

Abstract
Intercellular adhesion molecule 1 and the low-density lipoprotein receptor are used for cell entry by major and minor receptor group human rhinoviruses (HRVs), respectively. Whereas minor-group viruses, exemplified by HRV2, transfer their genomic RNA to the cytoplasm through a pore in the endosomal membrane (E. Prchla, C. Plank, E. Wagner, D. Blaas, and R. Fuchs, J. Cell Biol. 131:111–123, 1995), the mechanism of in vivo uncoating of major-group HRVs has not been elucidated so far. Using free-flow electrophoresis, we performed a comparative analysis of cell entry by HRV2 and the major group rhinovirus HRV14. Here we demonstrate that this technique allows the separation of free viral particles from those associated with early endosomes, late endosomes, and plasma membranes. Upon free-flow electrophoretic separation of microsomes, HRV14 was recovered from endosomes under conditions which prevent uncoating, whereas the proportion of free viral particles increased with time under conditions which promote uncoating. The remaining virus eluted within numerous fractions corresponding to membraneous material, with no clear endosomal peaks being discernible. This suggests that uncoating of HRV14 results in lysis of the endosomal membrane and release of subviral 135S and 80S particles into the cytoplasm.
The various members of the large family Picornaviridae use a number of different membrane receptors to attach to and ultimately infect their target cells. These surface proteins belong to families as diverse as the immunoglobulin (Ig) superfamily (14, 45, 47), integrins (4, 5), complement-inactivating proteins (24, 49), lipoprotein receptors (17, 20), and mucin-type glycoproteins (23). For several representative picornaviruses, data have been presented which strongly suggest that the virions are internalized by their respective receptors into endosomes (50), where it was shown for foot-and-mouth disease virus and for the minor-group rhinovirus human rhinovirus type 2 (HRV2) that the low intravesicular pH triggers RNA release (3, 39).
For poliovirus and major-group HRVs, it was demonstrated that their receptors, poliovirus receptor and intercellular adhesion molecule 1 (ICAM-1), respectively, are capable in a soluble form of uncoating their cognate viruses in vitro at physiologic pH (7, 11, 21, 22). This reaction results in so-called A particles (sedimenting at 135S) and RNA-free B particles (sedimenting at 80S) with altered conformation and with increased hydrophobicity. The effect exerted by isolated receptors raise the possibility that productive uncoating also takes place at the plasma membrane. For polioviruses, internalization into endosomes has been demonstrated by electron microscopy (50) and by cell fractionation techniques (26, 27). Nevertheless, the question whether endocytosis is required for productive infection is still controversial. For major-group rhinoviruses, receptor-mediated endocytosis has not been demonstrated so far.
For HRV2, a minor-group rhinovirus, we have recently shown internalization into endosomes by the low-density lipoprotein receptor and by the low-density lipoprotein receptor-related protein (20, 39). Moreover, infection was shown to be strictly dependent on the low endosomal pH. As demonstrated by the size-dependent escape of cointernalized dextran from isolated endosomes in vitro, virus-induced pores are opened in the vesicular membrane in response to the low intraendosomal pH. Although not explicitly shown, it is likely that the viral RNA is released through these pores into the cytoplasm (40). In contrast, major-group HRVs such as HRV14 bind to ICAM-1, an immunoglobulin-like molecule which lacks endocytosis signals and is involved in the interaction between various types of lymphocytes (for a review, see reference 10). Binding to AP2 adapter molecules of clathrin-coated pits thus seems to be dispensable, since the receptor remains functional for virus infection even upon replacement of its transmembrane domain by a glycosylphosphatidylinositol anchor (44). However, this does not necessarily exclude transport to endosomes, since glycosylphosphatidylinositol-linked proteins also enter the endosomal system via caveolin-coated membrane invaginations (for a recent review, see reference 35). Indeed, some degradation of the viral capsid starting at 60 min postinfection is indicative of endocytosis and transport to lysosomes (31). In addition, it was reported by Perez and Carrasco that bafilomycin A1, a specific inhibitor of vacuolar proton ATPases, which are present in endocytic and exocytic vesicles, inhibits the infection of HeLa cells by HRV14 (37). This strongly indicates that HRV14 also follows an endosomal entry route. We therefore decided to investigate whether HRV14 is indeed taken up into endosomes and whether it releases its RNA by the same mechanism as that used by the minor-group virus HRV2.
Employing free-flow electrophoretic vesicle separation techniques, we here demonstrate an endosomal localization of HRV14 and HRV2 at 20°C. Upon prolonged infection at 34°C, free subviral HRV14 particles accumulated as free particles. Concomitantly, the endosomal peaks disappeared, with viral counts spreading over many fractions. In contrast, HRV2 accumulated in early and late endosomes under all conditions. From these data, we conclude that HRV14 infection results in endosome rupture, thereby releasing subviral particles into the cytoplasm.
MATERIALS AND METHODS
Chemicals.
All chemicals were obtained from Sigma. Fluorescein isothiocyanate (FITC)-dextran (70 kDa) was extensively dialyzed against Tris-buffered saline (pH 7.3) followed by phosphate-buffered saline (PBS) (pH 7.4) before use. Horseradish peroxidase (HRP) P8250 and tolylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (TPCK-TXIII) were from Sigma. [35S]methionine (2,000 Ci/mmol) was purchased from American Radiochemicals. Bafilomycin A1 was kindly provided by K. H. Altendorf and was stored at 20 mM in dimethyl sulfoxide at −20°C. Methionine-free minimal essential medium (MEM) was from Sigma.
Cell culture.
HeLa cells (Ohio strain) were grown in monolayers in MEM-Eagle containing heat-inactivated 5% fetal calf serum and 5% calf serum; in suspension culture, Joklik’s MEM supplemented with 7% horse serum was used. Media and sera were purchased from GIBCO-BRL.
Preparation of A and B particles.
HRV2 and HRV14 were propagated and labeled with [35S]methionine in HeLa cells and purified as described elsewhere (34, 42). A particles were generated by mixing purified HRV with an equal volume of 1 M sodium acetate buffer (pH 5.0). The mixture was left at room temperature for 20 min, chilled, and neutralized with 1 volume of 0.5 M Tris (16). B particles were prepared by heating purified virions in medium containing 10% serum at 56°C for 30 min (25).
Internalization of fluid-phase markers.
Suspension HeLa cells (5 × 107 to 9 × 107 cells in 2 ml of MEM) were first incubated with 20 mg of FITC-dextran per ml for 5 min and then chased for 12 min with fresh medium at 34°C to label late endosomes. Early endosomes were then labeled by the addition of HRP at 10 mg/ml for 2 min at 34°C. Alternatively, HRP (10 mg/ml) was internalized for 2 h at 20°C to label both early and late endosomes. The cells were washed repeatedly with ice-cold TEA buffer (10 mM triethanolamine, 10 mM acetic acid, 1 mM EDTA [pH 7.4]) before homogenization.
Virus uptake.
For binding at 4°C, 106 HeLa cells in 2 ml of infection medium (MEM containing 2% fetal calf serum (FCS) and 30 mM MgCl2) were incubated with approximately 4 × 105 cpm of 35S-labeled HRV14 under slow rotation for 2 h. For uptake at 20°C, 5 × 107 cells in 2 ml of infection medium were incubated with approximately 106 cpm of 35S-labeled HRV2 or HRV14 for 2 h. For uptake at 34°C, 9 × 107 cells were incubated in 6 ml of infection medium with 3 × 106 cpm of 35S-labeled HRV2 or HRV14. Aliquots were removed at 20, 60, and 120 min. In all cases, the cells were pelleted and washed twice with ice-cold PBS containing 10 mM EDTA followed by 0.25 M sucrose in TEA buffer.
Preparation of microsomes for FFE.
Equal numbers of cells having internalized fluid-phase markers were mixed with cells which had been incubated with virus under various conditions. The cells were pelleted and resuspended in 5 volumes of 0.25 M sucrose in TEA buffer with respect to the volume of the cell pellet. Homogenization was carried out with a ball-bearing homogenizer (2), and a postnuclear supernatant was prepared by centrifugation at 1,000 × g for 10 min. A microsomal pellet was then obtained by centrifuging the postnuclear supernatant onto a 2.5 M sucrose cushion in a TST 60.4 swing-out rotor at 100,000 × g for 1 h. Material at the interphase was suspended in the sucrose cushion and adjusted to 1 mg of protein per ml and to 0.25 M sucrose with TEA buffer. The sample was then subjected to gentle trypsin treatment (32, 41) by incubation with 0.03 mg of trypsin/mg of protein for 5 min at 37°C. The reaction was halted by adding a 10-fold excess of soybean trypsin inhibitor and immediate cooling to 4°C. The sample was injected into the free-flow electrophoresis (FFE) apparatus (Elphor VaP22; Bender and Hobein) at 110 mA and 1,300 V with 0.25 M sucrose in TEA buffer in the chamber. Where indicated, trypsin treatment was omitted. Ninety-six fractions were collected and analyzed for protein (6), fluorescence (FITC-dextran), peroxidase (32, 41), alkaline phosphatase (36), and radioactivity by liquid scintillation counting.
Fluorescence microscopy.
HeLa cells were grown on eight-well tissue culture chamber slides (Nunc) overnight, and the medium was replaced with infection medium containing about 5 μg of sucrose gradient-purified HRV2 or HRV14, respectively. The cells were incubated for 2 h at 20°C or 20 min at 34°C, chilled, washed three times with cold PBS containing 1 mM CaCl2 and 1 mM MgCl2, fixed, and permeabilized for 15 min at 4°C with methanol precooled to −20°C. HRV2 was revealed with purified monoclonal antibody 3B10 (19), and HRV14 was revealed with purified monoclonal antibody 17-1A (43) at final concentrations of 25 and 50 μg/ml, respectively. After incubation for 1 h at room temperature, the cells were washed as before and incubated with FITC-conjugated rabbit anti-mouse IgG (Dako) at a dilution of 1:40. Control incubations were carried out in the absence of virus. The cells were mounted in Moviol and viewed with a Zeiss fluorescence microscope. Photographs were taken with a Quantix charge-coupled device camera (Photometrics). For control purposes, FITC-dextran (10 mg/ml) was internalized under the same conditions and the cells were fixed with 2% paraformaldehyde and mounted.
Infection assay.
A total of 2 × 105 HeLa suspension cells in Eppendorf vials were preincubated in 0.5 ml of methionine-free infection medium plus 2% dialyzed FCS with or without 200 nM bafilomycin for 30 min at 34°C under rotation. The medium was replaced with fresh medium containing 108 PFU of HRV2 or HRV14, and the cells were incubated for 4.5 h. Thereafter, 20 μCi of [35S]methionine was added, and incubation was continued overnight. Where indicated, bafilomycin A1 was present throughout. Cells and cell debris were removed by centrifugation, and 300 μl of RIPA buffer (9) was added to the supernatants. Virus in the supernatants was recovered by Staphylococcus aureus-aided immunoprecipitation with rabbit hyperimmune sera directed against the respective virus serotypes (34) (kindly provided by F. Heinz). Analysis was carried out with 12.5% polyacrylamide–sodium dodecyl sulfate (SDS) minigels followed by autoradiography.
RESULTS
FFE efficiently separates early and late endosomes and plasma membrane-derived vesicles of HeLa cells.
FFE has been demonstrated to be a powerful technique to separate early and late endosomes and plasma membranes of BHK and CHO cells (32, 41). To verify that this also applies to HeLa cells (Ohio strain [46]), which were used in the present study, endosomes were labeled by fluid-phase endocytosis at 37°C with FITC-dextran for 5 min followed by a chase of 12 min (to label late endosomes) and with HRP for 2 min (to label early endosomes), respectively. A microsomal pellet was then prepared, and an aliquot was directly injected into the FFE apparatus (Fig. 1A) whereas another aliquot was subjected to gentle trypsin treatment prior to injection (Fig. 1B). Trypsinization was previously shown to be required for efficient separation of endosomal subpopulations (41). The concentrations of the various markers in the FFE fractions were determined accordingly by fluorescence spectrometry or enzymatic analyses. No separation was apparent in the absence of trypsin treatment (Fig. 1A), whereas early endosomes were well separated from late endosomes and plasma membranes in the sample that had been incubated with trypsin (Fig. 1B). Under the latter condition, vesicle-enclosed markers were shifted to the anode whereas free markers were recovered in fractions eluting near the cathode. As a consequence, the total amounts of endosomal and free markers could be calculated. Although the incubation with the protease at 37°C was only for 5 min, substantial leakage of internalized markers (about 30%) was evident (compare the amount of free markers in Fig. 1A and B). This is presumably due to a limited leakage of endosomal content, which had to be taken into account in the quantification of internalized versus released viral material (see below).
FIG. 1.
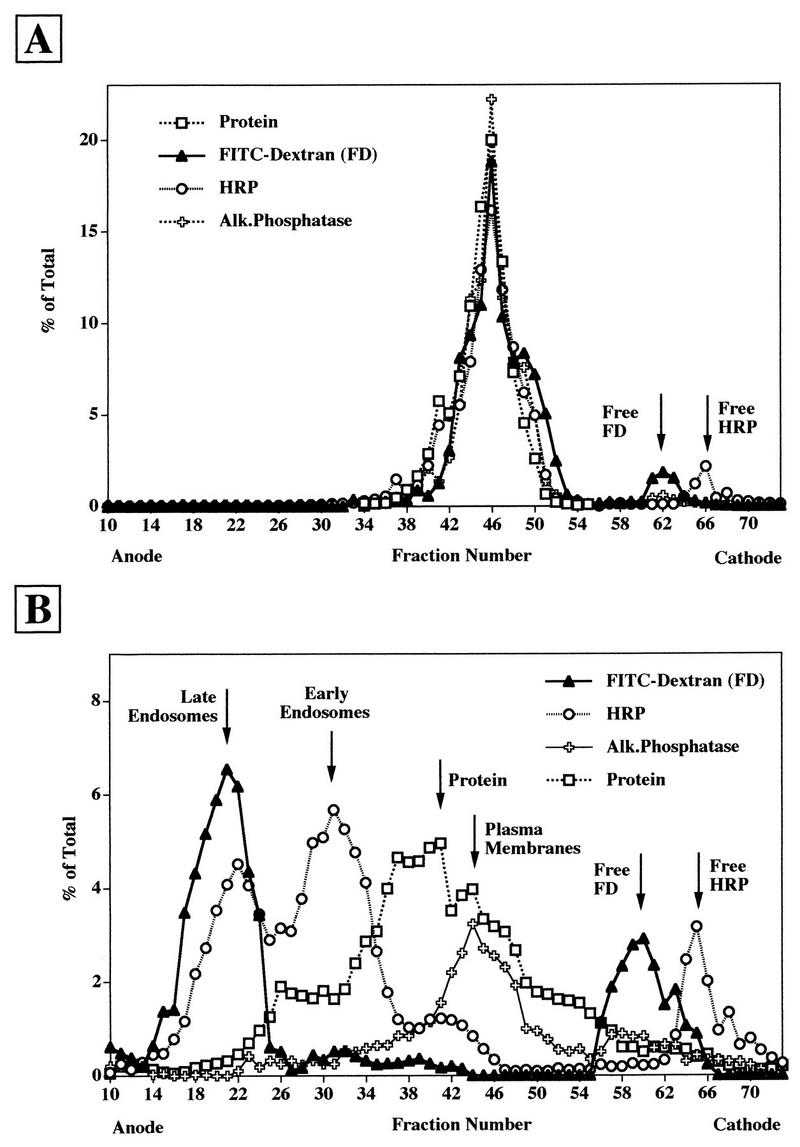
FFE profile of HeLa cell microsomes analyzed without (A) or with (B) trypsin treatment prior to analysis. HeLa cells were allowed to internalize FITC-dextran for 5 min followed by a chase of 10 min and HRP for 2 min at 37°C. The cells were homogenized, and microsomes were prepared and either injected as such (A) or treated with 3% trypsin for 5 min at 37°C prior to injection (B) into the FFE apparatus. In total, 96 fractions were collected, and the distribution of the markers was analyzed by fluorescence spectrometry (FITC-dextran, late endosomes), for HRP activity (early endosomes), for alkaline phosphatase (Alk.Phosphatase) activity (plasma membranes), and for total protein by the Bradford protein assay.
Estimation of nonspecific attachment of native and subviral particles of HRV2 and HRV14 to microsomal material.
Due to leakage of endosomal content during preparation (see above), the possible nonspecific attachment of native virus and of subviral particles to the outer surface of vesicular material was analyzed. 135S and 80S particles were prepared from the respective [35S]methionine-labeled HRVs by exposure to pH 5.0 and heating to 56°C, respectively, by published procedures (25, 29, 30). Homogenates from cells that had been allowed to internalize HRP and FITC-dextran to label early and late endosomes (see above) were then mixed with each of these preparations from HRV2 (Fig. 2A) and from HRV14 (Fig. 2B), respectively, and microsomes were prepared. Under the centrifugation conditions used, unattached input viral material was also recovered in the microsomal pellet. Microsomes were trypsin treated and subjected to FFE. The radioactivity in the fractions was determined by scintillation counting. The positions of late endosomes, early endosomes, plasma membranes, and the bulk of the protein were determined as in Fig. 1B. For the sake of clarity, the corresponding profiles are not shown in Fig. 2, but the positions of peak fractions are indicated by arrows.
FIG. 2.
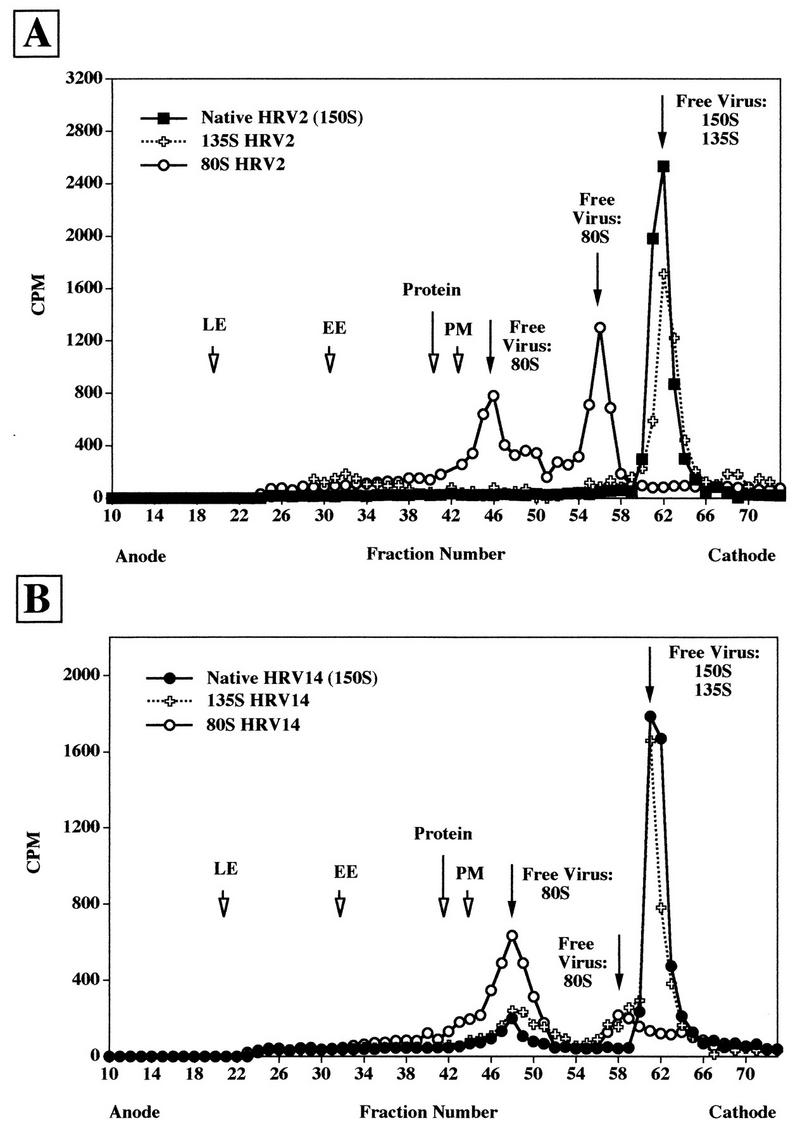
FFE profile of [35S]methionine-labeled HRV2 (A) and HRV14 (B) mixed with HeLa cell extracts. Endosomes were labeled with endocytic markers, and the cells were homogenized as in Fig. 1. 35S-labeled viruses were added, and microsomes were prepared and analyzed after trypsin treatment. The same experiments were also carried out with artificially produced 135S particles and with 80S particles. The position of viral material was determined by scintillation counting. The positions of endocytic markers were determined as in Fig. 1. The results of the runs with native 150S virus and 135S and 80S subviral particles of HRV2 (A) and HRV14 (B) are shown. The positions of late endosomes (LE), early endosomes (EE), plasma membranes (PM), and protein are also indicated.
For HRV2, native virus and 135S particles were deflected to the cathode, where they eluted in a single peak at the same position (Fig. 2A). The 80S particles eluted in two peaks (fraction 46 and 56). Thus, all viral counts were recovered in fraction numbers higher than fraction 40, well separated from membranous material. Only minor side fractions of plasma membranes and the major protein peak were found to coelute with the first peak of the 80S viral particles. Interestingly, whereas only native particles were recovered upon incubation of native HRV2 with the cell homogenate (Fig. 2A), about 14% of total native input HRV14 were converted to 80S particles, which eluted around fraction 48 (Fig. 2B). Apparently, the short incubation at 37°C during trypsin treatment is sufficient for a fraction of HRV14 to be uncoated by surface-accessible membrane receptors. Moreover, some 80S material was also recovered from the incubation of 135S particles. Upon analysis of the nonspecific association of 80S particles with cell homogenates, two peaks were observed; the first was in a region corresponding to side fractions of the bulk of plasma membranes, whereas the smaller one was essentially free material. These experiments demonstrate the separation of free virus and subviral particles from early and late endosomes and other cellular material by FFE; in addition, substantial nonspecific attachment to cellular membranes can be excluded. Although 135S particles cannot be separated from 150S native virus, the 80S particles eluted at a different position. However, for unknown reasons, two distinct peaks were seen, with the first presumably being associated with a subfraction of cellular material.
FFE analysis of HRV14 bound to HeLa cells at 4°C.
In contrast to HRV2, which is released from the plasma membrane upon chelation of Ca2+, HRV14 attaches to its receptor in a Ca2+-independent manner (31). Therefore, the localization of HRV14 bound to HeLa cells at 4°C can be analyzed by FFE, which requires an EDTA-containing buffer, whereas the method precludes the analysis of plasma membrane-bound HRV2. Since trypsinization must be carried out at 37°C, a temperature where receptor-mediated alteration of native HRV14 to subviral particles can take place, we investigated the distribution of the viral label as a function of trypsin treatment. [35S]methionine-labeled HRV14 was bound to HeLa cells for 2 h at 4°C, the cells were washed, and microsomes were prepared and analyzed either without (Fig. 3A) or with (Fig. 3B) trypsin treatment by FFE. In the absence of trypsin treatment, approximately 53% of the total input radioactivity colocalized with the bulk of nonseparated membranes (compare to Fig. 1A), whereas the remainder was recovered as free virus (around fraction 60). When the microsomes were subjected to trypsin treatment prior to FFE, about 30% of total HRV14 colocalized with plasma membranes. Free native virus (or 135S particles [see above]) amounted to 41%. Most noticeably, the remainder (29%) eluted at a position corresponding to 80S particles. In any case, no radioactivity was seen in the region corresponding to early or late endosomes. Taken together, these data indicate that even in the absence of trypsin treatment, a substantial amount of initially bound HRV14 dissociated from the membranes. Upon incubation with trypsin at 37°C, this amount was found to increase from 47 to 70%. In addition, this short incubation time was apparently sufficient to convert 29% of input virus to its 80S conformation.
FIG. 3.
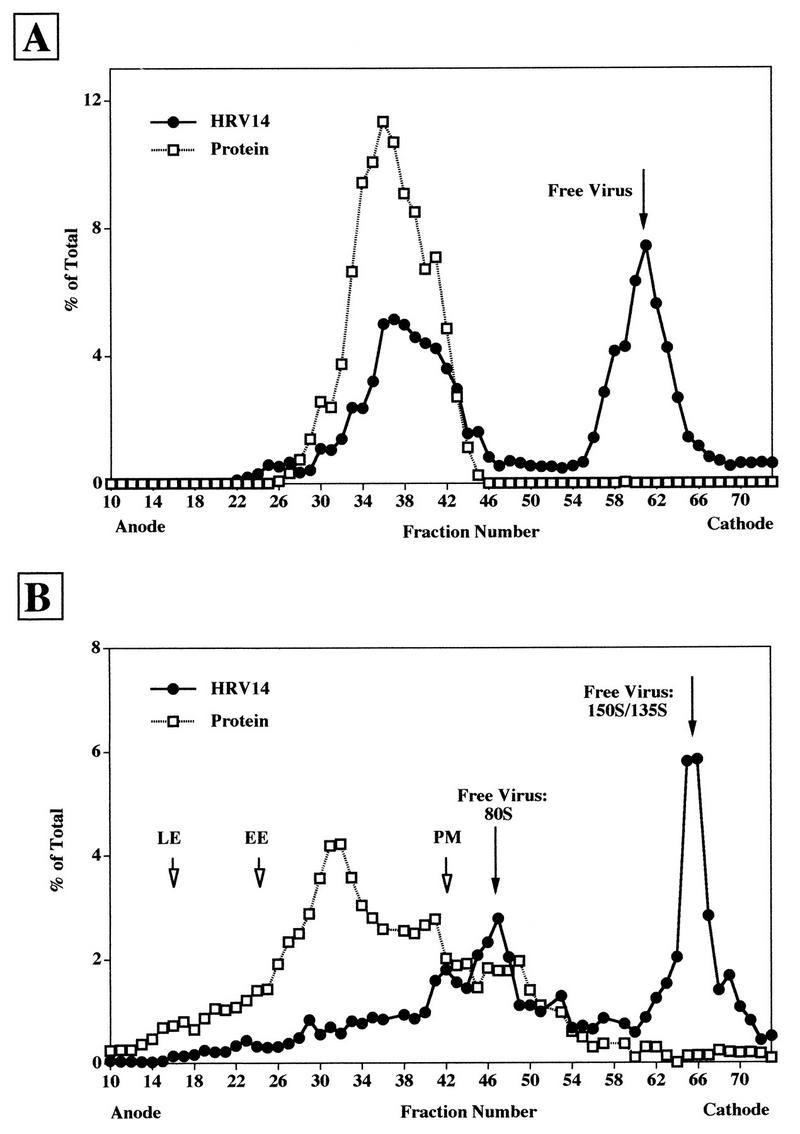
FFE profile of [35S]methionine-labeled HRV14 bound to HeLa cells at 4°C. [35S]methionine-labeled HRV14 was allowed to attach to HeLa cells for 2 h at 4°C. The cells were mixed with HeLa cells labeled with endosomal markers (as in Fig. 1), and microsomes were prepared and analyzed without (A) or with (B) trypsin treatment. Radioactivity was measured in each fraction by scintillation counting and is given as a percentage of the total recovered. For the sake of clarity, only the protein concentration in each fraction is depicted; positions of early (EE) and late (LE) endosomes and plasma membranes (PM) are indicated by arrows. Total counts in the respective cellular compartments were determined as follows. (A) Membrane-associated virus, fractions 20 to 49; free virus, fractions 50 to 70. (B) Plasma membranes, fractions 16 to 44; free 80S particles, fractions 45 to 60; free virus 150S/135S, fractions 60 to 74.
HRV14 is internalized into endosomes.
HRV14 is not uncoated below 26°C (21, 28). In contrast, uncoating of HRV2 also takes place at 20°C and appears to be solely low-pH dependent (34, 39). Therefore, internalization of [35S]methionine-labeled HRV2 and HRV14 was first conducted at 20°C for 2 h to prevent receptor-mediated uncoating of HRV14. Cells loaded with HRP under identical conditions were added prior to homogenization to allow the subsequent localization of endosomes in the fractions collected from the FFE run. The cells were washed and homogenized, and microsomes were prepared, treated with trypsin, and injected into the FFE apparatus. As seen in Fig. 4A, the majority of HRV2 localized to early and to late endosomes, with only a small amount being present as free particles (80S and 150S/135S). Under the same conditions, HRV14 clearly colocalized with HRP in early and late endosomes (Fig. 4B). Quantification revealed that the majority of viral counts (about 68%) was present as free virus around fractions 52 and 62 (80S and 150S/135S particles, respectively). Nevertheless, the results of these experiments strongly suggest that HRV14 reaches endosomal compartments. Warming to 37°C during trypsin treatment results in substantial release of membrane-associated virus and even in some receptor-mediated uncoating (Fig. 3). Therefore, the amount of endosome-associated viral material might be underestimated (Fig. 4B).
FIG. 4.
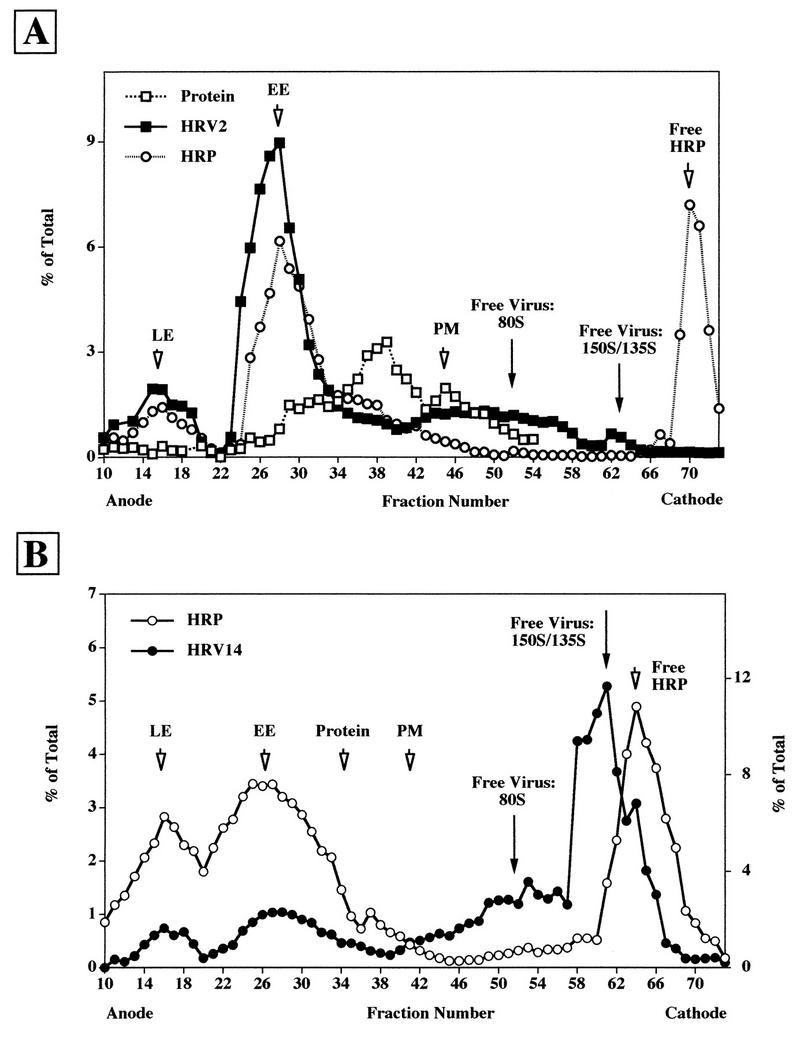
FFE profile of HeLa cell microsomes after challenge with [35S]methionine-labeled HRV2 (A) and HRV14 (B) at 20°C for 2 h. HeLa cells were incubated with HRP or with 35S-labeled virus for 2 h at 20°C, respectively. The cells were mixed, and microsomes were separated by FFE after trypsin treatment as in Fig. 3. For quantification, pools of fractions 10 to 44 (membrane-associated material) and fractions 45 to 73 (free 80S and 135S/150S particles) were counted. The positions of plasma membranes and of the protein peak are indicated by arrows.
To obtain additional evidence for the presence of HRV14 in endosomes upon infection at 20°C, HeLa cells were challenged with HRV2 or with HRV14 for 2 h at 20°C. The cells were fixed, and the respective serotype was detected by indirect immunofluorescence. In addition, FITC-dextran was internalized under identical conditions to reveal endocytic vesicles accessible to fluid-phase uptake. As shown in Fig. 5, FITC-dextran internalization resulted in labeling of vesicles throughout the cytoplasm. HRV2 and HRV14 also exhibited a clear endosomal staining pattern primarily in the perinuclear area. However, since high virus concentrations are required to allow detection by microscopy (18), we had to exclude virus uptake by a fluid-phase mechanisms. Therefore, parallel experiments were carried out with HEp-2 cells, which do not express ICAM-1 and thus do not bind major-group HRVs (48). Fluorescence microscopy revealed the presence of HRV2 in endosomes of HEp-2 cells, whereas no labeling was observed in cells incubated with HRV14 (data not shown). This control experiment clearly excludes fluid-phase uptake of HRV14.
FIG. 5.
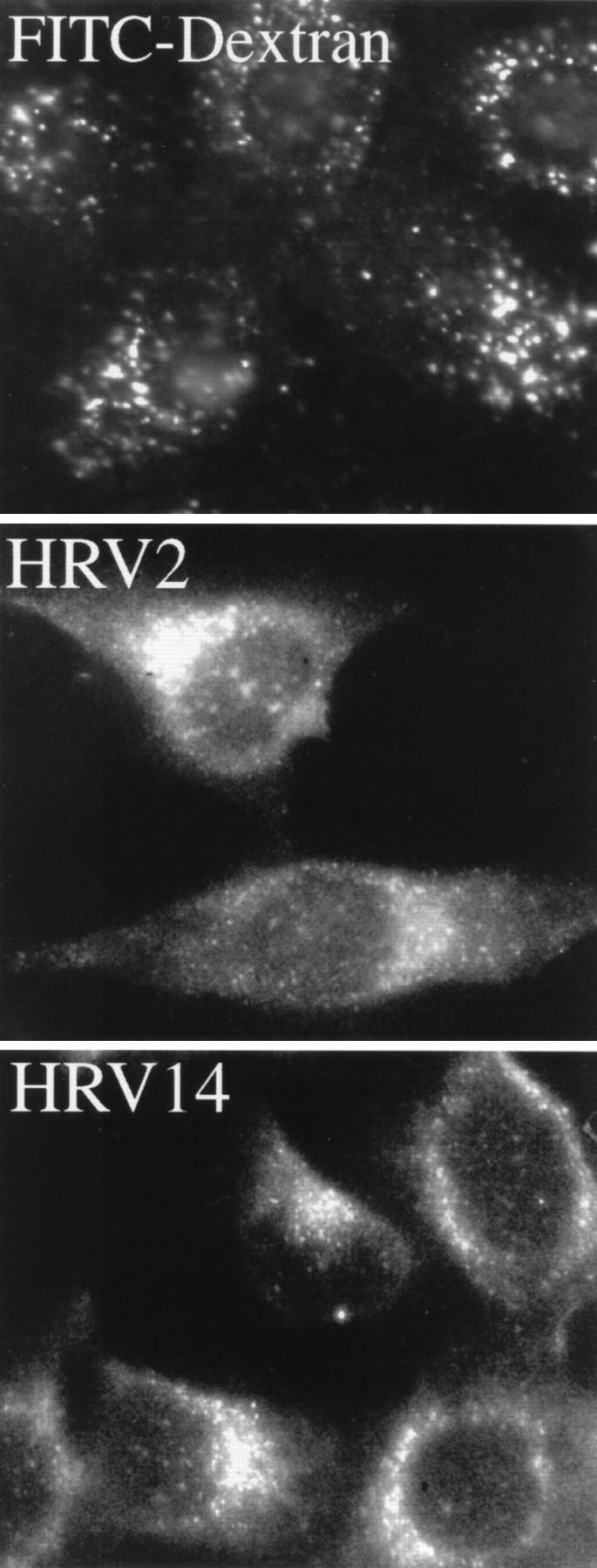
Immunofluorescence microscopy of HRVs internalized into HeLa cells at 20°C for 2 h. HeLa cells grown on chamber slides were incubated with FITC-dextran (10 mg/ml), HRV2 (5 μg/ml), or HRV14 (5 μg/ml). HRV2 was detected with monoclonal antibody 3B10, and HRV14 was detected with 17-1A followed by FITC-conjugated rabbit anti-mouse IgG.
Under infection conditions, HRV14 subviral particles are released by disruption of endosomes.
Incubation of HeLa cells with the radiolabeled viruses was then conducted at 34°C, conditions which lead to productive infection. At 20, 60, and 120 min, aliquots were withdrawn and microsomes were prepared and analyzed by FFE after trypsin treatment (Fig. 6). Under all incubation conditions, HRV2 accumulated in late endosomes at all incubation times, as exemplified by the experiment conducted for 20 min (Fig. 6A). For HRV14, essentially no clear virus peaks could be seen to be associated with endosomal material regardless of the incubation time. However, some radioactivity was equally distributed over the whole region corresponding to these cellular compartments (fractions 13 to 42 in Fig. 6B). Under all conditions, most of the viral counts eluted as free 80S and 135S/150S particles. Determination of total radioactivity in the various fractions disclosed a time-dependent increase in the amount of viral label in the region corresponding to early and late endosomes and to plasma membrane fractions. Whereas the viral label in these fractions increased by 60% when the incubation time was increased from 20 to 120 min, the increase in the number of free viral particles was almost 100% within this period. For HRV2, the relation between endosome-associated material and free virus (plus subviral particles) was about 80 and 20%, respectively, for all incubation times. For HRV14, quantification of viral counts within the broad region including endosomal and plasma membrane fractions gives a value of 42%, with 58% free virus. Consequently, the substantially larger amount of free HRV14 than of free HRV2 can certainly not be ascribed to leakage of endosomal material during preparation but, rather, is the product of endosome lysis in the process of HRV14 uncoating. It should be mentioned that cointernalization of HRV2 or HRV14 with endocytic markers had no effect on the electrophoretic migration behavior of endosomes (data not shown).
FIG. 6.
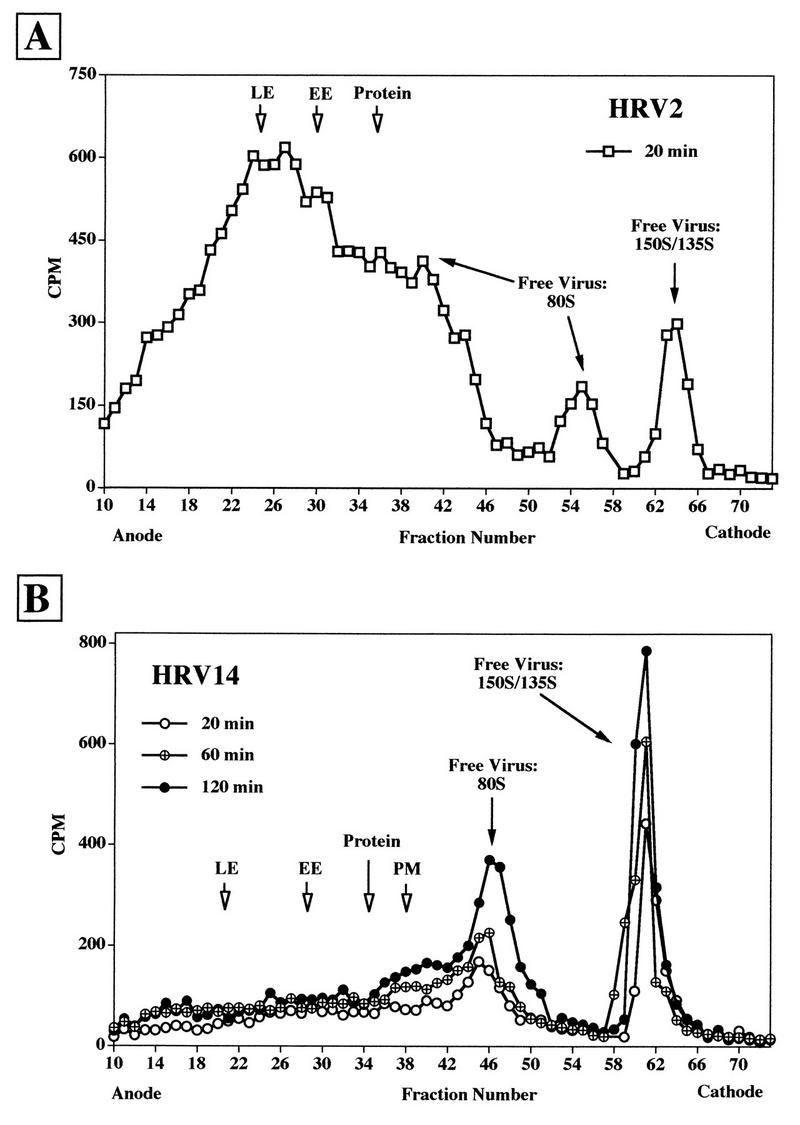
FFE profiles of [35S]methionine-labeled HRV2 (A) and HRV14 (B) incubated with HeLa cells at 34°C. HeLa cells were incubated with the respective labeled viruses at 34°C. Samples were withdrawn at 20, 60, and 120 min and mixed with cells which had been allowed to endocytose FITC-dextran and HRP (as in Fig. 1). Microsomes were prepared and analyzed as in Fig. 4. Only the profile of the incubation for 20 min is shown for HRV2, whereas all time points are depicted for HRV14. Positions of the peak fractions of the markers are indicated by arrows. For quantification of free versus membrane-associated virus, the fractions were pooled as follows: (A) endosome- and plasma membrane-associated HRV2, fractions 10 to 38; free virus, fractions 39 to 70; (B) endosome- and plasma membrane-associated HRV14, fractions 10 to 42; free virus, fractions 43 to 70.
Again, additional evidence for endosomal lysis by HRV14 was obtained by fluorescence microscopy. HeLa cells were infected at 34°C with HRV2 or HRV14. In accordance with the results from the FFE experiments, HRV2 was clearly present in endosomes; however, no fluorescence was evident for HRV14 with respect to background staining of uninfected cells (Fig. 7). This is most probably due to endosomal lysis, resulting in dilution of virions in the cytosol and thus precluding their detection.
FIG. 7.
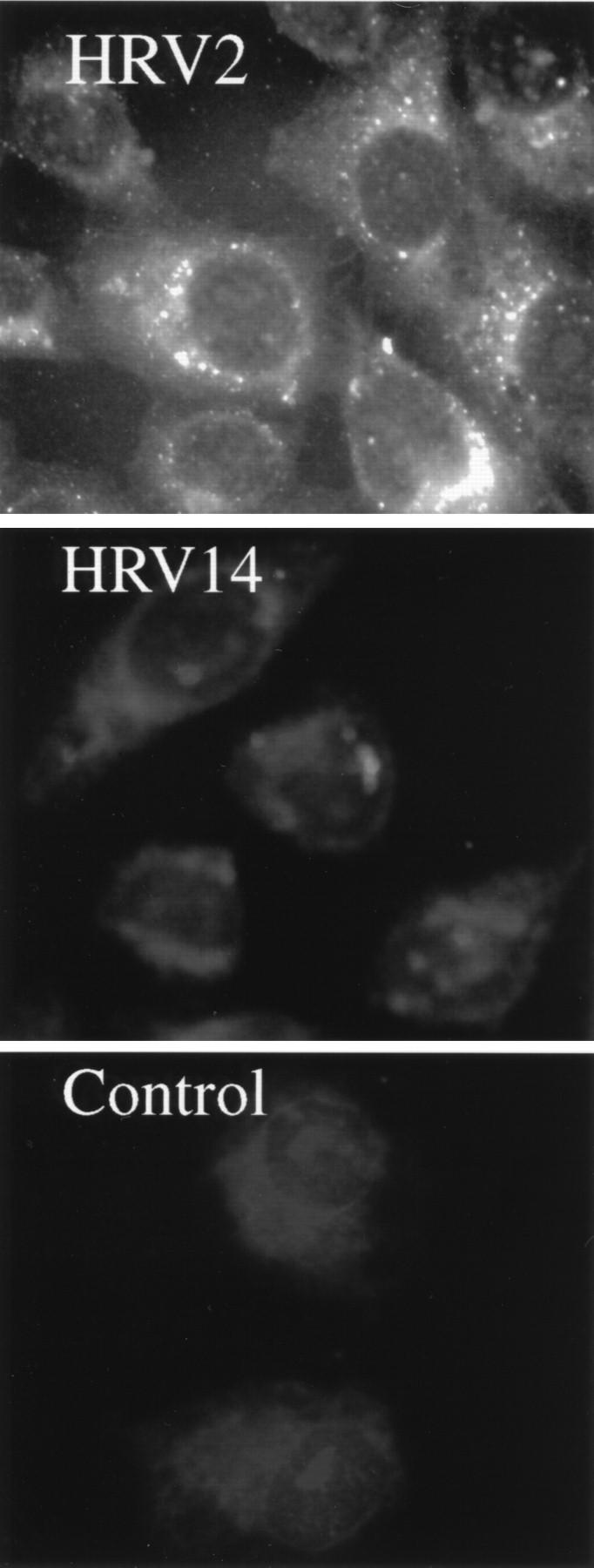
Immunofluorescence microscopy of HRV internalized into HeLa cells at 34°C for 20 min. HeLa cells grown on chamber slides were challenged with 5 μg of HRV2 or HRV14 per ml. HRV2 was detected with monoclonal antibody 3B10 and HRV14 was detected with 17-1A followed by FITC-conjugated rabbit anti-mouse IgG. Control incubations without HRV2 or HRV14 (not shown) exhibited background fluorescence.
Effect of bafilomycin A1 on HRV2 and HRV14 infection.
Perez and Carrasco (37) have previously used the specific vacuolar H+-ATPase inhibitor bafilomycin A1 at 2 μM to demonstrate the low-pH requirement for infection with HRV14. In fact, these workers showed that host cell shutoff was prevented in the presence of the drug. However, close inspection of the gel shown in Fig. 8 of their article clearly reveals the presence of viral bands under these conditions. Prchla et al. (39), using immunoprecipitation techniques, subsequently showed that HRV2 infection was prevented by bafilomycin A1 at concentrations as low as 200 nM. We thus decided to use this technique to reinvestigate whether the drug completely blocks HRV14 infection (38). Suspension HeLa cells were infected with HRV2 or HRV14 in the presence or absence of 200 nM bafilomycin A1. Progeny viruses were labeled with [35S]methionine, precipitated with specific antisera, and analyzed by SDS-polyacrylamide gel electrophoresis followed by autoradiography. As seen in Fig. 8, HRV2 infection was completely inhibited by the drug whereas HRV14 infection was only reduced.
FIG. 8.

Effects of bafilomycin A1 on infection of HeLa cells with HRV2 and with HRV14. Suspension HeLa cells were preincubated with or without 200 nM bafilomycin A1 and challenged with HRV2 or HRV14. [35S]methionine was added 4.5 h p.i., and incubation was continued overnight. Cells and debris were removed, and progeny virus present in the supernatant was immunoprecipitated with the respective antiserum aided by fixed S. aureus cells. Viral proteins were analyzed by SDS-polyacrylamide gel electrophoresis followed by autoradiography.
FFE was also carried out with microsomes prepared from cells which had been allowed to internalize HRV2 or HRV14 in the presence of 200 nM bafilomycin A1 for 20 min at 34°C. As shown in Fig. 9, under these conditions HRV2 accumulates in early endosomes, in contrast to the situation in the absence of bafilomycin A1 where virus is found in late endosomes (Fig. 6A). This is in agreement with inhibition of transport from early to late endosomes by bafilomycin A1 (8). In contrast, for HRV14 no clear endosomal peaks are visible and the profile resembles that observed in the absence of the drug (Fig. 6B). This is in line with the inability of bafilomycin A1 to completely inhibit HRV14 infection (compare with Fig. 8).
FIG. 9.

FFE profile of HeLa cell microsomes after challenge with [35S]methionine-labeled HRV2 and HRV14 at 34°C in the presence of bafilomycin A1. The cells were preincubated with 200 nM bafilomycin A1 for 30 min, whereupon the respective labeled viruses were added and internalized for 20 min. These cells were mixed with HeLa cells that had internalized FITC-dextran and HRP as described in Fig. 1. Microsomes were prepared and analyzed as in Fig. 6. The positions of early (EE) and late (LE) endosomes and plasma membranes (PM) are indicated. Data are expressed as percentage of the total radioactivity recovered after FFE.
DISCUSSION
Major-group HRVs attach to ICAM-1, a member of the Ig superfamily. This membrane receptor is devoid of any known internalization signal and is involved in intercellular communication rather than in endocytosis of ligands; this is in contrast to the receptors of minor-group HRVs, which are members of the low-density lipoprotein receptor family which, as typical recycling receptors, are rapidly clustered in coated pits, from where they carry their cargo to early and late endosomes. There, the ligands are dissociated and further transported to lysosomes. The receptors are recycled to the plasma membrane, from where they initiate another round of internalization.
Since we have shown that minor-group HRVs initiate infection from late endosomes (39), we asked whether major-group viruses would also follow this route, assuming that ICAM-1 was internalized either as a result of clustering around the multivalent virus capsid or as a consequence of its natural turnover (1). By FFE separation techniques, we could clearly demonstrate that at least a fraction of the major-group HRV14 localized to endosomal compartments upon infection of HeLa cells at 20°C (Fig. 4). At this temperature, receptor-mediated structural changes from native particles to 135S and ultimately to 80S RNA free particles do not take place (7, 15, 21). Apparently, virus thus becomes trapped in the endosomal compartment, from where it cannot escape at this temperature. Nevertheless, a large proportion of free particles (150S/135S and 80S) were observed under these conditions. These are due to the short exposure of the microsomes to 37°C, as required for trypsin treatment, since 80S particles were absent upon omission of this treatment. Moreover, the proportion of membrane-associated material was elevated (60 and 32% in the absence and presence, respectively, of trypsin treatment [data not shown]). Sucrose density gradient sedimentation analysis of viral particles generated by in vitro incubation of cell homogenates demonstrated that a 5-min incubation at 37°C without trypsin essentially yielded the same proportion of modified particles (data not shown). Therefore, it is not the trypsin which causes particle release and/or modification. Attempts to carry out trypsinization for prolonged periods at 4 or 20°C were unsuccessful in achieving endosome separation.
The viral particles observed upon FFE analysis of HRV14 internalized at 20°C (Fig. 4) could be set free either by release and/or alteration of plasma membrane associated virus or by limited endosome lysis. The large proportion of these particles already seen upon attachment at 4°C (Fig. 3B) makes it likely that they originate from the plasma membrane. When cells were infected at 34°C, no distinct peaks of viral label in endosomes were observed (Fig. 6B); instead, free viral material was found to accumulate in a time-dependent manner. In addition, viral label was distributed all over the region corresponding to early endosomes, late endosomes, and plasma membranes (Fig. 6B). Concomitantly, the proportion of free 80S particles was seen increased with time. This indicates that virus was transported into endosomes, where it most probably became modified by the uncoating activity of ICAM-1, resulting in endosome rupture and release of viral particles into the cytosol. This scenario is in sharp contrast to that of the minor-group rhinovirus HRV2, which was found to accumulate in endosomes regardless of temperature and incubation time (Fig. 4A and 6A).
The results of FFE are also supported by the data obtained from fluorescence microscopy, which clearly show that HRV14 is internalized into endosomes at 20°C (Fig. 5). If cells were challenged at 34°C, however, no fluorescence was seen, indicating that virions were released from the vesicles and were no longer detectable in the cytoplasm as a result of dilution. In contrast, HRV2 was seen in vesicular structures under all conditions (Fig. 5 and 7).
The results of our experiments thus suggest that major-group viruses are indeed internalized into endosomes, where they undergo receptor-mediated structural changes leading to an increase of hydrophobicity, which ultimately leads to disruption of the endosomal membrane. At least in this respect, major-group HRVs resemble adenoviruses, which are also released into the cytoplasm as modified particles following internalization and rupture of endosomes (12, 13). Currently, we use single-organelle flow analysis (33) to quantitate the endosomal content and pH of internalized markers (Cy5-FITC-dextran) under the influence of viral infection. Preliminary results indicate that adenovirus leads to a loss of 40% of the dextran-labeled endosomes. For HRV14, a 30% loss has been observed. Compared to HRV2 (0% loss), this is a clear indication for endosomal rupture being induced by HRV14.
Structural modifications of HRV14 from 150S to 135S also occur upon exposure to low pH in vitro. Apparently, this does not lead to rupture of the endosomal membrane, since virions were found in endosomes upon internalization at 20°C, a temperature where low endosomal pH is maintained but receptor function is inhibited (Fig. 4B and 5).
Bafilomycin A1 was reported to inhibit HRV14 infection at 34°C (37). However, our data indicate that this drug inhibits infection by HRV14 only to a small extent under the conditions used (Fig. 8) (38). This raises the question of the distinct roles of the receptor and the low pH in the process of endosome rupture by HRV14. Therefore, we are investigating the effect of bafilomycin A1 on virus-induced release of fluid-phase markers from endosomes by single-organelle flow analysis.
Although the results presented in this report do not identify the endosomal route as being the exclusive infection pathway, they demonstrate that HRV14 subviral particles are transferred into the cytosol whereas in the case of HRV2 most probably only the RNA is delivered into the cytoplasm, with the capsid proteins being degraded in lysosomes.
ACKNOWLEDGMENTS
D.S. and P.K. contributed equally to this work.
We thank A. Mosser and K. Altendorf for the kind gifts of monoclonal antibody 17-1A and bafilomycin A1, respectively. We particularly thank I. Gösler for virus preparations.
This work was supported by an EMBO fellowship to P.K. and by Austrian Science Foundation grants P10384-MOB to D.B. and P10618-MED to R.F.
REFERENCES
- 1.Almenar Queralt A, Duperray A, Miles L A, Felez J, Altieri D C. Apical topography and modulation of ICAM-1 expression on activated endothelium. Am J Pathol. 1995;147:1278–1288. [PMC free article] [PubMed] [Google Scholar]
- 2.Balch W E, Rothman J E. Characterization of protein transport between successive compartments of the Golgi apparatus: asymmetric properties of donor and acceptor activities in a cell-free system. Arch Biochem Biophys. 1985;240:413–425. doi: 10.1016/0003-9861(85)90046-3. [DOI] [PubMed] [Google Scholar]
- 3.Baxt B. Effect of lysosomotropic compounds on early events in foot-and-mouth disease virus replication. Virus Res. 1987;7:257–271. doi: 10.1016/0168-1702(87)90032-3. [DOI] [PubMed] [Google Scholar]
- 4.Bergelson J M, Shepley M P, Chan B M C, Hemler M E, Finberg R W. Identification of the integrin VLA-2 as a receptor for echovirus-1. Science. 1992;255:1718–1720. doi: 10.1126/science.1553561. [DOI] [PubMed] [Google Scholar]
- 5.Berinstein A, Roivainen M, Hovi T, Mason P W, Baxt B. Antibodies to the vitronectin receptor (integrin alpha(v)beta(3)) inhibit binding and infection of foot-and-mouth disease virus to cultured cells. J Virol. 1995;69:2664–2666. doi: 10.1128/jvi.69.4.2664-2666.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 7.Casasnovas J M, Springer T A. Pathway of rhinovirus disruption by soluble intercellular adhesion molecule 1 (ICAM-1): an intermediate in which ICAM-1 is bound and RNA is released. J Virol. 1994;68:5882–5889. doi: 10.1128/jvi.68.9.5882-5889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clague M J, Urbe S, Aniento F, Gruenberg J. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J Biol Chem. 1994;269:21–24. [PubMed] [Google Scholar]
- 9.Collett M S, Erikson R L. Protein kinase activity associated with the avian sarcoma virus src gene product. Proc Natl Acad Sci USA. 1978;75:2021–2024. doi: 10.1073/pnas.75.4.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dustin M L, Springer T A. Role of lymphocyte adhesion receptors in transient interactions and cell locomotion. Annu Rev Immunol. 1991;9:27–66. doi: 10.1146/annurev.iy.09.040191.000331. [DOI] [PubMed] [Google Scholar]
- 11.Gomez Yafal A, Kaplan G, Racaniello V R, Hogle J M. Characterization of poliovirus conformational alteration mediated by soluble cell receptors. Virology. 1993;197:501–505. doi: 10.1006/viro.1993.1621. [DOI] [PubMed] [Google Scholar]
- 12.Greber U F, Webster P, Weber J, Helenius A. The role of the adenovirus protease in virus entry into cells. EMBO J. 1996;15:1766–1777. [PMC free article] [PubMed] [Google Scholar]
- 13.Greber U F, Willetts M, Webster P, Helenius A. Stepwise dismantling of adenovirus-2 during entry into cells. Cell. 1993;75:477–486. doi: 10.1016/0092-8674(93)90382-z. [DOI] [PubMed] [Google Scholar]
- 14.Greve J M, Davis G, Meyer A M, Forte C P, Yost S C, Marlor C W, Kamarck M E, McClelland A. The major human rhinovirus receptor is ICAM-1. Cell. 1989;56:839–847. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 15.Greve J M, Forte C P, Marlor C W, Meyer A M, Hooverlitty H, Wunderlich D, Mcclelland A. Mechanisms of receptor-mediated rhinovirus neutralization defined by two soluble forms of ICAM-1. J Virol. 1991;65:6015–6023. doi: 10.1128/jvi.65.11.6015-6023.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gruenberger M, Pevear D, Diana G D, Kuechler E, Blaas D. Stabilization of human rhinovirus serotype-2 against pH-induced conformational change by antiviral compounds. J Gen Virol. 1991;72:431–433. doi: 10.1099/0022-1317-72-2-431. [DOI] [PubMed] [Google Scholar]
- 17.Gruenberger M, Wandl R, Nimpf J, Hiesberger T, Schneider W J, Kuechler E, Blaas D. Avian homologs of the mammalian low-density lipoprotein receptor family bind minor receptor group human rhinovirus. J Virol. 1995;69:7244–7247. doi: 10.1128/jvi.69.11.7244-7247.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grunert H P, Wolf K U, Langner K D, Sawitzky D, Habermehl K O, Zeichhardt H. Internalization of human rhinovirus 14 into HeLa and ICAM-1-transfected BHK cells. Med Microbiol Immunol. 1997;186:1–9. doi: 10.1007/s004300050039. [DOI] [PubMed] [Google Scholar]
- 19.Hewat, E. A., T. C. Marlovits, and D. Blaas. Structure of a neutralising antibody bound monovalently to human rhinovirus 2. Submitted for publication. [DOI] [PMC free article] [PubMed]
- 20.Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, Kuechler E, Blaas D. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc Natl Acad Sci USA. 1994;91:1839–1842. doi: 10.1073/pnas.91.5.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoover-Litty H, Greve J M. Formation of rhinovirus-soluble ICAM-1 complexes and conformational changes in the virion. J Virol. 1993;67:390–397. doi: 10.1128/jvi.67.1.390-397.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan G, Freistadt M S, Racaniello V R. Neutralization of poliovirus by cell receptors expressed in insect cells. J Virol. 1990;64:4697–4702. doi: 10.1128/jvi.64.10.4697-4702.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan G, Totsuka A, Thompson P, Akatsuka T, Moritsugu Y, Feinstone S M. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 1996;15:4282–4296. [PMC free article] [PubMed] [Google Scholar]
- 24.Karnauchow T M, Tolson D L, Harrison B A, Altman E, Lublin D M, Dimock K. The HeLa cell receptor for enterovirus 70 is decay-accelerating factor (CD55) J Virol. 1996;70:5143–5152. doi: 10.1128/jvi.70.8.5143-5152.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korant B D, Lonberg Holm K, Noble J, Stasny J T. Naturally occurring and artificially produced components of three rhinoviruses. Virology. 1972;48:71–86. doi: 10.1016/0042-6822(72)90115-8. [DOI] [PubMed] [Google Scholar]
- 26.Kronenberger P, Vrijsen R, Boeye A. Compartmentalization of subviral particles during poliovirus eclipse in HeLa cells. J Gen Virol. 1992;73:1739–1744. doi: 10.1099/0022-1317-73-7-1739. [DOI] [PubMed] [Google Scholar]
- 27.Kronenberger P, Vrijsen R, Geerts A, Boeye A. Internalization of intact poliovirus by HeLa cells as shown by subcellular fractionation in isoosmotic nycodenz gradients. J Gen Virol. 1992;73:597–605. doi: 10.1099/0022-1317-73-3-597. [DOI] [PubMed] [Google Scholar]
- 28.Lee W M, Monroe S S, Rueckert R R. Role of maturation cleavage in infectivity of picornaviruses: activation of an infectosome. J Virol. 1993;67:2110–2122. doi: 10.1128/jvi.67.4.2110-2122.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonberg Holm K, Gosser L B, Shimshick E J. Interaction of liposomes with subviral particles of poliovirus type 2 and rhinovirus type 2. J Virol. 1976;19:746–749. doi: 10.1128/jvi.19.2.746-749.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lonberg Holm K, Whiteley N M. Physical and metabolic requirements for early interaction of poliovirus and human rhinovirus with HeLa cells. J Virol. 1976;19:857–870. doi: 10.1128/jvi.19.3.857-870.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lonberg-Holm K, Korant B D. Early interaction of rhinoviruses with host cells. J Virol. 1972;9:29–40. doi: 10.1128/jvi.9.1.29-40.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marsh M, Schmid S, Kern H, Harms E, Male P, Mellman I, Helenius A. Rapid analytical and preparative isolation of functional endosomes by free flow electrophoresis. J Cell Biol. 1987;104:875–886. doi: 10.1083/jcb.104.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy R F. Analysis and isolation of endocytic vesicles by flow cytometry and sorting: demonstration of three kinetically distinct compartments involved in fluid-phase endocytosis. Proc Natl Acad Sci USA. 1985;82:8523–8526. doi: 10.1073/pnas.82.24.8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neubauer C, Frasel L, Kuechler E, Blaas D. Mechanism of entry of human rhinovirus 2 into HeLa cells. Virology. 1987;158:255–258. doi: 10.1016/0042-6822(87)90264-9. [DOI] [PubMed] [Google Scholar]
- 35.Parton R G. Caveolae and caveolins. Curr Opin Cell Biol. 1996;8:542–548. doi: 10.1016/s0955-0674(96)80033-0. [DOI] [PubMed] [Google Scholar]
- 36.Pekarthy J M, Short J, Lansing A I, Lieberman I. Function and control of liver alkaline phosphatase. J Biol Chem. 1972;247:1767–1774. [PubMed] [Google Scholar]
- 37.Perez L, Carrasco L. Entry of poliovirus into cells does not require a low-pH step. J Virol. 1993;67:4543–4548. doi: 10.1128/jvi.67.8.4543-4548.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prchla, E., N. Bayer, M. Schwab, D. Blaas, and R. Fuchs. Uncoating of human rhinovirus serotype 14 (HRV14) in vivo: role of viral receptor and low pH. Submitted for publication.
- 39.Prchla E, Kuechler E, Blaas D, Fuchs R. Uncoating of human rhinovirus serotype 2 from late endosomes. J Virol. 1994;68:3713–3723. doi: 10.1128/jvi.68.6.3713-3723.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prchla E, Plank C, Wagner E, Blaas D, Fuchs R. Virus-mediated release of endosomal content in vitro: different behavior of adenovirus and rhinovirus serotype 2. J Cell Biol. 1995;131:111–123. doi: 10.1083/jcb.131.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmid S L, Fuchs R, Male P, Mellman I. Two distinct endosome subpopulations involved in membrane recycling and transport to lysosomes. Cell. 1988;52:73–83. doi: 10.1016/0092-8674(88)90532-6. [DOI] [PubMed] [Google Scholar]
- 42.Skern T, Sommergruber W, Blaas D, Pieler C, Kuechler E. Relationship of human rhinovirus strain 2 and poliovirus as indicated by comparison of the polymerase gene regions. Virology. 1984;136:125–132. doi: 10.1016/0042-6822(84)90253-8. [DOI] [PubMed] [Google Scholar]
- 43.Smith T J, Olson N H, Cheng R H, Liu H, Chase E S, Lee W M, Leippe D M, Mosser A G, Rueckert R R, Baker T S. Structure of human rhinovirus complexed with Fab fragments from a neutralizing antibody. J Virol. 1993;67:1148–1158. doi: 10.1128/jvi.67.3.1148-1158.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Staunton D E, Gaur A, Chan P Y, Springer T A. Internalization of a major group human rhinovirus does not require cytoplasmic or transmembrane domains of ICAM-1. J Immunol. 1992;148:3271–3274. [PubMed] [Google Scholar]
- 45.Staunton D E, Merluzzi V J, Rothlein R, Barton R, Marlin S D, Springer T A. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell. 1989;56:849–853. doi: 10.1016/0092-8674(89)90689-2. [DOI] [PubMed] [Google Scholar]
- 46.Thomas D C, Conant R M, Hamparian V V. Rhinovirus replication in suspension cultures of HeLa cells. Proc Soc Exp Biol Med. 1970;133:62–65. doi: 10.3181/00379727-133-34407. [DOI] [PubMed] [Google Scholar]
- 47.Tomassini E, Graham T, DeWitt C, Lineberger D, Rodkey J, Colonno R. cDNA cloning reveals that the major group rhinovirus receptor on HeLa cells is intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1989;86:4907–4911. doi: 10.1073/pnas.86.13.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uncapher C R, Dewitt C M, Colonno R J. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology. 1991;180:814–817. doi: 10.1016/0042-6822(91)90098-v. [DOI] [PubMed] [Google Scholar]
- 49.Ward T, Pipkin P A, Clarkson N A, Stone D M, Minor P D, Almond J W. Decay-accelerating factor CD55 is identified as the receptor for echovirus 7 using CELICS, a rapid immune-focal cloning method. EMBO J. 1994;13:5070–5074. doi: 10.1002/j.1460-2075.1994.tb06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeichhardt H, Wetz K, Willingmann P, Habermehl K O. Entry of poliovirus type 1 and mouse Elberfeld (ME) virus into HEp-2 cells: receptor-mediated endocytosis and endosomal or lysosomal uncoating. J Gen Virol. 1985;66:483–492. doi: 10.1099/0022-1317-66-3-483. [DOI] [PubMed] [Google Scholar]