase N3 (PKN3), an AGC-family member, is often overexpressed in breast tumor cells. PKN3 plays a crucial role in tumor angiogenesis and metastasis.
2.1.3. Genes Associated with Chemotherapy Resistance
SOX4: SOX4 facilitates immune evasion in TNBC through the integrin αvβ6-TGFβ-SOX4 signaling axis. Clinical studies demonstrate that SOX4 expression significantly correlates with poor patient prognosis and treatment response, mediated by enhanced resistance to cytotoxic T cellsa finding particularly prominent in less-differentiated epithelial tumor cells.
WAVE3: WAVE3 drives chemoresistance in TNBC through a β-catenin-dependent signaling pathway. Genetic or phospho-deficiency targeting of WAVE3, when combined with conventional chemotherapy, significantly suppresses the malignant behavior of treatment-resistant TNBC cells in both preclinical models, highlighting WAVE3 inhibition as a promising strategy to overcome chemoresistance in TNBC.
CREIT: circRNA-CREIT exhibits tumor-suppressive properties in TNBC, where its downregulation correlates with doxorubicin resistance and poor patient prognosis. Notably, this circular RNA can be encapsulated into exosomes to systemically restore chemosensitivity in TNBC cells, revealing its dual role as both a prognostic biomarker and therapeutic vehicle for overcoming chemoresistance.
PRMT5: PRMT5 is highly expressed in breast cancer and enhances chemotherapy resistance through m6A RNA modification regulation. Research demonstrates that PRMT5 methylates and stabilizes KEAP1, suppressing the NRF2/HMOX1 pathway and consequently increasing resistance to both ferroptosis and immunotherapy.
BAG3: BAG3, a member of the BAG cochaperone family, promotes TNBC progression through multiple oncogenic mechanisms. It maintains breast cancer stem cell (BCSC) properties via CXCR4 signaling and enhances chemoresistance by stabilizing antiapoptotic Bcl-2 family proteins (Mcl-1, Bcl-2, Bcl-xL). These findings, combined with its roles in autophagy and stress response, establish BAG3 as a multifunctional regulator of TNBC aggressiveness and treatment resistance.
MYC: There is a strong correlation between MYC expression and the loss of immune signatures in human TNBC. MYC overexpression suppresses innate immunity and promotes tumor immune escape, leading to reduced immunogenicity of TNBC. Kyung-min Lee et al. demonstrated that MYC-driven immune evasion is mediated through STING suppression.
U2SURP: U2SURP (U2 snRNP-associated SURP motif-containing protein) is significantly upregulated in TNBC tissues, and its high expression correlates with poor patient prognosis. Mechanistically, MYC promotes U2SURP translation in an eIF3D-dependent manner, leading to its accumulation in TNBC. U2SURP facilitates the alternative splicing of SAT1 pre-mRNA by removing intron 3, resulting in increased mRNA stability and elevated protein expression. The spliced SAT1 enhances the oncogenic traits of TNBC cells. These findings highlight the role of the MYC–U2SURP–SAT1 axis in TNBC progression and suggest U2SURP as a potential therapeutic target.
2.2. Regulatory Genes in Non-Tumor Cells
COP1: COP1, an E3 ubiquitin ligase, promotes TNBC progression through dual tumor cell-intrinsic and immune-modulatory mechanisms. While known to ubiquitinate multiple oncogenic substrates (including p53, STAT3, and β-catenin) to regulate proliferation and survival, recent studies reveal its critical role in shaping an immunosuppressive microenvironment. COP1 knockout reduces macrophage chemokine secretion, decreases tumor-associated macrophage infiltration, and enhances response to immune checkpoint inhibitors, establishing COP1 as a multifaceted therapeutic target in TNBC.
RGS5: RGS5 exhibits context-dependent roles in vascular biology and cancer progression. While it normally suppresses inflammation and pyroptosis in vascular smooth muscle cells (VSMCs) to maintain vascular homeostasis, RGS5 undergoes functional switching in the tumor microenvironment to promote a pro-inflammatory VSMC phenotype that enhances tumor cell adhesion and facilitates metastasis in triple-negative breast cancer. This dichotomous function highlights the therapeutic complexity of targeting RGS5 in TNBC.
Lcn2: LCN2 is highly expressed in the urine and tissues of patients with TNBC, and elevated LCN2 levels are associated with poor prognosis. Secreted by TNBC cells, LCN2 promotes tumor progression by upregulating vascular endothelial growth factor (VEGF) to enhance angiogenesis, and by inducing epithelial–mesenchymal transition (EMT) to increase cancer cell invasiveness. , These findings highlight the crucial role of LCN2 in TNBC development and support its potential as a therapeutic target.
It should be noted that these mechanisms do not exist in isolation; they may interact with each other and collectively contribute to the occurrence and development of cancer. Further research will help to better understand the carcinogenic mechanisms of and guide the development of more effective therapeutic strategies.
3. Advances in Gene Editing Strategies for TNBC
3.1. Tools for Targeted Gene Editing
So far, gene editing technology has gone through three generations of development. The first generation of gene editing technology, Zinc Finger Nucleases (ZFN), is characterized by precise targeting and stability, enabling successful genome editing both in vitro and in vivo. ZFN is less likely to trigger immunogenicity and is compact enough to be used with adeno-associated viral (AAV) vectors. However, the process of designing ZFNs is time-consuming and requires substantial molecular biology expertise, and its interaction with DNA is context-dependent, adding to the complexity of design. The second generation of gene editing technology, Transcription Activator-Like Effector Nucleases (TALEN), allows for more flexible design with higher specificity and stability. While designing TALENs is much simpler than ZFNs, their design and construction still require considerable time and effort. The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system has revolutionized the field of gene editing. CRISPR/Cas9 offers higher accuracy and efficiency than other gene-editing technologies such as TALENs and zinc-finger nucleases. This system can not only identify and confirm genomic targets responsible for cancer but also edit, suppress, and epigenetically modify oncogenes in human cells96. As a powerful genome-editing tool, CRISPR/Cas9 can simultaneously edit multiple independent sites by merely altering the sgRNA sequence. Furthermore, CRISPR/Cas9 is capable of modifying chromosomal targets with high precision and low cytotoxicity. Its low design barrier and ease of use have significantly simplified previously complex genetic operations.
TNBC arises from genetic and epigenetic abnormalities. Using gene editing techniques like CRISPR/Cas9 to correct these genetic and epigenetic aberrations could be a viable therapeutic approach. Furthermore, transcription factors involved in cell-specific transcriptional regulation exhibit unique traits in cancer cells, suggesting that targeting transcriptional regulation could be an effective strategy for cancer therapy.
However, this technology still faces several challenges, one of which is reducing off-target effects. Strategies to address this issue include introducing tissue-specific promoters and carefully designing single guide RNAs (sgRNAs). Another challenge is to increase in vivo editing efficiency, which requires enhancing system activity and improving delivery efficiency to overcome varying editing rates at different gene targets. Additionally, the development of efficient nonviral vectors represents another significant challenge. The current nonviral vectors for CRISPR/Cas9 delivery remain quite limited, making it critical to research novel and effective nonviral vectors to meet this challenge.
The applications of RNAi can be mediated through two types of molecules; the chemically synthesized double-stranded small interfering RNA (siRNA) or vector based short hairpin RNA (shRNA). Small interfering RNAs, typically composed of approximately 21–23 nucleotides, are widely used for short-term gene silencing of protein-coding genes. In recent years, siRNA has gained considerable attention in the field of gene therapy due to its high specificity in downregulating gene expression and its ability to target a multitude of therapeutic sites, including those inaccessible by traditional drugs. siRNA achieves targeted gene silencing by complementary pairing with specific mRNA sequences, leading to the degradation or inhibition of translation of the target mRNA, making it a widely used tool in biological research for silencing or downregulating gene expression. However, siRNA faces some challenges in cancer therapy, including the development of effective delivery systems to protect siRNA from degradation, ensure its delivery to highly specific target cells, and promote its endosomal escape and efficient cytoplasmic distribution. shRNA is typically encoded in plasmids or viral vectors, where it is transcribed within the cell to produce siRNA, offering a more sustained gene silencing effect.
Additionally, certain small molecule compounds can impact gene expression and cellular function by modulating DNA methylation, histone modifications, and noncoding RNA expression, playing a role in the onset and progression of diseases. For instance, active compounds from medicinal plants, such as alkaloids, polyphenols, polysaccharides, terpenes, and anthocyanins, are widely believed to possess anticancer potential.
In summary, targeted gene editing tools are significant in the treatment of TNBC, offering the potential to develop precise therapeutic approaches that target specific genesfor example, by repairing or suppressing the expression of pathogenic genes and increasing tumor cell sensitivity to chemotherapy. This technology can also support personalized medicine, tailoring treatment plans based on the patient’s genomic characteristics to improve therapeutic outcomes and minimize adverse effects (Figure ).
1.
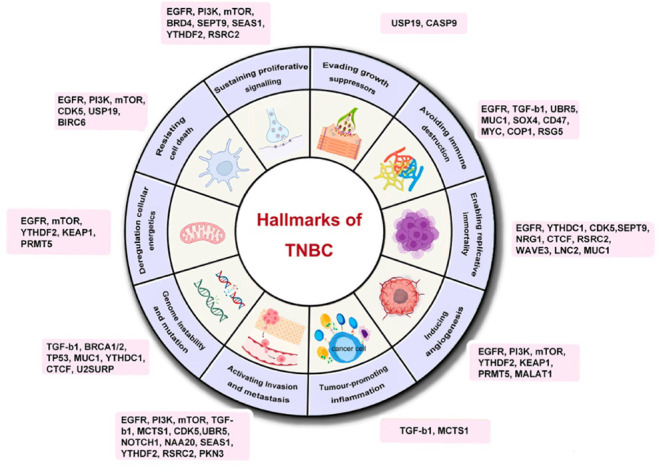
This review figure highlights key regulatory genes associated with TNBC and categorizes them based on their oncogenic mechanisms. The mechanisms include: 1. Evading growth suppressors, 2. Avoiding immune destruction, 3. Enabling replicative immortality, 4. Inducing angiogenesis, 5. Tumor-promoting inflammation, 6. Activating invasion and metastasis, 7. Genome instability and mutation, 8. Deregulating cellular energetics, 9. Resisting cell death, and 10. Sustaining proliferative signaling. By identifying and categorizing these genes, we can better understand the pathogenesis and progression of TNBC, providing a foundation for developing more effective therapeutic strategies. Further research into the interactions among these regulatory genes will help in designing comprehensive treatment plans to address the complexity and heterogeneity of TNBC.
3.2. Applications of Targeted Delivery Materials Encapsulating Different Gene Editing Tools in TNBC
An effective in vivo delivery vector must possess the following characteristics: (1) the ability to encapsulate and protect its payload to prevent degradation or fixation before entering the cell; (2) the capability to bind to target cells; (3) the ability to traverse the target cell membrane and enter the intracellular environment; and (4) the capacity to release its payload into the appropriate intracellular region (Figure ).
2.

This figure categorizes gene editing vectors used for TNBC into viral and nonviral vectors. Viral vectors include Adeno-Associated Virus (AAV), Lentivirus (LV), and Retrovirus (RV). Nonviral vectors consist of biologically derived vectors, lipid nanoparticles, and composite polymer vectors.
3.2.1. Current Viral Vectors for In Vivo Delivery
3.2.1.1. Adenovirus Vector System
Adeno-associated viruses (AAVs) are small, nonenveloped parvovirus, approximately 25 nm in size, and is nonpathogenic, having lost its pathogenicity through evolution. Its genome consists of approximately 4.7 kb of single-stranded DNA. The risk of insertional mutations with AAV vectors is relatively low, offering greater safety compared to other viral vectors. Additionally, recombinant AAV (rAAV) has improved packaging capacity while reducing immunogenicity and cytotoxicity during in vivo administration.
AAV also has a wide range of target cells and is highly biocompatible, capable of delivering effective payloads to various clinically relevant tissues. However, AAV shows poor organ specificity, which could limit its effectiveness in certain patient groups. Additionally, AAV can usually carry only small gene fragments, generally no more than 5.0kb, which constrains its use for larger or more complex gene constructs. Experiments on humans have shown that adaptive immune responses against the vector capsid can be an obstacle to sustained transgene expression, as they may activate and expand capsid-specific T cells.
3.2.1.2. Retroviral Vectors System
Retroviral vectors offer highly efficient gene delivery, enabling the introduction of exogenous genes into host cells and integrating them into the host genome for long-term stable expression of the target gene, making them an effective gene-editing tool. Retroviral replicating vectors (RRVs) have the potential for viral reservoir properties, allowing them to spontaneously reinfect and continue targeting growing tumor cells after initial infection. Additionally, RRVs can replicate in nonresectable normal tissues and subsequently destroy tumor-infiltrating cells, suggesting that they hold considerable potential for gene therapy in tumors.
3.2.1.3. Lentiviral Vectors System
Lentiviruses belong to the Retroviridae family. The name ″lentivirus″ derives from the prolonged interval between initial infection and the onset of disease. Lentiviral vectors (LVs) have the ability to transduce nondividing or slowly dividing cells, allowing for the delivery of a large gene payload while maintaining stable and long-term transgene expression. These attributes make lentiviral vectors an important tool in the field of gene delivery. Lentiviral vectors can stably integrate exogenous genes into the host cell genome, providing long-term, stable gene expression. In contrast, retroviral vectors usually require host cell division and DNA replication to maintain gene expression, leading to shorter expression periods. Additionally, lentiviral vectors can carry larger gene loads compared to retroviral vectors and are less likely to stimulate the host immune system.
Yongqiu Zeng et al. found that knocking down SEPT9 in MCF-7 cells using lentivirus-mediated shRNA significantly reduced breast tumor cell migration, focal adhesion (FA) maturation, and the expression levels of β-actin, β-tubulin, Cdc42, RhoA, and Rac.
Wei-Ying Kuo et al. designed a HSV-tk gene/GCV system driven by STAT3-NF-κB response elements. Utilizing a lentiviral vector, the HSV-TK gene was specifically delivered to the tumor interior, achieving the shrinkage of TNBC and reducing the expression of EMT phenotype and cancer stem cell (CSC) markers by targeting the STAT3/NF-κB signaling pathway. Additionally, the study found that this therapeutic strategy could enhance the sensitivity of TNBC to cisplatin, indicating that inhibiting STAT3/NF-κB might deplete chemotherapy-resistant cells in the tumor, thereby enhancing the efficacy of cisplatin (Figure C).
3.

A) Survival curves for different treatment groups. Reprinted with permission from: Rao, L.; Zhao, S. K.; Wen, C.; Tian, R.; Lin, L.; Cai, B.; Sun, Y.; Kang, F.; Yang, Z.; He, L.; et al. Activating Macrophage-Mediated Cancer Immunotherapy by Genetically Edited Nanoparticles. Adv Mater 2020, 32 (47), e2004853. DOI: 10.1002/adma.202004853. Copyright 2020 Wiley-VCH GmbH. B) GCa-NP/siEGFR-BRD4 significantly inhibited cell invasion.. Reprinted with permission from: Zhang, C.; Yuan, W.; Wu, Y.; Wan, X.; Gong, Y. Co-delivery of EGFR and BRD4 siRNA by cell-penetrating peptides-modified redox-responsive complex in triple negative breast cancer cells. Life Sci 2021, 266, 118886. DOI: 10.1016/j.lfs.2020.118886. Copyright 2021 Elsevier. C) Tumor burden was assessed by IVIS on days 0, 4, and 7. Lenti-STAT3-NF-κB-tk/GCV treatment showed the strongest tumor suppression among all groups. Reprinted with permission from: Kuo, W. Y.; Hwu, L.; Wu, C. Y.; Lee, J. S.; Chang, C. W.; Liu, R. S. STAT3/NF-κB-Regulated Lentiviral TK/GCV Suicide Gene Therapy for Cisplatin-Resistant Triple-Negative Breast Cancer. Theranostics 2017, 7 (3), 647–663. DOI: 10.7150/thno.16827. Copyright 2017 Ivyspring International Publisher. D) Representative images of the tumors dissected from MDA-MB-468 xenograft-bearing mice receiving various treatments (n = 5 mice in each group). Reprinted with permission from: Li, Y.; Tan, Y.; Wen, L.; Xing, Z.; Wang, C.; Zhang, L.; Wu, K.; Sun, H.; Li, Y.; Lei, Q.; et al. Overexpression of BIRC6 driven by EGF-JNK-HECTD1 signaling is a potential therapeutic target for triple-negative breast cancer. Mol Ther Nucleic Acids 2021, 26, 798–812. DOI: 10.1016/j.omtn.2021.09.011. Copyright 2021 Elsevier.
Additionally, Francesca Caccuri et al. used herpes simplex virus 1 (HSV-1) as a vector to deliver the U94 gene to TNBC cells. The expression of U94 successfully interfered with the proliferation and key steps of the metastatic cascade in TNBC cells, inducing a partial MET and leading to a less invasive phenotype. The study also found that U94, through the inhibition of DNA damage repair genes and cell cycle progression, ultimately leads to cell death via activation of the intrinsic apoptotic pathway. Notably, U94 was found to have a synergistic effect with certain DNA-damaging drugs, further enhancing its anticancer effect.
Although lentiviral vectors have potential in gene editing, their clinical application poses certain risks. These risks include integration-induced insertional mutagenesis and potential off-target effects associated with gene editing, which may affect safety for future generations.
3.2.2. Current Nonviral Vectors for In Vivo Delivery
Viral vectors have high gene delivery efficiency and provide long-term stable transgene expression. However, the inherent drawbacks of viral vectors, including oncogenic risk, insertion size limitations, immune response, and difficulty in large-scale production, significantly limit their application.
Nonviral gene delivery tools are considered a promising alternative for gene delivery due to their safety, scalability, and versatility. Compared to virus-derived vectors, nonviral vectors offer several advantages: safety in administration, low immunogenic response, high gene load capacity, flexible chemical design, almost no limitation on transgene size, and the possibility of repeated dosing.
3.2.2.1. Lipid Nanoparticles
Lipid nanoparticles (LNPs) are amphiphilic systems composed of various hydrophobic and hydrophilic components. They have gained attention for their low immunogenicity, high payload capacity, safety, and ease of preparation. LNPs typically consist of cationic or ionizable lipids, neutral lipids such as phospholipids or cholesterol, and polyethylene glycol (PEG) lipids. The addition of PEG helps create a system with longer circulation times and improved stability in vivo, increasing the half-life and stability in the bloodstream104. The inclusion of cholesterol and phospholipids can stabilize the LNP structure and enhance membrane fusion, improving the stability and transfection efficiency of lipid-based gene delivery systems. Because LNPs do not expose proteins or peptides externally, they are considered to have lower immunogenicity than viral vectors.
Di Zhang et al. packaged siRNA (siFAK), mRNA (Cas9), and targeted sgRNA together into self-assembled LNPs, resulting in decreased mechanical properties of tumor tissues (such as tumor hardness, elasticity, and deformation) as well as the ECM hardness. This treatment also increased the internalization of nanoparticles and enhanced their penetration into tumor tissues. This enabled the CRISPR-Cas9 technology to directly target the PD-L1 gene, significantly enhancing CRISPR gene editing efficacy in both in vitro and in vivo tumor cells. The results showed that siFAK+CRISPR-PD-L1-LNPs prolonged the survival of mice with MYC-induced carcinogenesis and provided significant survival benefits. Self-assembled LNPs can serve as effective carriers for delivering therapeutic drugs and gene vectors to TNBC tumor tissues, thereby reducing the mechanical properties of tumor tissues, enhancing gene editing efficacy, and offering new possibilities for TNBC treatment.
Peng Guo et al. synthesized and applied a noncationic, deformable, tumor-targeting lipid nanogel system (tNLGs) to achieve effective gene editing in TNBC cells using CRISPR technology. The tNLGs use noncationic lipids with a deformable core–shell structure, which not only eliminates cationic toxicity but also maintains high encapsulation efficiency. Additionally, the system delivers CRISPR plasmids directly into the cytoplasm of targeted TNBC cells through receptor-mediated membrane fusion, effectively avoiding the entrapment of CRISPR plasmids in endosomes within TNBC cells. The CRISPR mediated by tNLGs efficiently knocked out the Lipocalin 2 (Lcn2) gene, significantly suppressing Lcn2 protein synthesis and TNBC cell migration, thereby reducing TNBC invasiveness (Figure A). A novel lipid nanoparticle (SS-LNP/shPKN3) encapsulating a new shRNA for PKN3 with redox-responsiveness was developed. The material uses glutathione (GSH) to trigger the release of shPKN3 and has a protein inhibition rate and tumor suppression rate of over 60%.
3.2.2.2. Bioderived Vesicles
Bioderived vesicles are small vesicles released by cells, enveloped in cell membranes, and carry intracellular bioactive molecules such as proteins, nucleic acids, and lipids. Bioderived vesicles mainly consist of two types: exosomes and microvesicles. They are characterized by low immunogenicity, high cellular uptake, and excellent loading capacity. The targeting ability of bioderived vesicles depends on the type of their parent cells.
Cancer cell-derived exosomes, as natural carriers, efficiently deliver CRISPR/Cas9 plasmids into cancer cells. Seung Min Kim et al. found that compared to epithelial cell-derived exosomes, cancer cell-derived exosomes exhibit selective accumulation in ovarian cancer tumors. Moreover, tumor-derived exosomes are rich in CRISPR-Cas9 plasmids targeting poly(ADP-ribose) polymerase-1 (PARP-1), effectively inhibiting PARP-1 and inducing apoptosis in ovarian cancer cells, thereby enhancing sensitivity to cisplatin. Targeted inhibition of PARP-1 has shown significant efficacy in treating TNBC, as it can suppress EMT and restore TNBC cells’ sensitivity to chemotherapy. Cancer cell-derived exosomes, used as gene editing vectors, provide an effective means for delivering the CRISPR/Cas9 system in TNBC gene therapy. This approach allows precise gene editing and repair, reducing damage to healthy tissues and minimizing side effects, thereby enhancing the safety and efficacy of treatment. However, some studies have indicated that while bioderived vesicles offer high delivery efficiency, low cytotoxicity, and good target specificity, they may have certain limitations in terms of stability and payload capacity. This suggests that these issues require further investigation.
Lang Rao et al. recently developed genetically engineered cell-membrane-coated magnetic nanoparticles (gCM-MNs). By overexpressing a variant of SIRPα on the gCM shell through gene editing, they significantly increased its binding affinity with CD47, effectively blocking the CD47-SIRPα signaling pathway. In a TNBC 4T1 tumor model, gCM-MNs successfully suppressed cancer metastasis. Additionally, gCM-MNs induced tumor-associated macrophages (TAMs) to repolarize to the M1 phenotype, enhancing their ability to phagocytize tumor cells and stimulating T-cell-mediated antitumor immune responses. The magnetic core of gCM-MNs enables magnetic navigation, allowing gene-edited cell membranes to be precisely directed to the tumor microenvironment for targeted therapy. This targeting feature increases gCM-MNs accumulation in tumor tissues while reducing off-target immune activation (Figure B).
3.2.2.3. Polymer-Based Gene Editing Vectors
Polyethylene glycol (PEG) is a bioinert and biocompatible polymer composed of repeating units of ethylene oxide. It is known for its low toxicity, biocompatibility, and inert properties. Although PEG is considered to have low immunogenicity due to its favorable physicochemical characteristics, studies have indicated that when PEG is conjugated with other materials, it can trigger immune responses, particularly with proteins, lipid nanoparticles, and liposomes. , Anti-PEG antibodies can significantly impact the pharmacokinetics of PEGylated therapies. Repeated administration of PEGylated proteins and nanoparticles may lead to the accelerated blood clearance (ABC) phenomenon, causing nanomedicines to be cleared from the body at a faster rate, thereby undermining the stealth properties of PEGylation.
Poly(lactic-co-glycolic acid) (PLGA) is a versatile biomedical polymer formed by the copolymerization of lactic acid (LA) and glycolic acid (GA). PLGA properties are influenced by several factors, including the ratio of LA to GA and the molecular weight of the polymer. As the LA/GA ratio increases, the hydrophobicity of PLGA also increases, leading to slower degradation and a more gradual drug release. Conversely, a lower molecular weight accelerates PLGA’s degradation and drug release processes. Additionally, the size of PLGA nanoparticles significantly impacts degradation and drug release kinetics, with smaller particles better able to overcome biological barriers and improve drug delivery efficiency. Therefore, adjusting the LA/GA ratio is crucial for precisely controlling degradation times and drug release rates.
Yanhua Zhang et al. synthesized multifunctional tumor-targeted poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs-cRGD), comprising PLGA copolymers, cRGD targeting ligands, and Pt(IV). Through cRGD-mediated targeting, this nanocarrier selectively recognizes and localizes ovarian cancer cells. NPs-cRGD exhibits precise control and sustained release of drugs and genes, maintaining high concentrations in tumor tissues while reducing the toxicity of platinum-based drugs. The action of Pt(IV) in NPs-cRGD involves depleting intracellular glutathione (GSH), thereby enhancing the accumulation of Pt(II). Pt(II) binds to DNA, inhibiting gene expression and upregulating p53 expression, subsequently activating the mitochondrial apoptosis pathway. By reducing glutathione (GSH) activity and promoting Pt(II) generation, further stimulation of reactive oxygen species (ROS) production induces cell apoptosis, contributing to enhancing the therapeutic efficacy of platinum drugs.
As previously mentioned, TP53 gene mutations are common in TNBC, resulting in abnormal p53 protein function, which poses a challenge to treatment. By using a strategy that involves Pt(II) binding to DNA to upregulate p53, it is possible to activate the mitochondrial apoptosis pathway and increase TNBC sensitivity to treatment. Particularly in cases of drug resistance, incorporating strategies that regulate GSH and ROS levels may help overcome resistance and improve overall therapeutic outcomes. This study demonstrates that NPs-cRGD, with its precise targeting and multiple mechanisms of action, represents a promising therapeutic strategy, offering a new direction for TNBC treatment.
Chemical modification or bioconjugation of PLGA can significantly influence drug delivery and tissue regeneration in tumor studies. Additionally, precise control over PLGA’s characteristics and parameters allows for various customizable functions, yielding good mechanical properties and low human toxicity. This is crucial for expanding PLGA’s applications in the biomedical field.
Polyethylenimine (PEI) is a widely studied cationic polymer known for its high gene-loading capacity. PEI complexes remain stable at room temperature and can be stored for extended periods in a frozen state without losing activity, making them easy to handle and store.
Ruru Gao et al. developed a novel carrier by using polyethylenimine (PEI)-modified biosynthetic nanobubbles (GVs) to successfully knock out the EGFP and Cdh2 genes in 4T1 cells through the CRISPR/Cas9 system. This approach significantly inhibited tumor cell invasion and metastasis, effectively curbing tumor growth.
However, PEI still faces several challenges, including the instability of PEI/DNA complexes, the potential cytotoxicity resulting from unexpected interactions with host biomolecules, susceptibility to capture and clearance by the immune system, and short circulation time leading to low gene delivery efficiency.
3.2.2.4. Composite Polymer-Based Gene Editing Vectors
Although poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs), approved by the U.S. Food and Drug Administration (FDA), are widely used for encapsulating various antitumor drugs, they are prone to recognition and rapid clearance by the complement system following intravenous injection, which limits their application. Similarly, nanoparticles based on polyethylene glycol (PEG) and polyethylenimine (PEI) face related issues. These limitations highlight the need for new polymer-based gene editing vectors, especially composite polymer materials. Composite polymers, composed of two or more different polymers, can be combined through physical mixing, copolymerization, or cross-linking to create materials with new properties and functions. These materials can overcome the limitations of single polymers, such as enhancing biological stability, regulating drug release rates, and improving targeting, thus offering broader application prospects in drug delivery. The following discusses some recently developed composite polymer-based gene-editing vectors for cancer therapy. The following discusses some recently developed composite polymer-based gene-editing vectors for cancer therapy.
Alexander S. Timin et al. reported a study on polymer and hybrid microparticles made from degradable polymers such as polypeptides and polysaccharides, modified with a silicon shell, designed to deliver all CRISPR-Cas9 components. The study demonstrated that these microparticles achieve a higher transfection efficiency than commercial liposomal transfection reagents. Furthermore, the material can be broken down and metabolized after transfection, thus reducing potential toxic side effects and the risk of long-term accumulation. Polymer and hybrid microparticles made from degradable polymers, such as polypeptides and polysaccharides, and modified with a silicon shell, offer targeting, degradability, and protective properties, presenting a potential prospect for personalized therapy in TNBC. Polymer and hybrid microparticles made from degradable polymers, such as polypeptides and polysaccharides, and modified with a silicon shell, offer targeting, degradability, and protective properties, presenting a potential prospect for personalized therapy in TNBC.
Ji Li et al. developed multicomponent nanoparticles (SK/siTGF-β NP) comprising siTGF-β, shikonin (SK), folic acid (FA)-conjugated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-polyethylene glycol (DSPE-PEG-FA), and polyethyleneimine-polycaprolactone (PEI-PCL). These nanoparticles exhibited a uniform structure and good stability. The conjugated FA enhanced the nanoparticles’ uptake by tumor cells, while SK and siTGF-β achieved lysosomal escape through PEI protonation. SK can induce immunogenic cell death (ICD) in tumor cells, promote dendritic cell (DC) maturation, and enhance cytotoxic T-lymphocyte (CTL) tumor infiltration. Successful TGF-β silencing can mitigate the TGF-β-mediated immunosuppressive tumor microenvironment (ITM) and inhibit EMT, thereby facilitating CTL infiltration, suppressing regulatory T-lymphocyte (Treg) proliferation, and inhibiting lung metastasis. SK/siTGF-β nanoparticles (NPs), which are designed to enhance immune response and counteract the tumor microenvironment, could become a potential treatment for TNBC.
Yumi Sasayama et al. designed an innovative lipid nanoparticle formulation (LNPK15) to deliver siRNA for targeted gene inhibition in tumors. LNPK15 is highly PEGylated, containing 3.3% PEG-DSPE, a characteristic that contributes to its prolonged circulation in vivo. The research results indicate that LNPK15 acquires increased knockdown activity after undergoing PEG-DSPE hydrolysis in vivo and can achieve extended systemic circulation. LNPK15/KRAS demonstrated a strong antitumor efficacy in MIA PaCa-2 tumor xenograft mice following intravenous administration. The study also noted that KRAS mRNA and protein levels were significantly reduced in tumor tissues, indicating on-target antitumor efficacy. Using LNPK15 to deliver siRNA for targeting key genes in TNBC cells could offer a novel therapeutic approach.
Polymer complexes coated with PEG-functionalized lipids remain stable in biological fluids and have a prolonged circulation time, which facilitates their accumulation in tumor tissues. Vrinda Gote et al. designed a PEGylated liposomal system encapsulating Lipocalin 2 (Lcn2) small interfering RNA (Lcn2 siRNA) to selectively target metastatic breast cancer (MBC) cell line MCF-7 and cell line MDA-MB-231. The PEGylated liposomes were further modified with octreotide (OCT), a peptide that can bind to growth hormone receptors overexpressed on breast cancer cells. The experimental results indicated that this PEGylated liposomal system demonstrated superior cellular uptake both in vivo and in vitro, achieving approximately 55–60% silencing of Lcn2 mRNA. Furthermore, OCT-Lcn2-Lipo also inhibited tumor angiogenesis by reducing VEGF-A levels and decreasing the migration of endothelial cells. This approach shows promise for inhibiting angiogenesis in metastatic breast cancer.
To suppress BIRC6 expression in TNBC cells, Yongpeng Li et al. developed a PEGylated cationic lipid nanoparticle (pCLNs) designed for encapsulating BIRC6-siRNA. The study results showed that this carrier demonstrated high cellular uptake efficiency and could escape lysosomes in the cytoplasm, effectively silencing genes. After in vivo injection, pCLN/Cy3-siRNA was effectively delivered to the tumor site and exhibited good circulatory stability. Further studies indicated that pCLN/siBIRC6 complexes were more effective in inhibiting TNBC cell growth both in vitro and in vivo, compared to epidermal growth factor receptor (EGFR) inhibitors, offering a novel therapeutic strategy for treating TNBC (Figure D).
Pramod Kumar et al. recently developed a mesoporous silica-based multifunctional nanocarrier (MSNPs) that was sequentially modified with poly-l-arginine (PLR) and polyethylene glycol (PEG) for the targeted codelivery of doxorubicin (DOX), BCl-xL, and BCL-2 siRNA to drug-resistant TNBC. This nanocarrier not only protects siRNA in serum for up to 24 h, but also demonstrates good biocompatibility. The study results suggest that this material has significant potential for the targeted codelivery of drugs and siRNA to overcome drug resistance in TNBC cells.
Chi Zhang et al. developed GALA- and CREKA-modified PEG-SS-PEI for delivering small interfering RNAs (siRNAs) targeting EGFR and BRD4 for TNBC therapy. The study results showed that these modified PEG-SS-PEI/siRNA complexes exhibited excellent transfection efficiency, with redox-sensitive release characteristics and good biocompatibility. These complexes effectively protected siRNA from degradation by RNases. Additionally, they significantly inhibited the proliferation, invasion, and migration of MDA-MB-231 cells by synergistic inhibition of the EGFR/PI3K/Akt and BRD4/c-Myc pathways (Figure B).
Armin Mahmoud Salehi Khesht et al. designed a novel nanocarrier composed of chitosan lactate nanoparticles (NPs) functionalized with the transactivator of transcription (TAT) peptide and hyaluronate (HA) for delivering CD73 siRNA and doxorubicin to 4T1 and CT26 cancer cells. The study results indicated that the simultaneous transfer of CD73 siRNA and doxorubicin to 4T1 and CT26 cancer cells increased cancer cell death and inhibited the proliferation and spread of cancer cells. Additionally, the preferential aggregation of the NPs in the tumor microenvironment reduced tumor growth, promoted the survival of tumor-bearing mice, and induced antitumor immune responses. These findings suggest that CL-TAT-HA NPs are a promising candidate for targeted siRNA/drug delivery to cancer cells, and the simultaneous transfer of CD73 siRNA and doxorubicin using this nanocarrier may serve as a potential treatment for cancer.
Min Woo Kim et al. proposed a novel approach for treating TNBC using aptamer-coupled lipid nanocarriers encapsulating quantum dots (QDs) and siRNAs. These carriers incorporated hydrophobic quantum dots into lipid bilayers and then complexed with Bcl-2 and PKC-ι siRNAs to form quantum dot-lipid nanocarriers (QLs). Finally, anti-EGFR aptamer-lipid conjugates were inserted into the QLs to enable targeting for TNBC therapy (aptamo-QLs). After targeting tumor cells, these nanocarriers promoted siRNA escape from endosomal vesicles, releasing into the cytoplasm, allowing them to interfere with target genes. Additionally, they could protect siRNA and quantum dots from physical and chemical degradation in the intracellular environment. During blood circulation, these carriers showed sufficient stability and load efficiency, preventing exposure during blood circulation. Experiments demonstrated that these targeted carriers were more effective in inhibiting tumor growth compared to nontargeted nanocarriers, with even stronger suppression through combined interference with two genes.
Tian-Yan Han et al. developed a novel nonviral vector, PEI-SPDP-Man (PSM), which can simultaneously target cellular uptake pathways and intracellular responsive release for miR-34a. PSM is synthesized by connecting mannitol (Man) to branched polyethyleneimine (PEI) using a GSH-sensitive disulfide bond. The prepared PSM/miR-34a gene delivery system can induce and enter tumor cells through caveolae-mediated endocytosis, thereby reducing the degradation of miR-34a in lysosomes. Upon detecting high concentrations of glutathione (GSH) in tumor cells, PSM responds by breaking the disulfide bond to release miR-34a. This miRNA effectively inhibits and disrupts the translation of Bcl-2 and CD44 mRNA, reducing tumor cell proliferation and invasion, and demonstrating effective antitumor and antimetastatic characteristics in the treatment of orthotopic TNBC. This strategy of controlling intracellular transport pathways through targeted cellular uptake could represent a promising approach to highly effective cancer therapy.
Zhifeng Li et al. developed MUC1-C shRNA@Fe3O4 magnetic nanoparticles (MNPs) with magnetic hyperthermia and gene therapy capabilities. These nanoparticles could be internalized by TNBC cells and exhibited excellent magnetic hyperthermia efficiency. In alternating magnetic fields (AMF), they could rapidly heat up, exposing cancer cells to supraphysiological temperatures, which directly led to cell death while also modulating tumor immunity, reducing tumor heterogeneity, and increasing sensitivity to radiation and chemotherapy. Additionally, these nanoparticles could specifically silence the MUC1-C gene, significantly inhibiting the proliferation, migration, and invasion of TNBC tumor cells. Gene therapy and magnetic hyperthermia demonstrated a synergistic effect, offering a novel approach for TNBC therapy.
Ka Ming Wong et al. developed and constructed a dual plasmid (pHR-pCas9). The pCas9 plasmid is designed to induce double-strand breaks (DSB) at the 19q13.3-qter region on chromosome 19, while pHR allows for the insertion of the CTCF gene at the same location through homologous recombination. Subsequently, the dual plasmid (pHR-pCas9) was encapsulated with poly(acrylic acid) (PAA) and coated with polyglycolic acid (PGA), forming a targeted core–shell nanoparticle (NP) carrying the pHR-pCas9 system. This targeted core–shell NP facilitates precise insertion and stable expression of CTCF, thus inhibiting breast cancer cell migration, metastasis, and colonization.
Huan Deng et al. designed a biodegradable cationic polymer, poly(beta-amino ester)s (PBAEs), as a vector, utilizing the CRISPR-Cas9 system to target the Cyclin-dependent kinase 5 (Cdk5) gene for efficient gene knockout, aiming at the PD-1/PD-L1 pathway. This approach not only significantly inhibited tumor growth but also enhanced the immune response of CD8+ T cells, while reducing the number of regulatory T cells. The downregulation of PD-L1 can help transform the immunosuppressive tumor microenvironment into an immune-supportive one, making tumors more responsive to immunotherapy. Notably, reediting the endogenous Cdk5 gene can downregulate both the membrane-bound and cytosolic proteins of PD-L1, unlike antibody-based therapies, which neutralize only membrane proteins. This potentially reduces immune-related adverse effects. Overall, this combined approach of immune checkpoint genetic intervention and nanotechnology provides a powerful strategy for preclinical TNBC treatment.
Composite polymer materials play a crucial role in the field of gene editing, offering significant advantages for its application. By utilizing composite polymer materials as carriers, gene editing tools can be effectively encapsulated and delivered to target cells, thereby enhancing editing efficiency and precision. These materials offer tunability and multifunctionality, allowing for the adjustment of their structure and properties as needed to enable targeted gene editing. Additionally, composite polymer materials have good biocompatibility and stability, which helps to reduce the toxicity of gene editing tools and improve their bioavailability in vivo. Overall, composite polymer materials provide a reliable platform for the application of gene editing technology, with the potential to promote the widespread use of gene editing in medicine, offering new possibilities for disease treatment and personalized medicine.
3.3. Safety and Risk Mitigation Strategies of Gene Editing in TNBC
With the synergistic advancement of targeted delivery materials and gene editing technologies, the CRISPR/Cas9 system has shown immense potential in the treatment of TNBC. By optimizing the delivery efficiency of viral vectors (such as AAV and lentivirus) and nonviral vectors (such as lipid nanoparticles (LNP) and polymer nanoparticles), gene editing tools can more precisely target tumor tissues. However, gene editing technologies still face several safety challenges in clinical applications, particularly with regard to off-target effects, immune responses, genomic stability, and ethical concerns. To address these issues, researchers have developed systematic solutions, including high-fidelity Cas9 variants, optimized delivery systems, and strict ethical frameworks.
3.3.1. Off-Target Effects and Optimization Strategies
Off-target effects in gene editing are one of the major safety concerns associated with CRISPR/Cas9 technology. Off-target effects refer to the cleavage of genomic regions outside the intended target site by the Cas9 protein, which may lead to unintended genetic mutations that can adversely affect genomic stability and cellular function. Studies have shown that chromatin structure and epigenetic factors may influence off-target activity.
To reduce off-target effects, researchers have developed various high-fidelity Cas9 variants: 1) The first-generation high-fidelity variants (such as eSpCas9 and SpCas9-HF1) reduce off-target binding by introducing key mutations (e.g., K848A, N863A), which decreases off-target editing efficiency to less than 1/10 of the wild-type. , However, some variants exhibit reduced editing efficiency. For instance, eSpCas9 and LZ3 Cas9 show decreased homology-directed repair (HDR) efficiency in cell cycle-dependent editing, whereas SpCas9-HF1 balances efficiency and specificity in certain contexts. , 2) The second-generation optimized variants (such as SuperFi-Cas9 and Sniper2L) use structure-guided mutations (e.g., D147Y, P411T) to enhance both targeting precision and editing efficiency, with some variants even overcoming the trade-off between efficiency and specificity.
CRISPR/Cas9 technology mediates gene editing by inducing double-strand breaks (DSBs), but this repair process relies on the host cell’s DNA repair mechanisms, such as nonhomologous end joining (NHEJ) and homologous directed repair (HDR). Although these repair mechanisms can facilitate break repair, they may also induce chromosomal translocations, large-scale genomic deletions, or insertion mutations during the process. This remains a significant challenge in gene editing.
For example, yeast model studies have shown that approximately 20% of repair events following Cas9 cleavage involve genomic rearrangements. The combined use of HDR-enhancing strategies, such as inhibiting the key NHEJ factor DNA polymerase θ, can improve template-mediated precise insertion efficiency to 80% while reducing off-target mutations. Additionally, base editors, such as EvCas9, which integrate cytidine/adenosine deaminases, combine high fidelity with base-editing capabilities, thereby expanding the range of applications. Fusion of an efficiency-enhancing domain: Fusing the HMG-D domain at the N-terminus of SpCas9 to form eeCas9 results in a 1.4-fold increase in editing efficiency, particularly for non-NGG PAM variants and high-fidelity Cas9. The application of nuclear magnetic resonance (NMR) has revealed the interaction between Rec3 and RNA:DNA hybrids, and how it influences the catalytic active site of Cas9 by signal transduction. In this way, NMR provides deeper insights into understanding Cas9 specificity, aiding in the reduction of off-target effects and optimization of gene editing techniques. Computational prediction tools, such as CRISPRoff, can assess potential off-target sites in advance, further reducing risk. Improvements in delivery systems, such as the use of amphipathic ion polymers like ZEBRA for stable encapsulation of Cas9 ribonucleoproteins (RNPs), can significantly increase editing efficiency by 1.4-fold, especially for non-NGG PAM variants and high-fidelity Cas9. Optimization of delivery systems also includes LNP encapsulation of sgRNA/Cas9 complexes to reduce nonspecific delivery, while self-assembling polymer nanoparticles as carriers enhance genomic stability and reduce the risk of genomic rearrangements.
3.3.2. Immune Response Management
Gene editing tools often rely on viral vectors, such as lentiviruses and adenoviruses. Although the use of these vectors enhances editing efficiency, the associated risks of immune responses and insertional mutations remain significant challenges in clinical applications. For example, adenoviral vectors may activate innate immune responses (such as the TLR pathway) and induce the production of anti-Cas9 antibodies, potentially leading to adaptive immune rejection with prolonged use. Approximately 30% of the population has preexisting antibodies against adeno-associated viruses (AAVs), leading to vector clearance and treatment failure. Capsid engineering can reduce serum recognition, for instance, the AAV-DJ serotype, through directed evolution, selects capsid variants with low cross-reactivity, reducing preexisting antibody recognition by 40%. Recent studies have also developed nonviral vectors, such as double-stranded closed linear DNA (CELiD), which completely circumvent the issue of preexisting antibodies through structural optimization.
3.3.3. Ethical Considerations and Governance Framework
Ethical issues also pose a significant barrier to the clinical application of gene editing technologies. Although somatic gene editing does not directly affect germline genes, the risks associated with off-target effects and unpredictable genomic alterations still pose ethical concerns. Studies have shown that even with improved high-fidelity Cas9 variants, there remains an off-target mutation rate of approximately 1%, and single nucleotide variations can activate oncogenes or inactivate tumor suppressor genes This unpredictability can lead to treatments potentially causing secondary tumors or acquired genetic diseases.
3.3.4. Ethical Decisions and Gene Editing
The boundary between germline and somatic editing is increasingly becoming blurred in ethical decision-making. For example, animal model studies have shown that AAV vectors present a genomic editing risk of 3.7% outside the liver, suggesting that somatic editing vectors may induce transgenerational effects through germ cell homing mechanisms. The 2018 ″gene-edited babies″ incident exacerbated this issue, leading to strengthened global regulations, particularly regarding gene editing involving embryonic stem cells or gonadal tissue. Currently, there is considerable variation in the regulation of gene editing technologies across countries. For example, the US FDA has approved ZFN technology for the treatment of β-thalassemia, while the European EMA requires all gene editing therapies to undergo at least five years of reproductive toxicity tracking in preclinical stages. Furthermore, although CRISPR-mediated therapies such as photodynamic therapy (PDT) can effectively alleviate melanoma, accompanying genomic structural variations may lead to acquired resistance to treatment. More controversially, gene drive technologies, although capable of forcibly spreading anticancer genes, may disrupt genetic balance within ecosystems and threaten species diversity. Therefore, the Ethics Committee of the American Society of Clinical Oncology (ASCO) proposed the Triple Validation Principle, requiring any cancer gene editing therapy to undergo preclinical validation in primate models, patient-specific organoid models, and microhuman trials.
4. Conclusion
Based on the above examples, it can be inferred that using targeted delivery materials to encapsulate gene editing tools for TNBC offers many advantages. First, by efficiently delivering gene editing tools directly to tumor cells, the local concentration can be significantly increased, thus enhancing therapeutic effects while reducing systemic side effects. Second, targeted delivery systems can lower immunogenicity, avoiding activation of the immune system and reducing immune reactions, especially when compared to conventional delivery systems such as viral vectors that may trigger strong immune responses. Additionally, encapsulating materials can overcome biological barriers, protecting gene editing complexes from degradation by blood enzymes and facilitating their passage through biological barriers, like the dense extracellular matrix in the tumor microenvironment. Smart release mechanisms can respond to changes in the tumor microenvironment, such as pH or peroxide concentrations, to release the gene editing tools upon reaching the tumor site. Furthermore, targeted delivery improves editing precision and reduces off-target effects by minimizing the impact on nontarget cells, thereby increasing editing specificity.
The significance of using targeted delivery materials to encapsulate gene editing tools lies in its combination of high-precision gene editing technology with smart drug delivery systems, providing an unprecedented treatment strategy for cancer and other diseases. Despite the ongoing development and clinical testing of this method, it has opened new avenues in medical treatment with the potential to reverse or even cure previously incurable conditions.
However, current targeted gene editing technology has certain issues that need addressing. These include limited delivery efficiency and specificity, insufficient safety, limited editing efficiency and precision, and constraints with certain cell types. To further improve targeted gene editing, several areas require focus. First, more efficient, specific, and safe delivery systems need to be developed to increase delivery efficiency and specificity while reducing potential side effects. Second, improving the gene editing tools is crucial; by enhancing the CRISPR-Cas9 system or developing new gene editing tools, editing efficiency and precision can be increased while reducing nonspecific edits. Additionally, research should explore and develop targeted gene editing technologies suitable for different cell types to meet various cellular needs. Finally, fostering close collaboration between targeted gene editing technology and clinical practice will facilitate its application in clinical settings, allowing patients to benefit from these advances.
Future ethical governance should integrate artificial intelligence (AI) technologies and global collaboration to establish a more refined off-target prediction system and a unified ethical review framework, ensuring that the application of gene editing technologies in cancer treatment adheres to ethical standards. AI-assisted off-target prediction systems hold the potential to improve the accuracy of off-target risk prediction by integrating machine learning with life cycle assessment (LCA), thereby enhancing the safety of clinical treatments.
Through the synergistic development of technological innovation and ethical governance, the application of gene editing technologies in cancer treatment can achieve more precise and effective therapies while ensuring patient safety. Although gene editing technologies have demonstrated significant potential in the treatment of TNBC, safety concerns still need to be adequately addressed. To this end, the use of high-fidelity enzymes, nonviral vectors, and optimized genome stability management measures will be critical in ensuring the safety and efficacy of gene editing technologies. Furthermore, cross-national ethical review and a stringent regulatory framework on a global scale will provide the necessary safeguards for the widespread application of gene editing technologies. The establishment of rigorous risk assessment and management standards, along with strengthened ethical governance in clinical applications, will further ensure that gene editing technologies can be safely and controllably applied in cancer treatment.
Acknowledgments
This work was supported by the Outstanding Youth Science Fund Project of the Sichuan Natural Science Foundation (24NSFJQ0271), the Research Project of the Affiliated Hospital of North Sichuan Medical College (2023LC006, 2023ptzk022), and the Natural Science Foundation of Sichuan Province (23NSFSC1540, 23NSFSC1541).
△.
P.Q., X.L., and J.L. contributed equally.
The authors declare no competing financial interest.
References
- Sung H., Ferlay J., Siegel R. L., Laversanne M., Soerjomataram I., Jemal A., Bray F.. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Ca-Cancer J. Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Garrido-Castro A. C., Lin N. U., Polyak K.. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discovery. 2019;9(2):176–198. doi: 10.1158/2159-8290.CD-18-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zhang H., Merkher Y., Chen L., Liu N., Leonov S., Chen Y.. Recent advances in therapeutic strategies for triple-negative breast cancer. J. Hematol. Oncol. 2022;15(1):121. doi: 10.1186/s13045-022-01341-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchini G., Balko J. M., Mayer I. A., Sanders M. E., Gianni L.. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016;13(11):674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens K. N., Vachon C. M., Couch F. J.. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013;73(7):2025–2030. doi: 10.1158/0008-5472.CAN-12-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan M. E., Chiu J. W., Koller B. H., Jasin M.. Brca1 controls homology-directed DNA repair. Mol. Cell. 1999;4(4):511–518. doi: 10.1016/S1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S., Alexandrov L. B., Wedge D. C., Van Loo P., Greenman C. D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L. A.. et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149(5):979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Strasser A., Kelly G. L.. Should mutant TP53 be targeted for cancer therapy? Cell Death Differ. 2022;29(5):911–920. doi: 10.1038/s41418-022-00962-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derakhshan F., Reis-Filho J. S.. Pathogenesis of Triple-Negative Breast Cancer. Annu. Rev. Pathol. 2022;17:181–204. doi: 10.1146/annurev-pathol-042420-093238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Jin J., Ji W., Guan X.. Therapeutic landscape in mutational triple negative breast cancer. Mol. Cancer. 2018;17(1):99. doi: 10.1186/s12943-018-0850-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdeen S. K., Aqeilan R. I.. Decoding the link between WWOX and p53 in aggressive breast cancer. Cell Cycle. 2019;18(11):1177–1186. doi: 10.1080/15384101.2019.1616998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y. S., Tseng H. Y., Chen Y. A., Shen P. C., Al Haq A. T., Chen L. M., Tung Y. C., Hsu H. L.. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol. Cancer. 2019;18(1):42. doi: 10.1186/s12943-019-0988-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Haq A. T., Tseng H. Y., Chen L. M., Wang C. C., Hsu H. L.. Targeting prooxidant MnSOD effect inhibits triple-negative breast cancer (TNBC) progression and M2 macrophage functions under the oncogenic stress. Cell Death Dis. 2022;13(1):49. doi: 10.1038/s41419-021-04486-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Ye K., Li X., Liu X., Peng M., Chen F., Xiong W., Wang Y., Zhu L.. YTHDC1 is downregulated by the YY1/HDAC2 complex and controls the sensitivity of ccRCC to sunitinib by targeting the ANXA1-MAPK pathway. J. Exp. Clin. Cancer Res. 2022;41(1):250. doi: 10.1186/s13046-022-02460-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B., Zhou K., Liu W., Prince E., Qing Y., Li Y., Han L., Qin X., Su R., Pokharel S. P.. et al. RNA N(6) -methyladenosine reader YTHDC1 is essential for TGF-beta-mediated metastasis of triple negative breast cancer. Theranostics. 2022;12(13):5727–5743. doi: 10.7150/thno.71872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno Y., Derynck R.. Epithelial plasticity, epithelial-mesenchymal transition, and the TGF-β family. Dev. Cell. 2021;56(6):726–746. doi: 10.1016/j.devcel.2021.02.028. [DOI] [PubMed] [Google Scholar]; Mousset A., Lecorgne E., Bourget I., Lopez P., Jenovai K., Cherfils-Vicini J., Dominici C., Rios G., Girard-Riboulleau C., Liu B.. et al. Neutrophil extracellular traps formed during chemotherapy confer treatment resistance via TGF-β activation. Cancer Cell. 2023;41(4):757–775.e10. doi: 10.1016/j.ccell.2023.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Adewunmi O., Shen Y., Zhang X. H., Rosen J. M.. Targeted Inhibition of lncRNA Malat1 Alters the Tumor Immune Microenvironment in Preclinical Syngeneic Mouse Models of Triple-Negative Breast Cancer. Cancer Immunol Res. 2023;11(11):1462–1479. doi: 10.1158/2326-6066.CIR-23-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zuo Y., Li Y., Zhou Z., Ma M., Fu K.. Long non-coding RNA MALAT1 promotes proliferation and invasion via targeting miR-129–5p in triple-negative breast cancer. Biomed. Pharmacother. 2017;95(95):922–928. doi: 10.1016/j.biopha.2017.09.005. [DOI] [PubMed] [Google Scholar]; c Huang X. J., Xia Y., He G. F., Zheng L. L., Cai Y. P., Yin Y., Wu Q.. MALAT1 promotes angiogenesis of breast cancer. Oncol. Rep. 2018;40(5):2683–2689. doi: 10.3892/or.2018.6705. [DOI] [PubMed] [Google Scholar]
- Shaath H., Vishnubalaji R., Elango R., Khattak S., Alajez N. M.. Single-cell long noncoding RNA (lncRNA) transcriptome implicates MALAT1 in triple-negative breast cancer (TNBC) resistance to neoadjuvant chemotherapy. Cell Death Discovery. 2021;7(1):23. doi: 10.1038/s41420-020-00383-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhar M., Ebert A., Neumann T., Umkehrer C., Jude J., Wieshofer C., Rescheneder P., Lipp J. J., Herzog V. A., Reichholf B.. et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science. 2018;360(6390):800–805. doi: 10.1126/science.aao2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. Y., Lee C. F., Lai H. T., Yu C. T., Lee J. E., Zuo H., Tsai S. Y., Tsai M. J., Ge K., Wan Y.. et al. Opposing Functions of BRD4 Isoforms in Breast Cancer. Mol. Cell. 2020;78(6):1114–1132.e10. doi: 10.1016/j.molcel.2020.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Reguant L., Serra-Camprubí Q., Datta D., Cianferoni D., Kourtis S., Gañez-Zapater A., Cannatá C., Espinar L., Querol J., García-López L.. et al. Interactions between BRD4S, LOXL2, and MED1 drive cell cycle transcription in triple-negative breast cancer. EMBO Mol. Med. 2023;15(12):e18459. doi: 10.15252/emmm.202318459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. X., Agborbesong E., Li L. X., Li X.. Bromodomain Protein BRD4-Mediated Mutant p53 Transcription Promotes TNBC Progression. Int. J. Mol. Sci. 2022;23(23):15163. doi: 10.3390/ijms232315163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S., Oh C., Yoo K. H.. Functional roles of CTCF in breast cancer. BMB Rep. 2017;50(9):445–453. doi: 10.5483/BMBRep.2017.50.9.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. R., Wu Y. M., Lonigro R. J., Vats P., Cobain E., Everett J., Cao X., Rabban E., Kumar-Sinha C., Raymond V.. et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548(7667):297–303. doi: 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K. M., Song J., Wong Y. H.. CTCF and EGR1 suppress breast cancer cell migration through transcriptional control of Nm23-H1. Sci. Rep. 2021;11(1):491. doi: 10.1038/s41598-020-79869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J., Bao C., Xie Y., Guo H., Liu Y., Li J., Liu R., Li P., Bai J., Yan Y.. et al. Targeted core-shell nanoparticles for precise CTCF gene insert in treatment of metastatic breast cancer. Bioact. Mater. 2022;11:1–14. doi: 10.1016/j.bioactmat.2021.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikhil K., Shah K.. CDK5: an oncogene or an anti-oncogene: location location location. Mol. Cancer. 2023;22(1):186. doi: 10.1186/s12943-023-01895-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei Y., Cheng N., Chen T., Shu Y., Yang Y., Yang N., Zhou X., Liu B., Wei J., Liu Q.. et al. CDK5 Inhibition Abrogates TNBC Stem-Cell Property and Enhances Anti-PD-1 Therapy. Adv. Sci. 2020;7(22):2001417. doi: 10.1002/advs.202001417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q., Li L., Zhang J., Lei Y., Wang L., Liu D. X., Feng J., Hou P., Yao R., Zhang Y.. et al. CDK5 is essential for TGF-β1-induced epithelial-mesenchymal transition and breast cancer progression. Sci. Rep. 2013;3:2932. doi: 10.1038/srep02932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H., Tan S., Gao X., Zou C., Xu C., Tu K., Song Q., Fan F., Huang W., Zhang Z.. Cdk5 knocking out mediated by CRISPR-Cas9 genome editing for PD-L1 attenuation and enhanced antitumor immunity. Acta Pharm. Sin. B. 2020;10(2):358–373. doi: 10.1016/j.apsb.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorand R. D., Nthale J., Myers J. T., Barkauskas D. S., Avril S., Chirieleison S. M., Pareek T. K., Abbott D. W., Stearns D. S., Letterio J. J.. et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016;353(6297):399–403. doi: 10.1126/science.aae0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Okletey J., Angelis D., Jones T. M., Montagna C., Spiliotis E. T.. An oncogenic isoform of septin 9 promotes the formation of juxtanuclear invadopodia by reducing nuclear deformability. Cell Rep. 2023;42(8):112893. doi: 10.1016/j.celrep.2023.112893. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Connolly D., Hoang H. G., Adler E., Tazearslan C., Simmons N., Bernard V. V., Castaldi M., Oktay M. H., Montagna C.. Septin 9 amplification and isoform-specific expression in peritumoral and tumor breast tissue. Biol. Chem. 2014;395(2):157–167. doi: 10.1515/hsz-2013-0247. [DOI] [PubMed] [Google Scholar]
- Zeng Y., Cao Y., Liu L., Zhao J., Zhang T., Xiao L., Jia M., Tian Q., Yu H., Chen S.. et al. SEPT9_i1 regulates human breast cancer cell motility through cytoskeletal and RhoA/FAK signaling pathway regulation. Cell Death Dis. 2019;10(10):720. doi: 10.1038/s41419-019-1947-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang G., Wang S., Chen L., Song M., Song X., Wang H., Zhou P., Ma X., Yu J.. UBR5 targets tumor suppressor CDC73 proteolytically to promote aggressive breast cancer. Cell Death Dis. 2022;13(5):451. doi: 10.1038/s41419-022-04914-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L., Song M., Li X., Tang L., Zhang T., Zhang L., Pan Y., Chouchane L., Ma X.. E3 Ubiquitin Ligase UBR5 Drives the Growth and Metastasis of Triple-Negative Breast Cancer. Cancer Res. 2017;77(8):2090–2101. doi: 10.1158/0008-5472.CAN-16-2409. [DOI] [PubMed] [Google Scholar]
- Wu B., Song M., Dong Q., Xiang G., Li J., Ma X., Wei F.. UBR5 promotes tumor immune evasion through enhancing IFN-γ-induced PDL1 transcription in triple negative breast cancer. Theranostics. 2022;12(11):5086–5102. doi: 10.7150/thno.74989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Piwarski S. A., Thompson C., Chaudhry A. R., Denvir J., Primerano D. A., Fan J., Salisbury T. B.. The putative endogenous AHR ligand ITE reduces JAG1 and associated NOTCH1 signaling in triple negative breast cancer cells. Biochem. Pharmacol. 2020;174:113845. doi: 10.1016/j.bcp.2020.113845. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang K., Zhang Q., Li D., Ching K., Zhang C., Zheng X., Ozeck M., Shi S., Li X., Wang H.. et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a γ-secretase inhibitor. Clin. Cancer Res. 2015;21(6):1487–1496. doi: 10.1158/1078-0432.CCR-14-1348. [DOI] [PubMed] [Google Scholar]
- Miao K., Lei J. H., Valecha M. V., Zhang A., Xu J., Wang L., Lyu X., Chen S., Miao Z., Zhang X.. et al. NOTCH1 activation compensates BRCA1 deficiency and promotes triple-negative breast cancer formation. Nat. Commun. 2020;11(1):3256. doi: 10.1038/s41467-020-16936-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cravero K., Pantone M. V., Shin D. H., Bergman R., Cochran R., Chu D., Zabransky D. J., Karthikeyan S., Waters I. G., Hunter N.. et al. NOTCH1 PEST domain variants are responsive to standard of care treatments despite distinct transformative properties in a breast cancer model. Oncotarget. 2022;13:373–386. doi: 10.18632/oncotarget.28200. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Qi Y., Li M., Li S., Zeng D., Xiao Y., Li J., Ye Q., Bremer E., Zhang G. J.. Notch1 promotes resistance to cisplatin by up-regulating Ecto-5′-nucleotidase (CD73) in triple-negative breast cancer cells. Cell Death Dis. 2023;9(1):204. doi: 10.1038/s41420-023-01487-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong P., Chen L., Yu M., Tao J., Liu J., Wang Y., Pan H., Zhou W., Wang S.. miR-3178 inhibits cell proliferation and metastasis by targeting Notch1 in triple-negative breast cancer. Cell Death Dis. 2018;9(11):1059. doi: 10.1038/s41419-018-1091-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lin J., Xu Z., Xie J., Deng X., Jiang L., Chen H., Peng C., Li H., Zhang J., Shen B.. Oncogene APOL1 promotes proliferation and inhibits apoptosis via activating NOTCH1 signaling pathway in pancreatic cancer. Cell Death Dis. 2021;12(8):760. doi: 10.1038/s41419-021-03985-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hassan W. A., Yoshida R., Kudoh S., Hasegawa K., Niimori-Kita K., Ito T.. Notch1 controls cell invasion and metastasis in small cell lung carcinoma cell lines. Lung Cancer. 2014;86(3):304–310. doi: 10.1016/j.lungcan.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Qiao L., Dong C., Jia W., Ma B.. NAA20 recruits Rin2 and promotes triple-negative breast cancer progression by regulating Rab5A-mediated activation of EGFR signaling. Cell. Signalling. 2023;112:110922. doi: 10.1016/j.cellsig.2023.110922. [DOI] [PubMed] [Google Scholar]
- Shu L., Chen A., Li L., Yao L., He Y., Xu J., Gu W., Li Q., Wang K., Zhang T.. et al. NRG1 regulates Fra-1 transcription and metastasis of triple-negative breast cancer cells via the c-Myc ubiquitination as manipulated by ERK1/2-mediated Fbxw7 phosphorylation. Oncogene. 2022;41(6):907–919. doi: 10.1038/s41388-021-02142-4. [DOI] [PubMed] [Google Scholar]
- Casalino L., Talotta F., Matino I., Verde P.. FRA-1 as a Regulator of EMT and Metastasis in Breast Cancer. Int. J. Mol. Sci. 2023;24(9):8307. doi: 10.3390/ijms24098307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Huang H., Xi Z., Ma S., Ming J., Dong F., Guo H., Zhang H., Zhao E., Yao G.. et al. LncRNA SEMA3B-AS1 inhibits breast cancer progression by targeting miR-3940/KLLN axis. Cell Death Dis. 2022;13(9):800. doi: 10.1038/s41419-022-05189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing X., Liang H., Hao C., Yang X., Cui X.. Overexpression of MUC1 predicts poor prognosis in patients with breast cancer. Oncol. Rep. 2018;41(2):801–810. doi: 10.3892/or.2018.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Guo T., Zhao S., Lin M.. The Therapeutic Effects of MUC1-C shRNA@Fe(3)O(4) Magnetic Nanoparticles in Alternating Magnetic Fields on Triple-Negative Breast Cancer. Int. J. Nanomed. 2023;18:5651–5670. doi: 10.2147/IJN.S426849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N., Long M., Fushimi A., Yamamoto M., Hata T., Hagiwara M., Bhattacharya A., Hu Q., Wong K. K., Liu S.. et al. MUC1-C integrates activation of the IFN-γ pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. J. Immunother. Cancer. 2021;9(1):e002115. doi: 10.1136/jitc-2020-002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. Z., Liu Y., Xiao Y., Hu X., Jiang L., Zuo W. J., Ma D., Ding J., Zhu X., Zou J.. et al. Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic triple-negative breast cancer: the FUTURE trial. Cell Res. 2021;31(2):178–186. doi: 10.1038/s41422-020-0375-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual J., Turner N. C.. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019;30(7):1051–1060. doi: 10.1093/annonc/mdz133. [DOI] [PubMed] [Google Scholar]
- Dai M., Yan G., Wang N., Daliah G., Edick A. M., Poulet S., Boudreault J., Ali S., Burgos S. A., Lebrun J. J.. In vivo genome-wide CRISPR screen reveals breast cancer vulnerabilities and synergistic mTOR/Hippo targeted combination therapy. Nat. Commun. 2021;12(1):3055. doi: 10.1038/s41467-021-23316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Park J. M., Park S., Jung E., Ko D., Park M., Seo J., Nam K. D., Kang Y. K., Lee K.. et al. Suppression of TNBC metastasis by doxazosin, a novel dual inhibitor of c-MET/EGFR. J. Exp. Clin. Cancer Res. 2023;42(1):292. doi: 10.1186/s13046-023-02866-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicek E., Circir A., Oyken M., Akbulut Caliskan O., Dioken D. N., Guntekin Ergun S., Cetin-Atalay R., Sapmaz A., Ovaa H., Sahin O.. et al. EGF-SNX3-EGFR axis drives tumor progression and metastasis in triple-negative breast cancers. Oncogene. 2022;41(2):220–232. doi: 10.1038/s41388-021-02086-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi J., Wu Z., Zhang X., Zeng T., Dai W., Qiu N., Xu M., Qiao Y., Ke L., Zhao J.. et al. TMEM25 inhibits monomeric EGFR-mediated STAT3 activation in basal state to suppress triple-negative breast cancer progression. Nat. Commun. 2023;14(1):2342. doi: 10.1038/s41467-023-38115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hassink G. C., Zhao B., Sompallae R., Altun M., Gastaldello S., Zinin N. V., Masucci M. G., Lindsten K.. The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 2009;10(7):755–761. doi: 10.1038/embor.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang X., Chen X., Qian F., Zhu Y., He G., Yang J., Wu X., Zhang H., Yu X., Liu X.. Deubiquitinase USP19 modulates apoptotic calcium release and endoplasmic reticulum stress by deubiquitinating BAG6 in triple negative breast cancer. Clin. Transl. Med. 2023;13(9):e1398. doi: 10.1002/ctm2.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Tan Y., Wen L., Xing Z., Wang C., Zhang L., Wu K., Sun H., Li Y., Lei Q.. et al. Overexpression of BIRC6 driven by EGF-JNK-HECTD1 signaling is a potential therapeutic target for triple-negative breast cancer. Mol. Ther. Nucleic Acids. 2021;26:798–812. doi: 10.1016/j.omtn.2021.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einstein J. M., Perelis M., Chaim I. A., Meena J. K., Nussbacher J. K., Tankka A. T., Yee B. A., Li H., Madrigal A. A., Neill N. J.. et al. Inhibition of YTHDF2 triggers proteotoxic cell death in MYC-driven breast cancer. Mol. Cell. 2021;81(15):3048–3064.e9. doi: 10.1016/j.molcel.2021.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrutsky M. I., Troy C. M.. Caspase-9: A Multimodal Therapeutic Target With Diverse Cellular Expression in Human Disease. Front. Pharmacol. 2021;12:701301. doi: 10.3389/fphar.2021.701301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Wang J., Zhang Y., Zha W., Zhang H., Dong S., Xing H., Li X.. Efficient delivery of PKN3 shRNA for the treatment of breast cancer via lipid nanoparticles. Bioorg. Med. Chem. 2022;69:116884. doi: 10.1016/j.bmc.2022.116884. [DOI] [PubMed] [Google Scholar]
- Mukai H., Muramatsu A., Mashud R., Kubouchi K., Tsujimoto S., Hongu T., Kanaho Y., Tsubaki M., Nishida S., Shioi G.. et al. PKN3 is the major regulator of angiogenesis and tumor metastasis in mice. Sci. Rep. 2016;6:18979. doi: 10.1038/srep18979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagati A., Kumar S., Jiang P., Pyrdol J., Zou A. E., Godicelj A., Mathewson N. D., Cartwright A. N. R., Cejas P., Brown M.. et al. Integrin αvβ6–TGFβ–SOX4 Pathway Drives Immune Evasion in Triple-Negative Breast Cancer. Cancer Cell. 2021;39(1):54–67.e9. doi: 10.1016/j.ccell.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Rana P. S., Markovic V., Sossey-Alaoui K.. The WAVE3/β-catenin oncogenic signaling regulates chemoresistance in triple negative breast cancer. Breast Cancer Res. 2023;25(1):31. doi: 10.1186/s13058-023-01634-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Chen T., Li C., Li W., Zhou X., Li Y., Luo D., Zhang N., Chen B., Wang L.. et al. CircRNA-CREIT inhibits stress granule assembly and overcomes doxorubicin resistance in TNBC by destabilizing PKR. J. Hematol. Oncol. 2022;15(1):122. doi: 10.1186/s13045-022-01345-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Li R., Hou N., Zhang J., Wang T., Fan P., Ji C., Zhang B., Liu L., Wang Y.. et al. PRMT5 reduces immunotherapy efficacy in triple-negative breast cancer by methylating KEAP1 and inhibiting ferroptosis. J. Immunother. Cancer. 2023;11(6):e006890. doi: 10.1136/jitc-2023-006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B. Q., Zhang S., Li S., An M. X., Li C., Yan J., Wang J. M., Wang H. Q.. BAG3 promotes stem cell-like phenotype in breast cancer by upregulation of CXCR4 via interaction with its transcript. Cell Death Dis. 2017;8(7):e2933. doi: 10.1038/cddis.2017.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B., Klein C., Hoffmann M. E., Bonn F., Dikic I., Kögel D.. BAG3 is a negative regulator of ciliogenesis in glioblastoma and triple-negative breast cancer cells. J. Cell. Biochem. 2022;123(1):77–90. doi: 10.1002/jcb.30073. [DOI] [PubMed] [Google Scholar]
- Takayama S., Xie Z., Reed J. C.. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 1999;274(2):781–786. doi: 10.1074/jbc.274.2.781. [DOI] [PubMed] [Google Scholar]
- Zimmerli D., Brambillasca C. S., Talens F., Bhin J., Linstra R., Romanens L., Bhattacharya A., Joosten S. E. P., Da Silva A. M., Padrao N.. et al. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat. Commun. 2022;13(1):6579. doi: 10.1038/s41467-022-34000-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. M., Lin C. C., Servetto A., Bae J., Kandagatla V., Ye D., Kim G., Sudhan D. R., Mendiratta S., González Ericsson P. I.. et al. Epigenetic Repression of STING by MYC Promotes Immune Evasion and Resistance to Immune Checkpoint Inhibitors in Triple-Negative Breast Cancer. Cancer Immunol Res. 2022;10(7):829–843. doi: 10.1158/2326-6066.CIR-21-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L., Liao L., Zhang Y. L., Hu S. Y., Yang S. Y., Ma X. Y., Huang M. Y., Zhang F. L., Li D. Q.. MYC-driven U2SURP regulates alternative splicing of SAT1 to promote triple-negative breast cancer progression. Cancer Lett. 2023;560:216124. doi: 10.1016/j.canlet.2023.216124. [DOI] [PubMed] [Google Scholar]
- Choi H. H., Zou S., Wu J. L., Wang H., Phan L., Li K., Zhang P., Chen D., Liu Q., Qin B.. et al. EGF Relays Signals to COP1 and Facilitates FOXO4 Degradation to Promote Tumorigenesis. Adv. Sci. 2020;7(20):2000681. doi: 10.1002/advs.202000681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Tokheim C., Gu S. S., Wang B., Tang Q., Li Y., Traugh N., Zeng Z., Zhang Y., Li Z.. et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell. 2021;184(21):5357–5374.e22. doi: 10.1016/j.cell.2021.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong P., Wang X., Gao Y. K., Zhang D. D., Huang X. F., Song Y., Zhang W. D., Guo R. J., Li H., Han M.. RGS5 maintaining vascular homeostasis is altered by the tumor microenvironment. Biol. Direct. 2023;18(1):78. doi: 10.1186/s13062-023-00437-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farheen J., Hosmane N. S., Zhao R., Zhao Q., Iqbal M. Z., Kong X.. Nanomaterial-assisted CRISPR gene-engineering - A hallmark for triple-negative breast cancer therapeutics advancement. Mater. Today Bio. 2022;16:100450. doi: 10.1016/j.mtbio.2022.100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone M. K., Smrekar K., Park S., Blakely B., Walter A., Nasta N., Park J., Considine M., Danilova L. V., Pandey N. B.. et al. Cytokines secreted by stromal cells in TNBC microenvironment as potential targets for cancer therapy. Cancer Biol. Ther. 2020;21(6):560–569. doi: 10.1080/15384047.2020.1739484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gote V., Pal D.. Octreotide-Targeted Lcn2 siRNA PEGylated Liposomes as a Treatment for Metastatic Breast Cancer. Bioengineering. 2021;8(4):44. doi: 10.3390/bioengineering8040044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T., Yang Y., Qi H., Cui W., Zhang L., Fu X., He X., Liu M., Li P. F., Yu T.. CRISPR/Cas9 therapeutics: progress and prospects. Signal Transduction Targeted Ther. 2023;8(1):36. doi: 10.1038/s41392-023-01309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway A., Mendel M., Kim K., McGovern K., Boyko A., Zhang L., Miller J. C., DeKelver R. C., Paschon D. E., Mui B. L.. et al. Non-viral Delivery of Zinc Finger Nuclease mRNA Enables Highly Efficient In Vivo Genome Editing of Multiple Therapeutic Gene Targets. Mol. Ther. 2019;27(4):866–877. doi: 10.1016/j.ymthe.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschon D. E., Lussier S., Wangzor T., Xia D. F., Li P. W., Hinkley S. J., Scarlott N. A., Lam S. C., Waite A. J., Truong L. N.. et al. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat. Commun. 2019;10(1):1133. doi: 10.1038/s41467-019-08867-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamshirgaran Y., Liu J., Sumer H., Verma P. J., Taheri-Ghahfarokhi A.. Tools for Efficient Genome Editing; ZFN, TALEN, and CRISPR. Methods Mol. Biol. 2022;2495:29–46. doi: 10.1007/978-1-0716-2301-5_2. [DOI] [PubMed] [Google Scholar]
- Ahmed M., Daoud G. H., Mohamed A., Harati R.. New Insights into the Therapeutic Applications of CRISPR/Cas9 Genome Editing in Breast Cancer. Genes. 2021;12(5):723. doi: 10.3390/genes12050723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. W., Gao C., Zheng Y. M., Yi L., Lu J. C., Huang X. Y., Cai J. B., Zhang P. F., Cui Y. H., Ke A. W.. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol. Cancer. 2022;21(1):57. doi: 10.1186/s12943-022-01518-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Shen J., Li D., Cheng Y.. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics. 2021;11(2):614–648. doi: 10.7150/thno.47007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari P. K., Ko T. H., Dubey R., Chouhan M., Tsai L. W., Singh H. N., Chaubey K. K., Dayal D., Chiang C. W., Kumar S.. CRISPR/Cas9 as a therapeutic tool for triple negative breast cancer: from bench to clinics. Front. Mol. Biosci. 2023;10:1214489. doi: 10.3389/fmolb.2023.1214489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Hu S., Chen X.. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials. 2018;171:207–218. doi: 10.1016/j.biomaterials.2018.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z., Zhang Y., Propson N. E., Howden S. E., Chu L. F., Sontheimer E. J., Thomson J. A.. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. U. S. A. 2013;110(39):15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao D. D., Vorhies J. S., Senzer N., Nemunaitis J.. siRNA vs. shRNA: similarities and differences. Adv. Drug Delivery Rev. 2009;61(9):746–759. doi: 10.1016/j.addr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Liu H., Zhu X., Wei Y., Song C., Wang Y.. Recent advances in targeted gene silencing and cancer therapy by nanoparticle-based delivery systems. Biomed. Pharmacother. 2023;157:114065. doi: 10.1016/j.biopha.2022.114065. [DOI] [PubMed] [Google Scholar]
- Ming T., Tao Q., Tang S., Zhao H., Yang H., Liu M., Ren S., Xu H.. Curcumin: An epigenetic regulator and its application in cancer. Biomed. Pharmacother. 2022;156:113956. doi: 10.1016/j.biopha.2022.113956. [DOI] [PubMed] [Google Scholar]
- Raguram A., Banskota S., Liu D. R.. Therapeutic in vivo delivery of gene editing agents. Cell. 2022;185(15):2806–2827. doi: 10.1016/j.cell.2022.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A., Witzigmann D., Thomson S. B., Chen S., Leavitt B. R., Cullis P. R., van der Meel R.. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021;16(6):630–643. doi: 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- Wang D., Tai P. W. L., Gao G.. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discovery. 2019;18(5):358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pupo A., Fernández A., Low S. H., François A., Suárez-Amarán L., Samulski R. J.. AAV vectors: The Rubik’s cube of human gene therapy. Mol. Ther. 2022;30(12):3515–3541. doi: 10.1016/j.ymthe.2022.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinn E., Vandenberghe L. H.. Adeno-associated virus: fit to serve. Curr. Opin Virol. 2014;8:90–97. doi: 10.1016/j.coviro.2014.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Samulski R. J.. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020;21(4):255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- Perez O.D., Logg C.R., Hiraoka K., Diago O., Burnett R., Inagaki A., Jolson D., Amundson K., Buckley T., Lohse D.. et al. Design and Selection of Toca 511 for Clinical Use: Modified Retroviral Replicating Vector With Improved Stability and Gene Expression. Mol. Ther. 2013;21(2):494. doi: 10.1038/mt.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor B., Bailey R. M., Wimberly K., Kalburgi S. N., Gray S. J.. Methods for gene transfer to the central nervous system. Adv. Genet. 2014;87:125–197. doi: 10.1016/B978-0-12-800149-3.00003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Le Y., Zhang Z., Nian X., Liu B., Yang X.. Viral Vector-Based Gene Therapy. Int. J. Mol. Sci. 2023;24(9):7736. doi: 10.3390/ijms24097736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo W. Y., Hwu L., Wu C. Y., Lee J. S., Chang C. W., Liu R. S.. STAT3/NF-κB-Regulated Lentiviral TK/GCV Suicide Gene Therapy for Cisplatin-Resistant Triple-Negative Breast Cancer. Theranostics. 2017;7(3):647–663. doi: 10.7150/thno.16827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccuri F., Sommariva M., Marsico S., Giordano F., Zani A., Giacomini A., Fraefel C., Balsari A., Caruso A.. Inhibition of DNA Repair Mechanisms and Induction of Apoptosis in Triple Negative Breast Cancer Cells Expressing the Human Herpesvirus 6 U94. Cancers. 2019;11(7):1006. doi: 10.3390/cancers11071006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milone M. C., O’Doherty U.. Clinical use of lentiviral vectors. Leukemia. 2018;32(7):1529–1541. doi: 10.1038/s41375-018-0106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck J., Grossen P., Cullis P. R., Huwyler J., Witzigmann D.. Lipid-Based DNA Therapeutics: Hallmarks of Non-Viral Gene Delivery. ACS Nano. 2019;13(4):3754–3782. doi: 10.1021/acsnano.8b07858. [DOI] [PubMed] [Google Scholar]
- Piperno A., Sciortino M. T., Giusto E., Montesi M., Panseri S., Scala A.. Recent Advances and Challenges in Gene Delivery Mediated by Polyester-Based Nanoparticles. Int. J. Nanomed. 2021;16:5981–6002. doi: 10.2147/IJN.S321329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha E. A., Lee J., Hotta A.. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J. Controlled Release. 2022;342:345–361. doi: 10.1016/j.jconrel.2022.01.013. [DOI] [PubMed] [Google Scholar]
- Hong K., Zheng W., Baker A., Papahadjopoulos D.. Stabilization of cationic liposome-plasmid DNA complexes by polyamines and poly(ethylene glycol)-phospholipid conjugates for efficient in vivo gene delivery. FEBS Lett. 1997;400(2):233–237. doi: 10.1016/S0014-5793(96)01397-X. [DOI] [PubMed] [Google Scholar]
- Zhang D., Wang G., Yu X., Wei T., Farbiak L., Johnson L. T., Taylor A. M., Xu J., Hong Y., Zhu H.. et al. Enhancing CRISPR/Cas gene editing through modulating cellular mechanical properties for cancer therapy. Nat. Nanotechno.l. 2022;17(7):777–787. doi: 10.1038/s41565-022-01122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briolay T., Petithomme T., Fouet M., Nguyen-Pham N., Blanquart C., Boisgerault N.. Delivery of cancer therapies by synthetic and bio-inspired nanovectors. Mol. Cancer. 2021;20(1):55. doi: 10.1186/s12943-021-01346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijmans S. A. A., Schiffelers R. M., Zarovni N., Vago R.. Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharmacol. Res. 2016;111:487–500. doi: 10.1016/j.phrs.2016.07.006. [DOI] [PubMed] [Google Scholar]
- Kim S. M., Yang Y., Oh S. J., Hong Y., Seo M., Jang M.. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J. Controlled Release. 2017;266:8–16. doi: 10.1016/j.jconrel.2017.09.013. [DOI] [PubMed] [Google Scholar]
- Lin Y., Wagner E., Lächelt U.. Non-viral delivery of the CRISPR/Cas system: DNA versus RNA versus RNP. Biomater. Sci. 2022;10(5):1166–1192. doi: 10.1039/D1BM01658J. [DOI] [PubMed] [Google Scholar]
- Rao L., Zhao S. K., Wen C., Tian R., Lin L., Cai B., Sun Y., Kang F., Yang Z., He L.. et al. Activating Macrophage-Mediated Cancer Immunotherapy by Genetically Edited Nanoparticles. Adv. Mater. 2020;32(47):e2004853. doi: 10.1002/adma.202004853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M., Ramadan E., Elsadek N. E., Emam S. E., Shimizu T., Ando H., Ishima Y., Elgarhy O. H., Sarhan H. A., Hussein A. K.. et al. Polyethylene glycol (PEG): The nature, immunogenicity, and role in the hypersensitivity of PEGylated products. J. Controlled Release. 2022;351:215–230. doi: 10.1016/j.jconrel.2022.09.031. [DOI] [PubMed] [Google Scholar]
- Kozma G. T., Shimizu T., Ishida T., Szebeni J.. Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv. Drug Delivery Rev. 2020;154–155:163–175. doi: 10.1016/j.addr.2020.07.024. [DOI] [PubMed] [Google Scholar]
- a Wang F., Ye X., Wu Y., Wang H., Sheng C., Peng D., Chen W.. Time Interval of Two Injections and First-Dose Dependent of Accelerated Blood Clearance Phenomenon Induced by PEGylated Liposomal Gambogenic Acid: The Contribution of PEG-Specific IgM. J. Pharm. Sci. 2019;108(1):641–651. doi: 10.1016/j.xphs.2018.10.027. [DOI] [PubMed] [Google Scholar]; b Emam S. E., Elsadek N. E., Abu Lila A. S., Takata H., Kawaguchi Y., Shimizu T., Ando H., Ishima Y., Ishida T.. Anti-PEG IgM production and accelerated blood clearance phenomenon after the administration of PEGylated exosomes in mice. J. Controlled Release. 2021;334:327–334. doi: 10.1016/j.jconrel.2021.05.001. [DOI] [PubMed] [Google Scholar]
- Xu Y., Kim C. S., Saylor D. M., Koo D.. Polymer degradation and drug delivery in PLGA-based drug-polymer applications: A review of experiments and theories. J. Biomed Mater. Res. B Appl. Biomater. 2017;105(6):1692–1716. doi: 10.1002/jbm.b.33648. [DOI] [PubMed] [Google Scholar]
- Song X., Zhao Y., Wu W., Bi Y., Cai Z., Chen Q., Li Y., Hou S.. PLGA nanoparticles simultaneously loaded with vincristine sulfate and verapamil hydrochloride: systematic study of particle size and drug entrapment efficiency. Int. J. Pharm. 2008;350(1–2):320–329. doi: 10.1016/j.ijpharm.2007.08.034. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Dong Y., Fu H., Huang H., Wu Z., Zhao M., Yang X., Guo Q., Duan Y., Sun Y.. Multifunctional tumor-targeted PLGA nanoparticles delivering Pt(IV)/siBIRC5 for US/MRI imaging and overcoming ovarian cancer resistance. Biomaterials. 2021;269:120478. doi: 10.1016/j.biomaterials.2020.120478. [DOI] [PubMed] [Google Scholar]
- Hoxhaj G., Manning B. D.. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer. 2020;20(2):74–88. doi: 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins C., Sousa F., Araújo F., Sarmento B.. Functionalizing PLGA and PLGA Derivatives for Drug Delivery and Tissue Regeneration Applications. Adv. Healthcare Mater. 2018;7(1):1701035. doi: 10.1002/adhm.201701035. [DOI] [PubMed] [Google Scholar]
- Wang M., Huang H., Sun Y., Wang M., Yang Z., Shi Y., Liu L.. PEI functionalized cell membrane for tumor targeted and glutathione responsive gene delivery. Int. J. Biol. Macromol. 2024;255:128354. doi: 10.1016/j.ijbiomac.2023.128354. [DOI] [PubMed] [Google Scholar]
- Höbel S., Prinz R., Malek A., Urban-Klein B., Sitterberg J., Bakowsky U., Czubayko F., Aigner A.. Polyethylenimine PEI F25-LMW allows the long-term storage of frozen complexes as fully active reagents in siRNA-mediated gene targeting and DNA delivery. Eur. J. Pharm. Biopharm. 2008;70(1):29–41. doi: 10.1016/j.ejpb.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Gao R., Luo Q., Li Y., Song L., Cai J. S., Xiong Y., Yan F., Liu J.. Biosynthetic Nanobubble-Mediated CRISPR/Cas9 Gene Editing of Cdh2 Inhibits Breast Cancer Metastasis. Pharmaceutics. 2022;14(7):1382. doi: 10.3390/pharmaceutics14071382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Liu L., Wang J., Zhang H., Zhang Z., Xing G., Wang X., Liu M.. Drug-loaded PEG-PLGA nanoparticles for cancer treatment. Front. Pharmacol. 2022;13:990505. doi: 10.3389/fphar.2022.990505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timin A. S., Muslimov A. R., Lepik K. V., Epifanovskaya O. S., Shakirova A. I., Mock U., Riecken K., Okilova M. V., Sergeev V. S., Afanasyev B. V.. et al. Efficient gene editing via non-viral delivery of CRISPR-Cas9 system using polymeric and hybrid microcarriers. Nanomedicine. 2018;14(1):97–108. doi: 10.1016/j.nano.2017.09.001. [DOI] [PubMed] [Google Scholar]
- Li J., Zhao M., Liang W., Wu S., Wang Z., Wang D.. Codelivery of Shikonin and siTGF-β for enhanced triple negative breast cancer chemo-immunotherapy. J. Controlled Release. 2022;342:308–320. doi: 10.1016/j.jconrel.2022.01.015. [DOI] [PubMed] [Google Scholar]
- Sasayama Y., Hasegawa M., Taguchi E., Kubota K., Kuboyama T., Naoi T., Yabuuchi H., Shimai N., Asano M., Tokunaga A.. et al. In vivo activation of PEGylated long circulating lipid nanoparticle to achieve efficient siRNA delivery and target gene knock down in solid tumors. J. Controlled Release. 2019;311–312:245–256. doi: 10.1016/j.jconrel.2019.09.004. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Chu Y., Li C., Tang W., Qian J., Wei X., Lu W., Ying T., Zhan C.. Anti-PEG scFv corona ameliorates accelerated blood clearance phenomenon of PEGylated nanomedicines. J. Controlled Release. 2021;330:493–501. doi: 10.1016/j.jconrel.2020.12.047. [DOI] [PubMed] [Google Scholar]
- Kumar P., Salve R., Paknikar K. M., Gajbhiye V.. Nucleolin aptamer conjugated MSNPs-PLR-PEG multifunctional nanoconstructs for targeted co-delivery of anticancer drug and siRNA to counter drug resistance in TNBC. Int. J. Biol. Macromol. 2023;229:600–614. doi: 10.1016/j.ijbiomac.2022.12.266. [DOI] [PubMed] [Google Scholar]
- Zhang C., Yuan W., Wu Y., Wan X., Gong Y.. Co-delivery of EGFR and BRD4 siRNA by cell-penetrating peptides-modified redox-responsive complex in triple negative breast cancer cells. Life Sci. 2021;266:118886. doi: 10.1016/j.lfs.2020.118886. [DOI] [PubMed] [Google Scholar]
- Salehi Khesht A. M., Karpisheh V., Sahami Gilan P., Melnikova L. A., Olegovna Zekiy A., Mohammadi M., Hojjat-Farsangi M., Majidi Zolbanin N., Mahmoodpoor A., Hassannia H.. et al. Blockade of CD73 using siRNA loaded chitosan lactate nanoparticles functionalized with TAT-hyaluronate enhances doxorubicin mediated cytotoxicity in cancer cells both in vitro and in vivo. Int. J. Biol. Macromol. 2021;186:849–863. doi: 10.1016/j.ijbiomac.2021.07.034. [DOI] [PubMed] [Google Scholar]
- Kim M. W., Jeong H. Y., Kang S. J., Jeong I. H., Choi M. J., You Y. M., Im C. S., Song I. H., Lee T. S., Lee J. S.. et al. Anti-EGF Receptor Aptamer-Guided Co-Delivery of Anti-Cancer siRNAs and Quantum Dots for Theranostics of Triple-Negative Breast Cancer. Theranostics. 2019;9(3):837–852. doi: 10.7150/thno.30228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T. Y., Huan M. L., Cai Z., He W., Zhou S. Y., Zhang B. L.. Polymer-Initiating Caveolae-Mediated Endocytosis and GSH-Responsive MiR-34a Gene Delivery System for Enhanced Orthotopic Triple Negative Breast Cancer Therapy. Adv. Healthcare Mater. 2023;12(32):e2302094. doi: 10.1002/adhm.202302094. [DOI] [PubMed] [Google Scholar]
- Hermantara R., Richmond L., Taqi A. F., Chilaka S., Jeantet V., Guerrini I., West K., West A.. Improving CRISPR-Cas9 directed faithful transgene integration outcomes by reducing unwanted random DNA integration. J. Biomed. Sci. 2024;31(1):32. doi: 10.1186/s12929-024-01020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak J. K., Störtz F., Minary P.. Comprehensive computational analysis of epigenetic descriptors affecting CRISPR-Cas9 off-target activity. BMC Genomics. 2022;23(1):805. doi: 10.1186/s12864-022-09012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto D., Matsugi E., Kishi K., Inoue Y., Nigorikawa K., Nomura W.. SpCas9-HF1 enhances accuracy of cell cycle-dependent genome editing by increasing HDR efficiency, and by reducing off-target effects and indel rates. Mol. Ther. Nucleic Acids. 2024;35(1):102124. doi: 10.1016/j.omtn.2024.102124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., He W., Fu R., Wang S., Chen Y., Xu H.. Guide-specific loss of efficiency and off-target reduction with Cas9 variants. Nucleic Acids Res. 2023;51(18):9880–9893. doi: 10.1093/nar/gkad702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Panda G., Ray A.. Comparative structural and dynamics study of free and gRNA-bound FnCas9 and SpCas9 proteins. Comput. Struct. Biotechnol. J. 2022;20:4172–4184. doi: 10.1016/j.csbj.2022.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Huang X., Yang D., Zhang J., Xu J., Chen Y. E.. Recent Advances in Improving Gene-Editing Specificity through CRISPR-Cas9 Nuclease Engineering. Cells. 2022;11(14):2186. doi: 10.3390/cells11142186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulcsár P. I., Tálas A., Ligeti Z., Krausz S. L., Welker E.. SuperFi-Cas9 exhibits remarkable fidelity but severely reduced activity yet works effectively with ABE8e. Nat. Commun. 2022;13(1):6858. doi: 10.1038/s41467-022-34527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Kim Y. H., Kim N., Okafor I., Choi S., Min S., Lee J., Bae S. M., Choi K., Choi J., Harihar V.. et al. Sniper2L is a high-fidelity Cas9 variant with high activity. Nat. Chem. Biol. 2023;19(8):972–980. doi: 10.1038/s41589-023-01279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Spasskaya D. S., Davletshin A. I., Bachurin S. S., Tutyaeva V. V., Garbuz D. G., Karpov D. S.. Improving the on-target activity of high-fidelity Cas9 editors by combining rational design and random mutagenesis. Appl. Microbiol. Biotechnol. 2023;107(7–8):2385–2401. doi: 10.1007/s00253-023-12469-5. [DOI] [PubMed] [Google Scholar]
- Essawi K., Hakami W., Naeem Khan M. B., Martin R., Zeng J., Chu R., Uchida N., Bonifacino A. C., Krouse A. E., Linde N. S.. et al. Pre-existing immunity does not impair the engraftment of CRISPR-Cas9-edited cells in rhesus macaques conditioned with busulfan or radiation. Mol. Ther. Methods Clin. Dev. 2023;29:483–493. doi: 10.1016/j.omtm.2023.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J., Meng Z., Zhang Z., Su H., Fan Y., Huang R., Ding R., Zhang N., Li F., Wang S.. Comprehensive Analysis of CRISPR-Cas9 Editing Outcomes in Yeast Xanthophyllomyces dendrorhous. CRISPR J. 2022;5(4):558–570. doi: 10.1089/crispr.2021.0116. [DOI] [PubMed] [Google Scholar]
- Wimberger S., Akrap N., Firth M., Brengdahl J., Engberg S., Schwinn M. K., Slater M. R., Lundin A., Hsieh P. P., Li S.. et al. Simultaneous inhibition of DNA-PK and Pol⊖ improves integration efficiency and precision of genome editing. Nat. Commun. 2023;14(1):4761. doi: 10.1038/s41467-023-40344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang N., Wu Z., Gao Y., Chen W., Wang Z., Su M., Ji W., Ji Q.. Molecular Basis and Genome Editing Applications of a Compact Eubacterium ventriosum CRISPR-Cas9 System. ACS Synth. Biol. 2024;13(1):269–281. doi: 10.1021/acssynbio.3c00501. [DOI] [PubMed] [Google Scholar]
- Yin S., Zhang M., Liu Y., Sun X., Guan Y., Chen X., Yang L., Huo Y., Yang J., Zhang X.. et al. Engineering of efficiency-enhanced Cas9 and base editors with improved gene therapy efficacies. Mol. Ther. 2023;31(3):744–759. doi: 10.1016/j.ymthe.2022.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeens E., Sinha S., Ahsan M., D’Ordine A. M., Jogl G., Palermo G., Lisi G. P.. High-fidelity, hyper-accurate, and evolved mutants rewire atomic-level communication in CRISPR-Cas9. Sci. Adv. 2024;10(10):eadl1045. doi: 10.1126/sciadv.adl1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Moran Torres J. P., Li Y., Lugones L. G., Wösten H. A. B.. Inheritable CRISPR based epigenetic modification in a fungus. Microbiol. Res. 2023;272:127397. doi: 10.1016/j.micres.2023.127397. [DOI] [PubMed] [Google Scholar]
- a Zhang L., Yang L., Huang J., Chen S., Huang C., Lin Y., Shen A., Zheng Z., Zheng W., Tang S.. A zwitterionic polymer-inspired material mediated efficient CRISPR-Cas9 gene editing. Asian J. Pharm. Sci. 2022;17(5):666–678. doi: 10.1016/j.ajps.2022.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Han Y., Yuan Z., Zhong S. L., Xu H., Jiang S.. Minimizing the off-target frequency of the CRISPR/Cas9 system via zwitterionic polymer conjugation and peptide fusion. Chem. Sci. 2023;14(23):6375–6382. doi: 10.1039/D2SC07067G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Zhou S., Zeng J., Zhang S., Li M., Yue F., Chen Z., Dong Y., Zeng Y., Luo J.. A biodegradable lipid nanoparticle delivers a Cas9 ribonucleoprotein for efficient and safe in situ genome editing in melanoma. Acta Biomater. 2024;190:531–547. doi: 10.1016/j.actbio.2024.10.030. [DOI] [PubMed] [Google Scholar]
- a Kim Y. H., Jeon N., Park S., Choi S. Q., Lee E., Li S.. Complexation of Poly(ethylene glycol)-(ds)OligoDNA Conjugates with Ionic Liquids. ACS Macro Lett. 2024;13(5):528–536. doi: 10.1021/acsmacrolett.4c00028. [DOI] [PubMed] [Google Scholar]; b Eş I., Thakur A., Mousavi Khaneghah A., Foged C., de la Torre L. G.. Engineering aspects of lipid-based delivery systems: In vivo gene delivery, safety criteria, and translation strategies. Biotechnol. Adv. 2024;72:108342. doi: 10.1016/j.biotechadv.2024.108342. [DOI] [PubMed] [Google Scholar]
- Braun M., Lange C., Schatz P., Long B., Stanta J., Gorovits B., Tarcsa E., Jawa V., Yang T. Y., Lembke W.. et al. Preexisting antibody assays for gene therapy: Considerations on patient selection cutoffs and companion diagnostic requirements. Mol. Ther. Methods Clin. Dev. 2024;32(1):101217. doi: 10.1016/j.omtm.2024.101217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S., Yocum R. R., Silver P. A., Way J. C.. High spontaneous integration rates of end-modified linear DNAs upon mammalian cell transfection. Sci. Rep. 2023;13(1):6835. doi: 10.1038/s41598-023-33862-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assa G., Kalter N., Rosenberg M., Beck A., Markovich O., Gontmakher T., Hendel A., Yakhini Z.. Quantifying allele-specific CRISPR editing activity with CRISPECTOR2.0. Nucleic Acids Res. 2024;52(16):e78. doi: 10.1093/nar/gkae651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K., Fronza R., Evens H., Chuah M. K., VandenDriessche T.. Comprehensive analysis of off-target and on-target effects resulting from liver-directed CRISPR-Cas9-mediated gene targeting with AAV vectors. Mol. Ther. Methods Clin. Dev. 2024;32(4):101365. doi: 10.1016/j.omtm.2024.101365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Peng Y., Lv J., Ding L., Gong X., Zhou Q.. Responsible governance of human germline genome editing in China†. Biol. Reprod. 2022;107(1):261–268. doi: 10.1093/biolre/ioac114. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Astarăstoae V., Ioan B. G., Rogozea L. M., Hanganu B.. Advances in Genetic Editing of the Human Embryo. Am. J. Ther. 2023;30(2):e126–e133. doi: 10.1097/MJT.0000000000001604. [DOI] [PubMed] [Google Scholar]
- Cetin B., Erendor F., Eksi Y. E., Sanlioglu A. D., Sanlioglu S.. Gene and cell therapy of human genetic diseases: Recent advances and future directions. J. Cell. Mol. Med. 2024;28(17):e70056. doi: 10.1111/jcmm.70056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin J. P. J., Abrahamse H.. Optimizing CRISPR/Cas9 precision: Mitigating off-target effects for safe integration with photodynamic and stem cell therapies in cancer treatment. Biomed. Pharmacother. 2024;180:117516. doi: 10.1016/j.biopha.2024.117516. [DOI] [PubMed] [Google Scholar]
- Garg P., Singhal G., Pareek S., Kulkarni P., Horne D., Nath A., Salgia R., Singhal S. S.. Unveiling the potential of gene editing techniques in revolutionizing Cancer treatment: A comprehensive overview. Biochim. Biophys. Acta, Rev. Cancer. 2025;1880(1):189233. doi: 10.1016/j.bbcan.2024.189233. [DOI] [PubMed] [Google Scholar]
- Padilha V. A., Alkhnbashi O. S., Shah S. A., de Carvalho A., Backofen R.. CRISPRcasIdentifier: Machine learning for accurate identification and classification of CRISPR-Cas systems. Gigascience. 2020;9(6):giaa062. doi: 10.1093/gigascience/giaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]