

Abstract
95-19 and US11cl19.3 are BHK(TK−)-derived cell lines that are highly resistant to postattachment entry of herpes simplex virus type 1 (HSV-1) and HSV-2 but not to later steps in single-step replication. The resistance properties of these two cell types are not identical. US11cl19.3 cells are fully susceptible to pseudorabies virus (PRV), as shown by single-step growth experiments, whereas 95-19 cells are resistant to entry of free PRV but not to entry by cell-cell spread. We have tested the ability of HVEM to overcome the block to infection in both cell lines following transient and stable transfection. HVEM was able to mediate entry of free HSV-1 into both cell lines, as shown by an increase in the number of β-galactosidase-expressing cells in cultures transiently transfected with an HVEM expression plasmid and infected with lacZ-expressing HSV-1. In stably transfected 95-19 cells, HVEM enhanced infection by free HSV-1, as shown by an increase in the number of infectious centers obtained following infection. In both cell types, HVEM strongly enhanced entry of HSV-1 and HSV-2 by cell-cell spread, suggesting that HVEM can function as an entry mediator both in entry of free virus and in entry by cell-cell spread.
Herpes simplex virus (HSV) entry requires the coordinated function of multiple virus envelope proteins and at least two host cell factors. For most cell types studied, the entry process begins with a low-affinity attachment to the cell surface, mediated by an interaction between cell-surface heparan sulfate proteoglycan and virion glycoprotein gB, gC, or both (10, 11, 27, 32). Following this initial, low-affinity attachment, there may be secondary, higher-affinity binding events that lead to viral fusion. One such interaction is thought to be mediated by the virion glycoprotein gD. gD-lacking (gD−) viruses attach to but fail to penetrate susceptible cells, and anti-gD antibodies block entry following attachment but before membrane fusion (6, 7, 12, 16). Soluble gD can block HSV infection, and it binds to a saturable cell surface molecule, suggesting that gD makes an essential interaction with a host cell receptor (14). Fusion of the virus envelope with the cell surface absolutely requires at least four virion glycoproteins, including gD (gB, gD, gH, and gL), and may involve other, unidentified cellular factors (2, 5–8, 12, 13, 16, 17, 24, 25).
One host cell surface molecule, HVEM, that can mediate postattachment entry of HSV into CHO cells and is a member of the tumor necrosis factor-nerve growth factor receptor family (19) has been identified. HVEM is expressed in various tissues, including liver, lung, and kidney, and lymphocyte-rich tissue like spleen, and in peripheral blood leukocytes (18, 19). Transient transfection with HVEM can cause activation of transcriptional regulators including nuclear factor κB, Jun N-terminal kinase, and Jun containing transcription factor AP-1, suggesting that HVEM is associated with signal transduction pathways that activate the immune response (18).
In addition to its physiological role, HVEM mediates the fusion of viral and cellular membranes, presumably through interactions with one or more of the virion envelope glycoproteins essential for entry (gB, gD, gH, and gL). HVEM may not be a universal mediator of HSV entry, since anti-HVEM serum only weakly blocks HSV type 1 (HSV-1) infection in some susceptible cells (19). Two lines of evidence suggest that HVEM mediates entry by way of interaction with virion gD and is, in fact, a receptor for gD. First, soluble forms of HVEM and HSV-1 gD can form a specific complex in vitro (31). Second, the ability of HSV to use HVEM as a mediator is at least partly determined by mutations in the gD gene (19). Furthermore, the ability of specific gD sequences to bind to HVEM is correlated with the ability of viruses encoding those gD sequences to use HVEM as a mediator of entry (31).
We have characterized two cell lines which are highly resistant to HSV infection at a point postattachment but at or prior to penetration (22, 23). US11cl19.3 is a clonal cell line derived from BHK(TK−) cells. These cells are stably transfected with genes encoding HSV-1(F) proteins ICP4 and US11. The block to entry in these cells is exercised at a step following attachment but before fusion. This step is apparently mediated by gD, since viruses carrying mutations in the gD gene can at least partially overcome the block to infection. These cells are also partially susceptible to viruses selected for the ability to grow on gD-expressing cells (1, 3). These cells may be deficient in a receptor for gD. The second cell line, named 95-19, is a spontaneously resistant clonal cell line derived from BHK(TK−) cells. These cells are also resistant to entry of free HSV at a step after attachment, as well as to entry of HSV by cell-cell spread (23). These cells differ from US11cl19.3 cells in that they are resistant to mutant HSVs that can enter US11cl19.3 cells and gD-expressing cells. They are also resistant to the closely related alphaherpesvirus pseudorabies virus (PRV). Resistance in both cell lines can be overcome by exposure to the fusogen polyethylene glycol, and in cells so infected, normal viral replication ensues, suggesting that the only significant block to replication occurs at entry.
Though US11cl19.3 cells express both the ICP4 and US11 regulatory proteins of HSV-1(F), it seems likely for several reasons that their resistance to entry is a spontaneously arising property unrelated to their expression of viral genes. US11cl19.3 cells are derived from an ICP4-expressing BHK(TK−) cell line that is susceptible to infection, demonstrating that ICP4 expression does not lead to resistance. We have constructed other cell lines expressing US11 at high levels in a variety of cell line backgrounds and found no evidence for resistance to HSV entry (9a), suggesting that US11 expression does not suffice to induce resistance to entry. Finally, the isolation of cell lines like 95-19, having mechanisms of resistance to entry fundamentally similar to those of spontaneously arising clones from the BHK(TK−) cell line (22), suggests that US11 expression is unnecessary for the resistance phenotype.
Two types of HSV entry—entry of free virus and entry by cell-cell spread—can be distinguished by differences in the viral and cellular factors required. Cell surface heparan sulfate is required for attachment of free virus but may be dispensable for cell-cell spread (9). The virion glycoproteins gE and gI, which form a complex, are dispensable for entry of free virus but are essential for efficient cell-cell spread (4). The role of gD in cell-cell spread is uncertain but clearly species dependent. In HSV and PRV, gD or gp50 is required for entry of free virus, but where HSV gD is essential for cell-cell spread, PRV gp50 is not (16, 20, 21). The human alphaherpesvirus varicella-zoster virus has no gD homolog and spreads from cell to cell. Finally, wild-type bovine herpesvirus requires gD for cell-cell spread, but a point mutation in gH can eliminate the gD requirement altogether (26). Though HSV gD is essential for efficient cell-cell spread, it is unclear whether it plays the same role in cell-cell spread as in entry of free virus.
In this publication we further characterize the US11cl19.3 and 95-19 cell lines and we demonstrate the ability of HVEM to partially overcome their resistance both to entry of free virus and to entry by cell-cell spread.
MATERIALS AND METHODS
Cells and viruses.
US11cl19.3 cells were derived by limiting dilution cloning of cells from the US11cl19 population described in reference 22. Their properties of resistance to HSV-1 are the same as that for the parent population, except that the resistance phenotype is stable over at least 20 serial passages. BHK(TK−), 95-19, and US11cl19.3 cells were maintained in Dulbecco modified Eagle medium (DMEM) (high glucose) supplemented with 5% fetal bovine serum. Vero cells were maintained in DMEM (high glucose) supplemented with 5% newborn calf serum. Wild-type virus strains used were the Kaplan strain of PRV, HSV-1(F), HSV-2(G), and HSV-2(333). Recombinant HSV-1(17)(dUTPase/LAT) (gift of Ed Wagner, University of California, Irvine) contains the Escherichia coli β-galactosidase gene under control of the viral dUTPase promoter in place of both copies of the LAT genes and has been previously described (28).
Measurement of virus replication in single-step growth.
Cultures of BHK(TK−) and US11cl19.3 cells were exposed to virus at a multiplicity of infection of 10 for 90 min at 4°C to allow attachment of virus. The inoculum was then aspirated, and cells were washed three times in 37°C phosphate-buffered saline (PBS) and placed at 37°C under growth medium. This was designated time zero of infection. After incubation for 90 min to allow virus entry and initiation of infection, cells were washed once with citrate buffer (50 mM sodium citrate–4 mM KCl, adjusted to pH 3.0 with HCl) and then incubated in a second wash of citrate buffer for 1 min to inactivate most of the residual virus. Monolayers were then washed twice in PBS and incubated in growth medium for the remainder of the infection period. At various times, cultures were frozen at −80°C and then thawed to lyse the cells, diluted 1:1 with autoclaved skim milk, and sonicated with a Fisher Sonic Dismembrator at power level 0 for 20 s to fully disrupt the cells and release virus particles. The virus stocks were then titrated on Vero cell monolayers, and plaques were counted after immunostaining (HSV-1 and HSV-2) or staining with amido black (PRV).
Plaque assays.
Cultures of BHK(TK−) or 95-19 cells that were 50% confluent were exposed to virus at 37°C for 90 min and then incubated in growth medium containing 0.01% pooled human immunoglobulin (Gammar; Armour Pharmaceutical) for 72 h to permit virus plaque formation. Cultures were then washed with PBS and fixed in methanol at −20°C for 20 min. Virus plaques were detected by immunoassay with monoclonal antibody directed against HSV-1 glycoprotein D (Goodman Cancer Research Labs) as previously described (22).
Infectious-center assay.
Duplicate cultures of BHK(TK−) and 95-19 or US11cl19.3 cells in six-well cultures (10 cm2) at 50% of confluence were exposed to virus at various multiplicities of infection at 37°C. The time of addition of virus was designated time zero of the infection. After 90 min of incubation to allow initiation of infection, cells were washed once with PBS and once more rapidly with citrate buffer (50 mM sodium citrate–4 mM KCl, adjusted to pH 3.0 with HCl) and then incubated in a second wash of citrate buffer for 1 min to inactivate most of the residual virus. Monolayers were then washed twice in PBS to remove the low pH buffer and placed in growth medium containing pooled human immunoglobulin to neutralize any extracellular virus. At 4 h of infection, one culture from each set of duplicates was treated with trypsin to detach the cells and one-half of the cell suspension was seeded into a six-well culture of Vero cells cultured in growth medium containing 0.1% pooled human immunoglobulin (Gammar; Armour Pharmaceutical). All cultures were then incubated at 37°C until 48 h after infection. Cultures were then fixed and plaques were visualized by immunostaining as previously described (22).
Construction of stably transfected cell lines.
US11cl19.3/pcDNA cells were generated by transfecting 10-cm2 cultures of US11cl19.3 cells with 1.5 μg of RSV5.hyg and 12 μg of pcDNA3 using 15 μl of Lipofectamine (Gibco/BRL) in DMEM without serum or antibiotics. Two days after transfection, cells were seeded into medium containing 200 μg of hygromycin B (Sigma) per ml. After passage for several weeks in selective medium, the cell population was used for infectious center assays. US11cl19.3/BEC cells were generated in the same way, except that the transfecting plasmids were RSV5.hyg and pBEC10 (gift of P. G. Spear). 95-19/pcDNA and 95-19/BEC cells were generated by transfection of 10-cm2 cultures of 95-19 cells with 2.5 μg of either pcDNA3 or pBEC10 using 7.5 μl of Lipofectamine in DMEM without serum or antibiotics. Two days after transfection, cells were seeded into medium containing 400 μg of Geneticin (Gibco/BRL) per ml. Cells were passed for several weeks in selective medium and then used for infectious-center assays.
Assay for HVEM expression.
Cells transfected with pBEC10 or vector control were washed twice with PBS containing 0.5 mM EDTA and then incubated in a third wash until the cells detached from the substrate. Detached cells were pelleted at low speed in a clinical centrifuge and then fixed by resuspension in PBS containing 1.9% formaldehyde and incubation for 10 min. Fixed cells were washed by three cycles of pelleting and resuspension in PBS. The washed cell pellet was resuspended in PBS containing 1% bovine serum albumin and a 1:600 dilution of anti-HVEM antiserum R133 (gift of Gary Cohen and Roselyn Eisenberg) (31), and antibody was allowed to bind for 1 h at room temperature. Cells were then washed with another three cycles of pelleting and resuspension in PBS. The washed cell pellet was resuspended in PBS containing 10% normal goat serum (Sigma Chemical Co.) and a 1:200 dilution of fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit immunoglobulin G (Life Technologies), and the secondary antibody was allowed to bind for 1 h in the dark at room temperature. Cells were then washed three times with PBS and analyzed with a fluorescence-activated cell sorter (FACS) (FACScan; Becton Dickinson).
RESULTS
Infection of US11cl19.3 cells with PRV.
PRV and HSV can compete with each other for binding to susceptible cell surfaces; this ability to compete is dependent on the presence of gD (15). Subramanian et al., however, have shown that ST cells, which are resistant to HSV, are fully susceptible to PRV, suggesting that PRV can use a cellular receptor that HSV can not use (29, 30). To determine whether US11cl19.3 cells are deficient in cellular factors required for entry of both HSV and PRV and whether these cells are also resistant to HSV-2, they were tested for the ability to support PRV and HSV-2(G) infection in a single-step growth assay, as described in Materials and Methods. Monolayer cultures of BHK(TK−) and US11cl19.3 cells were infected with HSV-2 or PRV at a multiplicity of 10, and virus yield was determined at various times after infection (Fig. 1). Replication of HSV-2(G) and PRV on BHK(TK−) cells showed typical kinetics, with an increase in PFU of more than 3 log orders of magnitude over the residual virus (i.e., virus present at the earliest time point). On US11cl19.3 cells, in contrast, HSV-2(G) showed no indication of replication, indicating that this cell line is highly resistant to HSV-2 in addition to HSV-1. The viral titer in the infected cultures dropped continuously with time after infection, probably reflecting the loss of residual infecting virus. In contrast to the results with HSV, PRV replicated nearly as efficiently on US11cl19.3 cells as on BHK(TK−) cells, indicating that these cells are not significantly resistant to PRV entry.
FIG. 1.

Replication of HSV-2(G) and PRV on BHK(TK−) and US11cl19.3 cells. Shown are plots of the logarithms of PFU of virus accumulated in cultures of BHK(TK−) cells or US11cl19.3 cells versus time after infection. Cultures were infected, citrate treated, harvested, and titrated on Vero cells, as described in Materials and Methods. (A) The infecting virus is HSV-2, strain G. (B) The infecting virus is the PRV Kaplan strain. Data points represent means of three independent experiments. Error bars indicate sample ranges.
HVEM expression makes both US11cl19.3 and 95-19 cells susceptible to initial infection by HSV.
Monolayer cultures of US11cl19.3 and 95-19 cells were transfected with the HVEM-expressing plasmid pBEC10 or the expression vector pcDNA3, as described in Materials and Methods. Two days after transfection, cultures were infected with 10 PFU of HSV-1(17)(dUTPase/LAT) per cell for 4 h and then fixed and stained for β-galactosidase activity (Fig. 2). Cells transfected with vector pcDNA3 (Fig. 2A and C) were resistant to HSV infection and showed very few infected cells. The fields shown in Fig. 2A and C are typical and show no infected cells, but several hundred isolated infected cells were observed in examination of the entire culture. Transfection of either cell line with pBEC10 (Fig. 2B and D) greatly increased the number of infected cells observed. The frequency of infected cells was similar to the frequency of transfection as assessed in a parallel transfection with a marker plasmid expressing β-galactosidase (not shown), suggesting that the block to entry of free virus can largely be overcome by expression of HVEM.
FIG. 2.
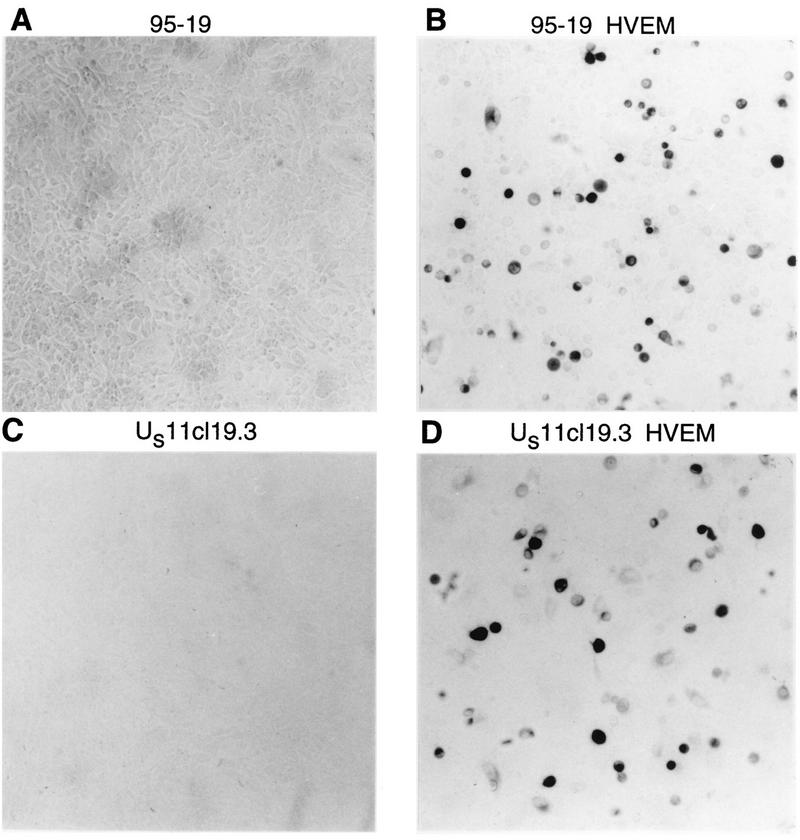
Susceptibility to infection of HVEM-transfected cells. Shown are photographic images of monolayers of 95-19 cells (A and B) and US11cl19.3 cells (C and D) transiently transfected with the expression vector pcDNA3 (A and C) or with pBEC10 (B and D), which expresses HVEM under the control of the human cytomegalovirus major immediate-early promoter, superinfected with HSV-1(17)(dUTPase/LAT), and stained for β-galactosidase activity.
HVEM expression renders both US11cl19.3 and 95-19 cells susceptible to cell-cell spread of HSV-1 and HSV-2.
We have previously shown that 95-19 cells are resistant to entry by cell-cell spread. An infectious-center assay was used to determine whether US11cl19.3 cells show a similar block and whether the block can be overcome in either cell type by expression of HVEM. Resistant cells were transfected with pBEC10 or pcDNA3 and then grown in the presence of selective agent to select for those cells that had stably integrated the plasmid. The 95-19 and US11cl19.3 cell populations selected for stable integration of pcDNA3 were designated 95-19/pcDNA and US11cl19.3/pcDNA, respectively. The populations selected for stable integration of pBEC10 were designated 95-19/BEC and US11cl19.3/BEC. Infectious-center assays were then performed on the stably transfected cell lines and on susceptible BHK(TK−) cells. The rationale for this assay is depicted in Fig. 3. Duplicate monolayer six-well cultures (10 cm2) of BHK(TK−) cells and the transfected cell lines were exposed to various amounts of virus to allow entry and initiation of infection. After removal of residual virus, infection was allowed to proceed at 37°C in the presence of neutralizing anti-HSV antibodies. At 4 h after infection, the cells from one culture from each set of duplicates were detached by trypsinization and seeded into a monolayer of susceptible Vero cells. All cultures were then incubated in the presence of neutralizing antibody to prevent infection by free virus. At 48 h after infection, all of the cultures were fixed and immunostained to allow visualization of plaques. Plaques developing on the susceptible Vero cells indicated the presence of infectious centers [i.e., BHK(TK−) or 95-19 cells that had become infected and supported virus replication and egress to a degree that permitted infection of an adjacent susceptible cell by cell-cell spread]. Plaques forming on the test cells indicated the spread of virus from cell to cell. If test cells are fully susceptible to infection by cell-cell spread, then there should be no difference between the number of infectious centers evident on Vero cells and the number of plaques on the test cells themselves, since each infectious center will be surrounded by susceptible cells (outcome 1 in Fig. 3). If test cells are resistant to infection by cell-cell spread, then the number of infectious centers will exceed the number of plaques formed on the test cells themselves, since infectious centers in the test cell monolayer will be surrounded by resistant cells. Efficiency of cell-cell spread was also assessed by examining plaque morphology after immunostaining. Representative results are presented in Tables 1 through 3. The results shown in Tables 1 through 3 are representative of three (Table 1) or two (Tables 2 and 3) independent experiments.
FIG. 3.
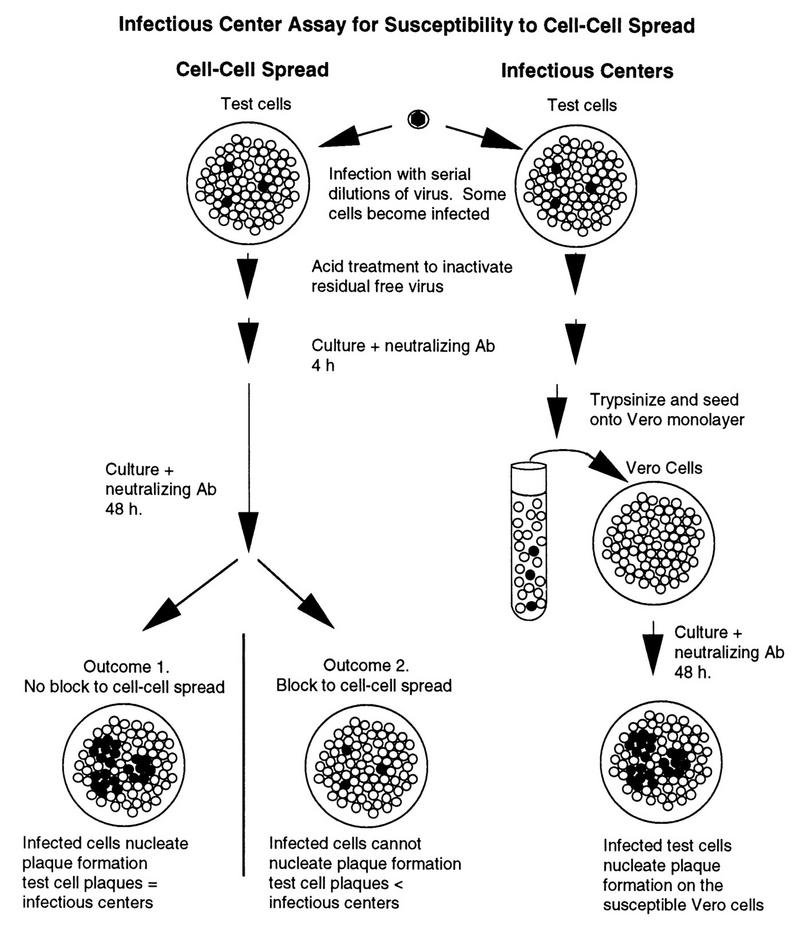
Infectious-center assay strategy. Open circles indicate uninfected cells; filled circles indicate infected cells. Ab, antibody.
TABLE 1.
Formation of HSV infectious centers and plaques on susceptible BHK(TK−) cells, resistant 95-19/pcDNA cells, and HVEM-expressing 95-19/BEC cells
| Test cells | Input PFUa | HSV-1(F)
|
HSV-2(G)
|
||
|---|---|---|---|---|---|
| Infectious centersb | Test cell plaquesc | Infectious centers | Test cell plaques | ||
| BHK(TK−) | 5 × 104 | TNTCd | TNTC | TNTC | TNTC |
| 5 × 103 | 211 | 238 | 186 | 264 | |
| 5 × 102 | 19 | 24 | 18 | 26 | |
| 95-19/pcDNA | 5 × 107 | TNTC | 2 | TNTC | 1 |
| 5 × 106 | 130 | 1 | 117 | 0 | |
| 5 × 105 | 15 | 0 | 6 | 0 | |
| 95-19/BEC | 5 × 107 | TNTC | TNTC | TNTC | TNTC |
| 5 × 106 | TNTC | TNTC | 167 | 135 | |
| 5 × 105 | 64 | 52 | 9 | 18 | |
| 5 × 104 | 7 | 4 | 1 | 1 | |
| 5 × 103 | 0 | 0 | 0 | 0 | |
| 5 × 102 | 0 | 0 | 0 | 0 | |
Determined by titration of virus stock on Vero cells.
Number of plaques obtained following seeding of infected test cells onto a Vero cell monolayer in the presence of neutralizing antibody.
Number of plaques observed on the infected test cell monolayer. Plaques are defined as two or more adjacent infected cells. Macroscopic plaques were rarely formed on 95-19 cells.
TNTC, too numerous to count.
TABLE 3.
Formation of PRV(Kaplan) infectious centers and plaques on susceptible BHK(TK−) cells, resistant 95-19 cells, and HVEM-expressing 95-19 cells
| Test cells | Input PFUa | Infectious centersb | Test cell plaquesc |
|---|---|---|---|
| BHK(TK−) | 5 × 103 | 39 | 50 |
| 5 × 102 | 4 | 10 | |
| 5 × 101 | 1 | 0 | |
| 95-19/pcDNA3 | 5 × 106 | TNTCd | TNTC |
| 5 × 105 | 39 | 51 | |
| 5 × 104 | 4 | 4 | |
| 95-19/HVEM | 5 × 106 | TNTC | TNTC |
| 5 × 105 | 27 | 31 | |
| 5 × 104 | 1 | 4 | |
| 5 × 103 | 0 | 0 |
Determined by titration of virus stock on Vero cells.
Number of plaques obtained following seeding of infected test cells onto a Vero cell monolayer.
Number of plaques observed on the infected test cell monolayer.
TNTC, too numerous to count.
TABLE 2.
Formation of HSV infectious centers and plaques on susceptible BHK(TK−) cells, resistant US11cl19.3/pcDNA cells, and HVEM-expressing US11cl19.3/BEC cells
| Test cells | Input PFUa | HSV-1(F)
|
HSV-2(G)
|
||
|---|---|---|---|---|---|
| Infectious centersb | Test cell plaquesc | Infectious centers | Test cell plaques | ||
| BHK(TK−) | 5 × 104 | TNTCd | TNTC | TNTC | TNTC |
| 5 × 103 | 128 | 232 | 84 | 80 | |
| 5 × 102 | 14 | 22 | 16 | 28 | |
| US11cl19.3/pcDNA | 5 × 107 | TNTC | 0 | TNTC | 1 |
| 5 × 106 | 98 | 0 | 56 | 0 | |
| 5 × 105 | 6 | 0 | 2 | 0 | |
| US11cl19.3/BEC | 5 × 107 | TNTC | 32 | TNTC | 76 |
| 5 × 106 | 58 | 2 | 36 | 10 | |
| 5 × 105 | 6 | 0 | 0 | 0 | |
| 5 × 104 | 0 | 0 | 0 | 0 | |
Determined by titration of virus stock on Vero cells.
Number of plaques obtained following seeding of infected test cells onto a Vero cell monolayer in the presence of neutralizing antibody.
Number of plaques observed on the infected test cell monolayer. Plaques are defined as two or more adjacent infected cells. Macroscopic plaques were rarely formed on US11cl19.3 cells.
TNTC, too numerous to count.
In 95-19 cells (Table 1), stable transfection with the HVEM-expressing plasmid pBEC10 rendered the population more susceptible to infection by free HSV-1 of either strain, as shown by an increase in the number of infectious centers. In the experiment shown, the HVEM-transfected cell population had about fourfold more HSV-1(F)-induced infectious centers than the control population. A slightly greater increase was observed with HSV-1(17)(dUTPase/LAT) (not shown). No increase in HSV-2-induced infectious centers was observed. For all HSV strains tested, the HVEM-expressing cell population was rendered much more susceptible to virus entry by cell-cell spread. For each virus strain tested, the number of test cell plaques observed was increased to be roughly equivalent to the number of infectious centers. The block to cell-cell spread was not completely overcome in these cells, however. Plaques formed on 95-19 cells were generally microscopic and composed of relatively few infected cells (Fig. 4B). Cells transfected with pBEC10 formed many more plaques than untransfected or vector-transfected 95-19 cells, but the plaque size was not substantially increased (Fig. 4C).
FIG. 4.

Plaque sizes on BHK(TK−), 95-19, and 95-19/BEC cells. (A to C) Photographic images of representative plaques formed by HSV-1(F) on monolayers of BHK(TK−) (A), 95-19 (B), and 95-19/BEC (C) cells immunostained with antibody directed against gD, as described in Materials and Methods. (D) Plaque formed by PRV on 95-19 cells stained with amido black.
In US11cl19.3 cells (Table 2), in contrast to what was observed for 95-19 cells, no more infectious centers were observed on pBEC10-transfected cells than on cells transfected with the vector control with any of the viral strains tested. However, the US11cl19.3/BEC cells were rendered much more susceptible to cell-cell spread than the vector-transfected controls, demonstrating that in these cells also, HVEM can mediate cell-cell spread. The HVEM-dependent recovery of cell-cell spread was not complete, since the number of test cell plaques was lower than the number of infectious centers for all strains tested and since the plaques formed on US11cl19.3/BEC cells were composed of very few cells (not shown).
The low enhancement of infectious-center formation in 95-19 cells and the absence of enhancement of infectious-center formation on US11cl19.3 cells were somewhat surprising given the enhancement of susceptibility in transient transfection. Since these experiments were done with stably transfected cell populations and not with clonal lines, this might have been due to a low frequency of HVEM expression in the population and to variable expression among the HVEM-expressing members of the population. To assess this, FACS analysis was performed on fixed, nonpermeabilized cells with anti-HVEM polyclonal antiserum and FITC-conjugated secondary antibody (Fig. 5). Both stably transfected cell populations (shaded regions of the histograms in Fig. 5) contained a minority subpopulation that expressed HVEM on the surface and only a relatively small number of cells that showed high-level HVEM expression (i.e., greater than 1 log10 unit greater than the mean background). This may account for the small plaque size observed on the stably transfected cells, since any infected cell will be surrounded by very few cells expressing detectable HVEM.
FIG. 5.
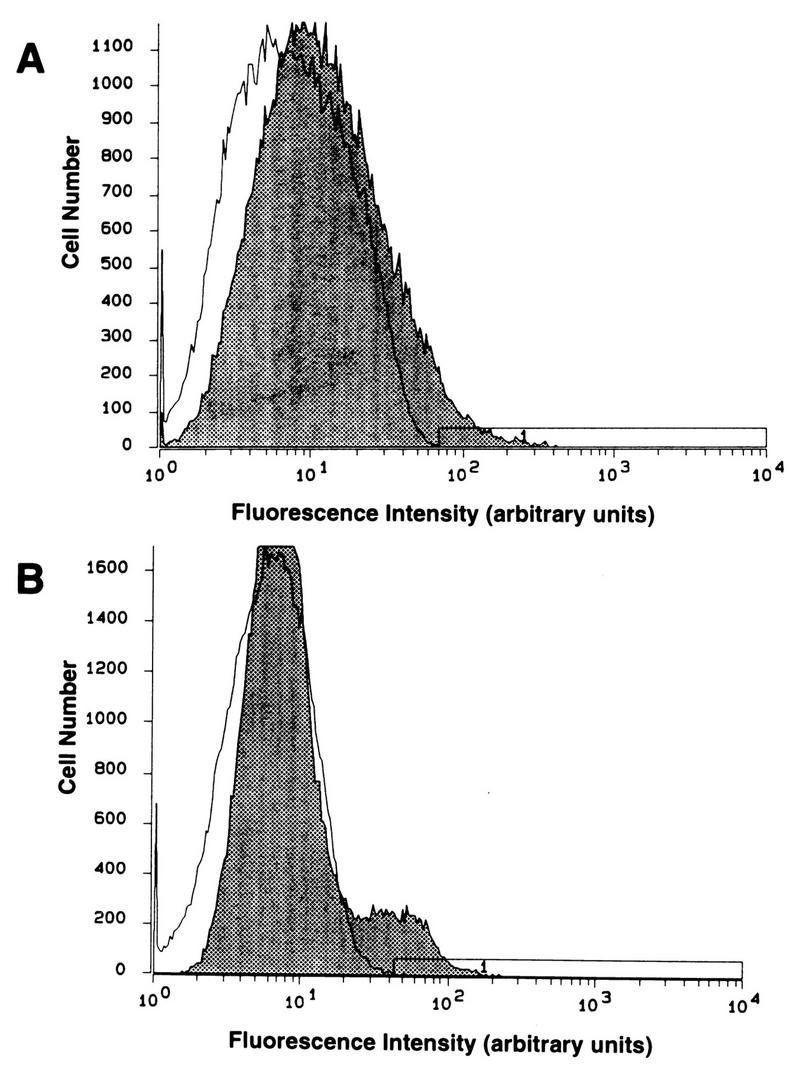
FACS analysis of HVEM expression in stably transfected cell populations. Untransfected (open curves) and pBEC10-transfected (shaded curves) 95-19 cells (A) or US11cl19.3 cells (B) were reacted with anti-HVEM antibody and FITC-conjugated secondary antibody, as described in Materials and Methods.
PRV, unlike HSV, does not require its gD homolog for efficient cell-cell spread. If 95-19 cells are indeed missing a cell surface factor that interacts with PRV gD, then there should be no resistance to PRV cell-cell spread. Infectious-center assays were performed to test for resistance to entry of free virus and cell-cell spread (Table 3). 95-19 cells yielded roughly 100-fold fewer infectious centers than did BHK(TK−) cells, confirming that these cells are resistant to infection by free PRV. Stable transfection of an HVEM-expressing plasmid did not increase susceptibility to PRV. 95-19 cells, however, do support efficient cell-cell spread of PRV, since the number of test cell plaques was equivalent to the number of infectious centers and since PRV formed large plaques on 95-19 cells (Fig. 4D).
DISCUSSION
HVEM mediates entry of free virus into both 95-19 and US11cl19.3 cells.
Two lines of evidence suggest that HVEM renders either 95-19 cells or US11cl19.3 cells susceptible to infection by free HSV. (i) Transient transfection of an HVEM-expressing plasmid into either line increases the number of β-galactosidase-positive cells following superinfection with β-galactosidase-expressing HSV-1. (ii) Stable transfection of an HVEM-expressing plasmid into 95-19 cells increases the number of infectious centers observed following infection with HSV-1. No such increase in the number of infectious centers was observed in stably transfected US11cl19.3 cells even though these cells were found to be more susceptible to virus entry by cell-cell spread. This difference between the two stably transfected cell types is apparently not due to differences in the level of HVEM expression or in the proportion of the population that expresses detectable HVEM, since FACS analysis of cell-surface HVEM shows that the cell populations used contain a similar fraction of expressing cells and that the level of HVEM expression is similar. Since neither cell line shows any significant block to infection following entry, this result suggests that HVEM may mediate entry more efficiently in some cell surface contexts than in others.
The observation that expression of a single molecule, HVEM, renders both 95-19 and US11cl19.3 cells susceptible to HSV infection suggests that the basic mechanism of resistance is the same in the two lines. Since the evidence to date suggests that HVEM can act as a receptor for gD (19, 31), the most economical hypothesis is that each of the cell lines is missing a receptor for HSV gD, for which HVEM can act as a substitute. Several aspects of our observations are consistent with this hypothesis. First, both cell lines are resistant to infection at a postattachment entry step, and studies with anti-gD neutralizing antibodies and gD− virus suggest that gD exercises its essential function at this stage of entry. Second, US11cl19.3 cells are at least partially susceptible to viruses that carry mutations in the gD gene (22). Third, 95-19 cells, though resistant to entry of free PRV, are not resistant to entry via cell-cell spread. The PRV homolog of gD, gp50, is required for entry of free virus, but is dispensable for cell-cell spread. Since gp50 is dispensable for cell-cell spread, it follows that its cellular interaction partner should be dispensable for this process also.
The hypothesis that the basic mechanism of resistance observed in US11cl19.3 cells and 95-19 cells is the same, however, must be reconciled with the different properties of resistance shown by these two cell lines. Both cell lines are highly resistant to infection with HSV-1 and HSV-2, but 95-19 cells are, in addition, resistant to infection by free PRV, by mutant viruses that can enter US11cl19.3 cells, and by mutant viruses that enter resistant gD-expressing cells (22, 23). Furthermore, the responses of the cell lines to HVEM expression are not identical. HVEM strongly increased the susceptibility to cell-cell spread in both lines, but similar levels of stable HVEM expression had different effects on the susceptibility to free virus. Stably expressed HVEM failed to function in entry of free virus in US11cl19.3 cells. Mediation of cell-cell spread may require less HVEM on the cell surface than does entry of free virus. It seems most likely that on susceptible BHK(TK−) cells there are several factors that can mediate efficient herpesvirus entry: (i) a factor that can mediate entry of wild-type HSV-1, HSV-2, and possibly PRV (this factor is likely missing on 95-19 and US11cl19.3 cells, accounting for their resistance to HSV); (ii) a factor, separate from that described in i, that can mediate entry of free PRV but not wild-type HSV (this factor is evidently present on US11cl19.3 cells but is absent or substantially less active on 95-19 cells); and (iii) a factor, separate from that described in i, that can mediate entry of viruses like R5000 (22) and U21 (1), which carry specific mutations in the gD coding sequence, but not wild-type HSV (this factor is evidently present on US11cl19.3 cells but not on 95-19 cells). It is possible that the factors described in ii and iii are the same. The lack of multiple entry functions in 95-19 cells might be explained in several ways: (i) multiple point or deletion mutations, each of which causes the loss of a different entry mediator; (ii) a single deletion which causes the loss of multiple entry mediators (this implies a clustered arrangement of such mediators in the genome); or (iii) a single point or deletion mutation that disrupts the function of a single molecule required for proper expression of multiple entry mediators.
On Vero cells, HSV and PRV can compete with each other for a saturable entry factor, and the ability of each virus to compete with the other is dependent on expression of gD (15), suggesting that the gD homologs recognize the same receptor and that HSV blocks all of the receptors available to PRV. The results presented here suggest that PRV and HSV do not recognize the same receptor or a completely overlapping set of receptors. The susceptibility of US11cl19.3 cells to PRV and their resistance to HSV suggest that these cells express an entry mediator that PRV can use and that HSV cannot use. Subramanian et al. observed a similar phenomenon in swine testis ST cells and suggested that the ability of PRV to enter ST cells was reflective of PRV tropism for swine cells (29). The properties of US11cl19.3 cells suggest that this property is shared by at least some non-swine cell types.
HVEM mediates entry of HSV by cell-cell spread into both 95-19 and US11cl19.3 cells.
95-19 and US11cl19.3 cells stably transfected with an HVEM-expressing plasmid are substantially less resistant to cell-cell spread of both HSV-1 and HSV-2, as shown by an increase in the number of plaques formed on these cells. In neither of the stably transfected cell lines is the block to cell-cell spread completely overcome, since plaque sizes are much smaller than those on permissive BHK(TK−) cells, and in US11cl19.3 cells the number of plaques, though increased, is smaller than the number of infectious centers. Again, we suspect that this reflects low levels of HVEM expression in the stably transfected cells. The failure of gD− virus to spread from cell to cell in permissive cells (16) shows that some function of gD is essential for this type of entry. The observation that HVEM overcomes a block to cell-cell spread in resistant cells suggests that gD mediates its function in cell-cell spread at least in part by binding to the same receptor used in entry of free virus. This further suggests that both types of entry may be susceptible to therapeutic manipulations that alter gD-receptor interaction.
ACKNOWLEDGMENTS
We are grateful to Ed Wagner for providing HSV-1(17)(dUTPase/LAT), to Gary Cohen and Roselyn Eisenberg for providing anti-HVEM antisera, to Pat Spear for providing pBEC10, and to Pat Spear and Betsy Herold for helpful discussions.
This work was supported by the University of Iowa and by research project grant RPG-97-070-01-VM from the American Cancer Society.
REFERENCES
- 1.Brandimarti R, Huang T, Roizman B, Campadelli-Fiume G. Mapping of herpes simplex virus 1 genes with mutations which overcome host restrictions to infection. Proc Natl Acad Sci USA. 1994;91:5406–5410. doi: 10.1073/pnas.91.12.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cai W, Gu B, Person S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J Virol. 1988;62:2596–2604. doi: 10.1128/jvi.62.8.2596-2604.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campadelli-Fiume G, Qi S, Avitabile E, Foà-Tomasi L, Brandimarti R, Roizman B. Glycoprotein D of herpes simplex virus encodes a domain which precludes penetration of cells expressing the glycoprotein by superinfecting herpes simplex virus. J Virol. 1990;64:6070–6079. doi: 10.1128/jvi.64.12.6070-6079.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dingwell K S, Doering L C, Johnson D C. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J Virol. 1995;69:7087–7098. doi: 10.1128/jvi.69.11.7087-7098.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Forrester A, Farrell H, Wilkinson G, Kaye J, Davis-Poynter N, Minson T. Construction and properties of a mutant herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J Virol. 1992;66:341–348. doi: 10.1128/jvi.66.1.341-348.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuller A O, Lee W-C. Herpes simplex virus type 1 entry through a cascade of virus-cell interactions requires different roles of gD and gH in penetration. J Virol. 1992;66:5002–5012. doi: 10.1128/jvi.66.8.5002-5012.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuller A O, Santos R, Spear P G. Potent neutralizing antibodies to gH of herpes simplex virus do not block attachment but prevent penetration of virus. J Virol. 1989;63:3535–3543. doi: 10.1128/jvi.63.8.3435-3443.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuller A O, Spear P G. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc Natl Acad Sci USA. 1987;84:5454–5458. doi: 10.1073/pnas.84.15.5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gruenheid S, Gatzke L, Meadows H, Tufaro F. Herpes simplex virus infection and propagation in a mouse L cell mutant lacking heparan sulfate proteoglycans. J Virol. 1993;67:93–100. doi: 10.1128/jvi.67.1.93-100.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9a.Heil, G., and R. Roller. Unpublished observations.
- 10.Herold B C, Visalli R J, Susmarski N, Brandt C R, Spear P G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J Gen Virol. 1994;75:1211–1222. doi: 10.1099/0022-1317-75-6-1211. [DOI] [PubMed] [Google Scholar]
- 11.Herold B C, WuDunn D, Soltys N, Spear P G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J Virol. 1991;65:1090–1098. doi: 10.1128/jvi.65.3.1090-1098.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Highlander S, Sutherland S L, Gage P J, Johnson D C, Levine M, Glorioso J C. Neutralizing monoclonal antibodies specific for herpes simplex virus glycoprotein D inhibit virus penetration. J Virol. 1987;61:3356–3364. doi: 10.1128/jvi.61.11.3356-3364.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hutchinson L, Graham F L, Cai W, Debroy C, Person S, Johnson D C. Herpes simplex virus (HSV) glycoproteins B and K inhibit cell fusion induced by HSV syncytial mutants. Virology. 1993;196:514–531. doi: 10.1006/viro.1993.1507. [DOI] [PubMed] [Google Scholar]
- 14.Johnson D C, Burke R L, Gregory T. Soluble forms of herpes simplex virus glycoprotein D bind to a limited number of cell surface receptors and inhibit virus entry into cells. J Virol. 1990;64:2569–2576. doi: 10.1128/jvi.64.6.2569-2576.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee W C, Fuller A O. Herpes simplex virus type 1 and pseudorabies virus bind to a common saturable receptor on Vero cells that is not heparan sulfate. J Virol. 1993;67:5088–5097. doi: 10.1128/jvi.67.9.5088-5097.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ligas M W, Johnson D C. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J Virol. 1988;62:1486–1494. doi: 10.1128/jvi.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacLean C A, Efstathiou S, Elliott M L, Jamieson F E, McGeoch D J. Investigation of herpes simplex virus type 1 genes encoding multiply inserted membrane proteins. J Gen Virol. 1991;72:897–906. doi: 10.1099/0022-1317-72-4-897. [DOI] [PubMed] [Google Scholar]
- 18.Marsters S A, Ayres T M, Skubatch M, Gray C L, Rothe M, Ashkenazi A. Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates transcription factors NF-κB and AP-1. J Biol Chem. 1997;272:14029–14032. doi: 10.1074/jbc.272.22.14029. [DOI] [PubMed] [Google Scholar]
- 19.Montgomery R I, Warner M S, Lum B J, Spear P G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 20.Peeters B, de Wind N, Hooisma M, Wagenaar F, Gielkens A, Moormann R. Pseudorabies virus envelope glycoproteins gp50 and gII are essential for virus penetration, but only gII is involved in membrane fusion. J Virol. 1992;66:894–905. doi: 10.1128/jvi.66.2.894-905.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rauh I, Mettenleiter T C. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J Virol. 1991;65:5348–5356. doi: 10.1128/jvi.65.10.5348-5356.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roller R J, Roizman B. A herpes simplex virus-1 US11-expressing cell line is resistant to herpes simplex virus infection at a step in viral entry mediated by glycoprotein D. J Virol. 1994;68:2830–2839. doi: 10.1128/jvi.68.5.2830-2839.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roller R J, Herold B C. Characterization of a BHK(TK−) cell clone resistant to postattachment entry by herpes simplex virus types 1 and 2. J Virol. 1997;71:5805–5813. doi: 10.1128/jvi.71.8.5805-5813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roop C, Hutchinson L, Johnson D C. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol. 1993;67:2285–2297. doi: 10.1128/jvi.67.4.2285-2297.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarmiento M, Haffey M, Spear P G. Membrane proteins specified by herpes simplex viruses. III. Role of glycoprotein VP7 (B2) in virion infectivity. J Virol. 1979;29:1149–1158. doi: 10.1128/jvi.29.3.1149-1158.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroder C, Linde G, Fehler F, Keil G M. From essential to beneficial: glycoprotein D loses importance for replication of bovine herpesvirus 1 in cell culture. J Virol. 1997;71:25–33. doi: 10.1128/jvi.71.1.25-33.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shieh M-T, WuDunn D, Montgomery R I, Esko J, Spear P G. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J Cell Biol. 1992;116:1273–1281. doi: 10.1083/jcb.116.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh J, Wagner E K. Herpes simplex virus recombination vectors designed to allow insertion of modified promoters into transcriptionally “neutral” segments of the viral genome. Virus Genes. 1995;10:127–136. doi: 10.1007/BF01702593. [DOI] [PubMed] [Google Scholar]
- 29.Subramanian G, LeBlanc R A, Wardley R C, Fuller A O. Defective entry of herpes simplex virus types 1 and 2 into porcine cells and lack of infection in infant pigs indicate species tropism. J Gen Virol. 1995;76:2375–2379. doi: 10.1099/0022-1317-76-9-2375. [DOI] [PubMed] [Google Scholar]
- 30.Subramanian G, McClain D S, Perez A, Fuller A O. Swine testis cells contain functional heparan sulfate but are defective in entry of herpes simplex virus. J Virol. 1994;68:5667–5676. doi: 10.1128/jvi.68.9.5667-5676.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitbeck J C, Peng C, Lou H, Xu R, Willis S H, Ponce De Leon M, Peng T, Nicola A V, Montgomery R I, Warner M S, Soulika A M, Spruce L A, Moore W T, Lambris J D, Spear P G, Cohen G H, Eisenberg R J. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J Virol. 1997;71:6083–6093. doi: 10.1128/jvi.71.8.6083-6093.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.WuDunn D, Spear P G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J Virol. 1989;63:52–58. doi: 10.1128/jvi.63.1.52-58.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]