

Abstract
The human parvovirus adeno-associated virus (AAV) infects a broad range of cell types, including human, nonhuman primate, canine, murine, and avian. Although little is known about the initial events of virus infection, AAV is currently being developed as a vector for human gene therapy. Using defined mutant CHO cell lines and standard biochemical assays, we demonstrate that heparan sulfate proteoglycans mediate both AAV attachment to and infection of target cells. Competition experiments using heparin, a soluble receptor analog, demonstrated dose-dependent inhibition of AAV attachment and infection. Enzymatic removal of heparan but not chondroitin sulfate moieties from the cell surface greatly reduced AAV attachment and infectivity. Finally, mutant cell lines that do not produce heparan sulfate proteoglycans were significantly impaired for both AAV binding and infection. This is the first report that proteoglycan has a role in cellular attachment of a parvovirus. Together, these results demonstrate that membrane-associated heparan sulfate proteoglycan serves as the viral receptor for AAV type 2, and provide an explanation for the broad host range of AAV. Identification of heparan sulfate proteoglycan as a viral receptor should facilitate development of new reagents for virus purification and provide critical information on the use of AAV as a gene therapy vector.
The primary event of any viral infection is attachment of virus to the host cell. A wide variety of cell surface molecules are now known to serve as viral attachment receptors. Such receptors range from cell-specific transmembrane proteins of well-defined receptor superfamilies (such as CD4 as a receptor for human immunodeficiency virus [HIV]) (55) to the more ubiquitous cell surface-associated carbohydrate moieties (such as the common carbohydrate moiety sialic acid for influenza virus) (23, 46). The mechanism by which adeno-associated virus (AAV) attaches to its host cell has not been delineated. A member of the Parvoviridae family, AAV is a small nonenveloped single-stranded DNA virus of 20 to 25 nm which has a unique requirement for a helper virus (e.g., adenovirus [Ad] or herpes simplex virus [HSV]) to complete its lytic cycle (2, 24, 39). In the absence of helper virus, AAV still infects the target cell, but it integrates into the host genome and establishes latency. Unique among eukaryotic DNA viruses, the AAV genome can integrate site specifically into human chromosome 19 (21, 22, 30, 51, 54). This property has drawn considerable attention to the potential use of AAV as a gene therapy vector (36, 38, 50, 52). Despite this growing interest in AAV, the events that govern the initial AAV infection remain poorly understood.
Viral receptors are often involved in defining the host range and specific tissue tropism of a virus. To date, among the 31 known eukaryotic parvoviruses, only one receptor, the erythrocyte P antigen, which serves as the receptor for human parvovirus B19 (11), has been identified. Identification of the receptor for B19 helps explain the tropism of this virus for erythrocytes. Unlike human parvovirus B19, AAV has a very broad host range and infects a wide variety of cell types, suggesting that the virus uses a ubiquitous receptor to mediate infection. Identification of the initial virus-host cell interactions necessary for efficient AAV infection is important not only for the general understanding of parvovirus infection but also for the effective use of AAV as a gene therapy vector.
Although the initial events in the life cycle of AAV are not well understood, previous studies suggest that AAV infects cells through interaction with a specific host cellular receptor (41, 45). AAV appears to exhibit saturation binding to HeLa cells. In addition, cellular attachment of AAV is sensitive to trypsin treatment, suggesting that a protein component is responsible for binding (41, 45). Further, AAV appears to bind to a 150-kDa glycoprotein. Although AAV binds this glycoprotein in a viral overlay protein binding assay, the evidence that the protein mediates virus binding to the cell surface remains indirect (41). Here we provide both biochemical and genetic evidence that cell surface heparan sulfate (HS) proteoglycan serves as a receptor for AAV.
Proteoglycans are proteins classified by a posttranslational attachment of polysaccharide glycosaminoglycan (GAG) moieties each comprised of repeating disaccharide units (for reviews see references 27 and 29). They can be found associated with both the extracellular matrix and plasma membranes. The four main widely distributed membrane-associated GAGs include heparin/HS and chondroitin sulfates A through C. These unbranched sulfated GAGs are defined by the repeating disaccharide units that comprise their chains, by their specific sites of sulfation, and by their susceptibility to bacterial enzymes known to cleave distinct GAG linkages (34). All have various degrees of sulfation which result in a high density of negative charge. Proteoglycans can be modified by more than one type of GAG and have a diversity of functions, including roles in cellular adhesion, differentiation, and growth. In addition, cell surface proteoglycans are known to act as cellular receptors for some bacteria and several animal viruses (48), including; foot-and-mouth disease type O virus (28), HSV types 1 and 2 (57, 61), and dengue virus (12).
In this report, we identify HS proteoglycans as the primary receptor for AAV. Our results show that cell surface HS GAGs, and not other GAGs, serve as a principal attachment receptor for AAV type 2 (AAV-2). Further, our results indicate that the presence of HS GAG on the cell surface directly correlates with the efficiency by which AAV can infect cells.
MATERIALS AND METHODS
Reagents.
The GAG lyases chondroitinase ABC, heparinase I, and heparinase III (heparitinase) were purchased from Sigma. Chondroitinase AC was obtained from Seikagagku America Inc. Soluble GAGs (heparin, from porcine intestinal mucosa; chondroitin sulfate A, from bovine trachea; chondroitin sulfate B, from porcine intestinal mucosa; and chondroitin sulfate C, from shark cartilage) were purchased from Sigma.
Cells and virus.
HeLa, CHO-K1, and CHO-K1 mutants deficient in proteoglycan biosynthesis were obtained from the American Type Culture Collection (Rockville, Md.). HeLa cells were maintained in Dulbecco modified Eagle medium (DMEM-H) supplemented with 10% fetal calf serum (FCS), and CHO cells were grown in Ham’s F-12 medium supplemented with 10% FCS. Viruses (wild-type [wt] AAV-2, recombinant AAV [rAAV]-LacZ, Ad dl309, and rAd-LacZ) were prepared as previously described (53, 58). All virus preparations were purified by two successive bandings on CsCl gradients to ensure purity. Wild-type AAV particle numbers were determined by protein quantitation (bicinchoninic acid reagent; Pierce), considering that the molecular mass of an AAV virion is 4.5 × 106 g/mol. Recombinant virus titers were determined as described previously (58). For preparation of radiolabeled wt AAV, 3 × 108 HeLa cells were infected with wt AAV and Ad dl309 at multiplicities of infection (MOIs) of 10 and 5, respectively. [methyl-3H]thymidine (Amersham) was added 7 h postinfection to a final concentration of 1 μCi/ml (9). Labeled virus was purified 48 h after infection as described above for wt AAV. 3H-AAV specific activity was approximately 4.0 × 10−8 cpm/virion. Fluorescent Cy3-labeled AAV-2 was a gift from Jeff Bartlett (Gene Therapy Center, University of North Carolina at Chapel Hill).
Binding assays.
All binding assays were done in a buffer which was determined to result in maximum cell viability, HEPES-buffered saline (HBS) containing 1% bovine serum albumin (HBSB; virus binding buffer). For direct binding assays, 3H-labeled wt AAV-2 was incubated with cells either attached to plates or in suspension, as indicated, at a ratio of 4 × 1011 particles/3 × 105 cells for 90 min at 4°C in HBSB. Cells were then washed three times in ice-cold HBSB to remove unbound virus and solubilized in 0.3 N NaOH. After neutralization with glacial acetic acid, cell-associated radioactivity was quantitated in a scintillation counter. For inhibition studies and after enzymatic treatments, 3H-AAV was bound to adherent HeLa cells. Binding to wt CHO and CHO cell mutants was done with suspended cells. Cells were first detached with 10 mM EDTA and then washed twice in phosphate-buffered saline (PBS) containing 8.8 mM CaCl2 and 0.5 mM MgCl2 and once in HBSB before binding of 3H-AAV. Nonspecific binding was determined in the presence of 100-fold excess unlabeled wt AAV (previously dialyzed in HBS–10% glycerol). Binding of Cy3-labeled virus was done on wt CHO and pgsA-745 cells grown on coverslips. After fixation in 4% paraformaldehyde for 20 min at room temperature, cells were mounted on slides, and bound Cy3-labeled virus was detected by confocal microscopy. Images were obtained with an argon-krypton laser at an excitation of 565 nm, a pinhole setting of 0.97, a 585-nm dichroic reflector, and a 590-nm long-pass barrier filter, using a Bio-Rad MRC-600 laser scanning attachment.
Viral infection assays. (i) Inhibition assay.
rAAV-LacZ at an MOI of 2 was incubated in DMEM-H in the presence or absence of indicated soluble GAGs at 1.0, 5.0, 10, 20, 30 μg/ml for 1 h at 37°C. For infection, rAAV-LacZ or the virus-GAG mixture was incubated with 2 × 105 HeLa cells for 1 h at 37°C in a 5% CO2 chamber. To stop the adsorption process, cells were washed thoroughly and overlaid with DMEM-H supplemented with 2% FCS. After 44 h, cells were washed in PBS and lysed with 100 mM potassium phosphate (pH 7.8)–0.2% Triton X-100. β-Galactosidase activity was then assayed by using a Galacto-Light Plus kit (Tropix Inc.) as described by the manufacturer. Data was collected in a luminometer within the linear range of the assay, and enzyme activity is expressed as relative light units (RLU)/microgram of protein. Each experimental condition was assayed in duplicate, and independent experiments yielded similar results. Preincubation studies were performed as described above except that HeLa cells were incubated with or without indicated concentrations of heparin in DMEM-H for 1 h at 37°C and washed thoroughly prior to rAAV-LacZ infection.
(ii) Enzymatic treatment.
GAG lyases were reconstituted in PBS. For enzymatic digestion of cell surface GAGs, 3 × 104 HeLa cells were washed and incubated with the indicated concentrations of GAG lyases in PBS containing 0.1% bovine serum albumin, 0.2% gelatin, and 0.1% glucose (digestion buffer) for 1 h at 37°C in a 5% CO2 chamber. Cells were then washed four times with digestion buffer and subjected to rAAV infection as described above. Prior to use, each enzyme was tested for activity by a standard method used to determine specific activity of GAG lyases except that enzyme activity was monitored under the conditions used for digestion, PBS (pH 7.4) at 37°C (33). Cells remained attached to the plate after all enzymatic treatments. Each experimental condition was performed in triplicate. β-Galactosidase activity was assayed as described above and is expressed in RLU. Enzyme concentrations are expressed in international units per milliliter (1 IU is equivalent to 600 Sigma units).
(iii) CHO cell infection and UV treatment.
For UV treatment, 3 × 104 CHO cells were washed, overlaid with PBS, and treated with UV irradiation (45 J/m2) in a UV Stratalinker (Stratagene, La Jolla, Calif.) prior to rAAV infection (18). rAAV infections, at an MOI of 10, for both UV-treated and non-UV-treated CHO cells were performed as described above. β-Galactosidase activity is expressed as the average RLU from rAAV infections performed in triplicate.
RESULTS
AAV binding and infection are inhibited by soluble GAGs.
Several observations led us to postulate that AAV-2 may use cell surface proteoglycans as a receptor. First, we demonstrated that AAV-2 binds to a cellufine sulfate column (47a). Other viruses known to interact with such columns bind to negatively charged surface molecules (for example, several members of the Herpesviridae family known to use HS proteoglycans as attachment receptors [1, 13, 40, 57, 61]). Second, AAV can infect a wide variety of human, rodent, and simian cell lines (8, 44), suggesting that it uses a ubiquitous cell surface molecule for infection. Since proteoglycans are present on numerous cell types and it appears that proteoglycans are ubiquitously expressed throughout the animal kingdom (14, 29), we tested the possibility that AAV-2 uses cell surface proteoglycans to mediate infection.
If AAV infection initiates through interaction with cell surface proteoglycans, one or more of the major GAGs found on membrane-associated proteoglycans should act as competitive inhibitors of AAV infection and binding. To test this possibility, we performed competition experiments with several known GAGs found on membranes. In this assay, we analyzed the ability of soluble GAGs to inhibit rAAV-LacZ reporter gene transduction in HeLa cells. Increasing concentrations of GAGs were incubated with rAAV prior to adsorption to cells at 37°C for 1 h. Cells were harvested 44 h postinfection and assayed for β-galactosidase activity. Of the four GAGs tested, heparin, a molecule chemically very similar to HS GAG, inhibited AAV infection maximally and in a dose-dependent manner (Fig. 1A). Heparin concentrations as low as 5 μg/ml resulted in nearly 100% inhibition. Chondroitin sulfate B, which shares the most structural similarity to heparin/HS, exhibited 71% inhibition at 30 μg/ml. In contrast, chondroitin sulfate A and chondroitin sulfate C at concentrations of up to 30 μg/ml exhibited no significant effect, with less than 20% inhibition. The observed inhibition was specific for AAV since similar studies showed no effect on infection with Ad, another nonenveloped DNA virus whose receptor is the coxsackievirus-Ad receptor (CAR) (data not shown) (6).
FIG. 1.

Inhibition of AAV infection by various GAGs. (A) rAAV was incubated with the indicated concentrations of heparin (▪), chondroitin sulfate B (dermatan sulfate) (○), chondroitin sulfate A (◊), or chondroitin sulfate C (▵) for 1 h at 37°C prior to a 1-h adsorption of the virus-GAG mixture to HeLa cells for infection. β-Galactosidase activity was assayed 44 h postinfection by using a Galacto-Light Plus kit (Tropix Inc.) and measured in a luminometer. Each point represents the average percent decrease in RLU per microgram of protein relative to the maximum level obtained in experiments without GAG. (B) HeLa cells were preincubated with increasing concentrations of heparin at 37°C for 1 h. After thorough washing, cells were infected with rAAV as described above. Data points represent the average percent maximum RLU/microgram of protein obtained without heparin preincubation.
To rule out the possibility that significant inhibition of AAV infection by heparin was due to an induced cellular effect of this molecule on HeLa cells, preincubation studies were performed. HeLa cells were incubated with the indicated concentrations of heparin (Fig. 1B), washed extensively, and then infected with rAAV as described above. Unlike competition experiments, preincubation of HeLa cells with heparin had little effect on the ability of rAAV to transduce cells (Fig. 1B). While the presence of heparin (5 μg/ml) during viral adsorption demonstrated 100% inhibition (Fig. 1A), preincubation demonstrated less than 20% inhibition at concentrations up to 20 μg/ml (Fig. 1B). These data suggest that heparin interacts directly with AAV and inhibits an early event of viral infection.
To further examine the observed specificity and to determine whether GAGs were inhibiting binding of the AAV virions to the cell surface, 3H-labeled wt AAV was incubated with GAGs at various concentrations. After incubation with cells, the mixture was washed and cells were solubilized to quantify bound virus. By this assay, heparin (5 μg/ml) inhibited 90% of AAV binding (Fig. 2), correlating with rAAV transduction data (Fig. 1). Furthermore, chondroitin sulfate B (dermatan sulfate; 30 μg/ml) inhibited binding by 51% (Fig. 2). As expected, chondroitin sulfates A and C at similar concentrations did not significantly affect the ability of virus to bind to HeLa cells, exhibiting no more than 20% inhibition. These data suggest that heparin inhibits AAV infection by interfering with virion binding to the cell surface, presumably by competing for structurally related HS moieties.
FIG. 2.

Soluble heparin inhibits binding of AAV to the cell surface. After preincubation of 3H-labeled wt AAV-2 with increasing concentrations of the indicated GAGs or the GAG analog dextran sulfate, labeled virus was adsorbed to HeLa cells for 90 min at 4°C. Unbound virus was removed by three washes with ice-cold binding buffer, and radioactivity was quantitated as described in Materials and Methods. Data are represented as the average percent inhibition relative to the counts per minute bound in the absence of soluble GAG.
Since heparin is known to be modified by more sulfate groups and has a higher charge density than the chondroitin sulfates (49), it was important to determine whether inhibition reflected specificity or was simply a function of charge. We examined the ability of a highly sulfated GAG analog, dextran sulfate (molecular weight, 5,000), to act as a competitive inhibitor. At the maximum concentration, 30 μg/ml, the observed inhibition by dextran sulfate was only 36% (Fig. 2), indicating that more than charge ratio is responsible for the inhibition of AAV by heparin and chondroitin sulfate B. Together, the above data support the hypothesis that AAV binds to cell surface proteoglycan, that this interaction is important for a productive infection, and that AAV exhibits specificity for particular GAG moieties.
AAV requires GAGs on the cell surface for infection.
Since some GAGs are known to bind to specific cell surface receptors (27), the foregoing experiments could not completely rule out competition for a common receptor as a mechanism for inhibiting virus binding. To address this issue, we used enzymes that are known to specifically digest the GAGs present on the cell surface before assaying virus specific binding. Heparinase I and heparitinase cleave distinct linkages found in HS. Chondroitinase ABC cleaves at a linkage found in all chondroitin sulfates, including dermatan sulfate (chondroitin sulfate B), and chondroitinase AC cleaves only chondroitin sulfates A and C (33). All enzymes were tested prior to use for activity and assayed under the conditions described in Materials and Methods. Subconfluent HeLa cells were treated with various concentrations of each enzyme and assessed for 3H-AAV binding. Consistent with early experiments, enzymatic treatment with either heparitinase or heparinase greatly reduced the ability of virus to bind the cell surface: 73 or 66%, respectively, at the maximum concentration of enzyme tested (Fig. 3A). Further, chondroitinase ABC and chondroitinase AC treatment did not result in any reduction of AAV binding to HeLa cells (Fig. 3A). These data indicate that HS proteoglycan mediates attachment of AAV to the cell surface.
FIG. 3.

Effect of enzymatic digestion of cell surface GAGs on AAV binding and infection. (A) HeLa cells were treated with the indicated concentrations of the GAG lyase heparitinase, heparinase I (▪), heparinase I (◊), chondroitinase ABC (○), or chondroitinase AC (▵) as described in Materials and Methods. After thorough washing, the ability of AAV to bind the cell surface was assessed as described for Fig. 2. Data points represent the average percent reduction in AAV binding relative to AAV binding obtained without enzymatic treatment. (B) HeLa cells were treated with heparitinase or heparinase I as described in Materials and Methods. After thorough washing, rAAV was incubated with cells for a 1-h adsorption period at 37°C. Cells were harvested 44 h postinfection and assayed for β-galactosidase activity. Results are graphed as the average percent reduction in AAV transduction relative to transduction observed in the absence of enzymatic treatment. Data points represent the mean and standard deviation of experiments performed in triplicate.
The fact that 10-fold more heparinase than heparitinase was required to obtain similar reduction of AAV binding was likely due to its known lower enzymatic activity under the conditions used (33). Further, to ensure that protease contaminants were not responsible for reduction in AAV binding, digestion in the presence of soluble substrate was performed. Exogenous addition of soluble HS reversed the effect of heparitinase treatment on AAV binding to HeLa cells (data not shown). Therefore, specific removal of plasma membrane-associated HS moieties results in a diminished ability of AAV to bind the cell surface. It was unexpected that chondroitinase ABC did not have any effect on AAV binding since the presence of soluble chondroitin sulfate B (dermatan sulfate) was able to inhibit AAV transduction and binding to HeLa cells, albeit much less efficiently than heparin. The lack of reduction in binding after enzymatic treatment with chondroitinase ABC suggests that AAV does not efficiently bind to dermatan sulfate present on the cell surface. It is possible that AAV exhibits an interaction with chondroitin sulfate B (dermatan sulfate) only when it is present in excess amounts in solution.
The above data ruled out the possibility that in the coincubation experiments, soluble heparin and AAV were competing for a similar receptor or that heparin was sterically hindering binding to something other than membrane-associated GAGs. Instead, the data suggest that AAV binds to cell surface proteoglycan and further suggest that this interaction is specific for HS, not chondroitin sulfate, moieties.
To demonstrate the biological relevance of AAV binding to cell surface HS, we determined whether removal of HS moieties rendered cells less susceptible to AAV infection. We examined the ability of rAAV-LacZ to transduce HeLa cells after treatment with various concentrations of heparitinase or heparinase. rAAV transduction was reduced by 80% compared to untreated control cells (Fig. 3B). Importantly, as little as 0.425 mIU of heparitinase per ml resulted in up to 72% reduction of AAV infection. Consistent with inefficient heparinase enzyme activity under these conditions, approximately 10-fold more enzyme was required for a similar reduction in AAV transduction. The reduced susceptibility of HeLa cells to AAV infection after enzymatic removal of membrane-associated HS GAG indicates a significant role for HS proteoglycan in AAV infection.
Mutants of the GAG synthesis pathway inhibit AAV binding.
CHO cell derivatives defective in GAG synthesis were used to further define the requirement for HS moieties for AAV infection. These mutant cell lines have defined deficiencies in the production of specific GAGs. Cell line pgsA-745 lacks xylosyltransferase, an enzyme necessary for the initiation of all GAG synthesis, and does not produce detectable levels of proteoglycans (16). Mutant pgsB-618 has a defect in the galactosyltransferase I gene and makes about 15% of the normal amount of proteoglycan synthesized by wt cells (15, 17). Cell line pgsE-606 is partially deficient in HS N-sulfotransferase and produces an undersulfated form of HS proteoglycan (4, 5). Finally, mutant pgsD-677 has a single mutation that affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities that are necessary for the polymerization of HS disaccharide chains and does not synthesize any HS proteoglycan. This mutant cell line also produces approximately three times more chondroitin sulfate than wt cells (15, 31).
To assess AAV binding to wt and mutant CHO cell lines, cells were incubated with Cy3-labeled virions or 3H-AAV-2 as described in Materials and Methods. Bound 3H-labeled virus was collected by centrifugation, and fluorescence virus was visualized by confocal microscopy (Fig. 4). Although binding of Cy3-AAV to wt CHO cells was easily detectable (Fig. 4A, I), no significant virus binding to pgsA mutant CHO cells was observed (Fig. 4A, II). These binding observations were quantified by using 3H-AAV-2. Compared to wt CHO cells, there were 7.0- and 6.4-fold reductions in AAV binding to cell lines pgsA-745 and pgsD-677, respectively. The poor attachment of AAV to the HS GAG deficient mutant cells pgsA and pgsD (Fig. 4B) provides genetic data indicating that the presence of HS proteoglycan is a principal requirement for AAV attachment to the cell surface. Further, the inefficient binding to pgsD-677 (HS GAG deficient and three times more chondroitin sulfates) demonstrates that AAV exhibits specificity for heparan. The 4.6-fold reduction in AAV binding to pgsB-618 (15% of the level of wt proteoglycans) was slightly higher than the binding to proteoglycan-deficient pgsA and pgsD cells. This result correlates well with the 85% overall lower production of GAGs by the pgsB-618 mutant cell line. Interestingly, AAV binding to the mutant pgsE-606 cell line, which produces an undersulfated form of HS, was also diminished, albeit to a lesser extent (2.6-fold). Since the pgsE-606 cell line is partially deficient in N-sulfotransferase, a reduction in AAV binding to this cell line suggests that N-sulfation of HS may be an important determinant influencing AAV attachment. The observed differences in virus binding to wt and GAG-deficient cells was specific for AAV, since no effect was observed when binding of labeled Ad was used (data not shown).
FIG. 4.
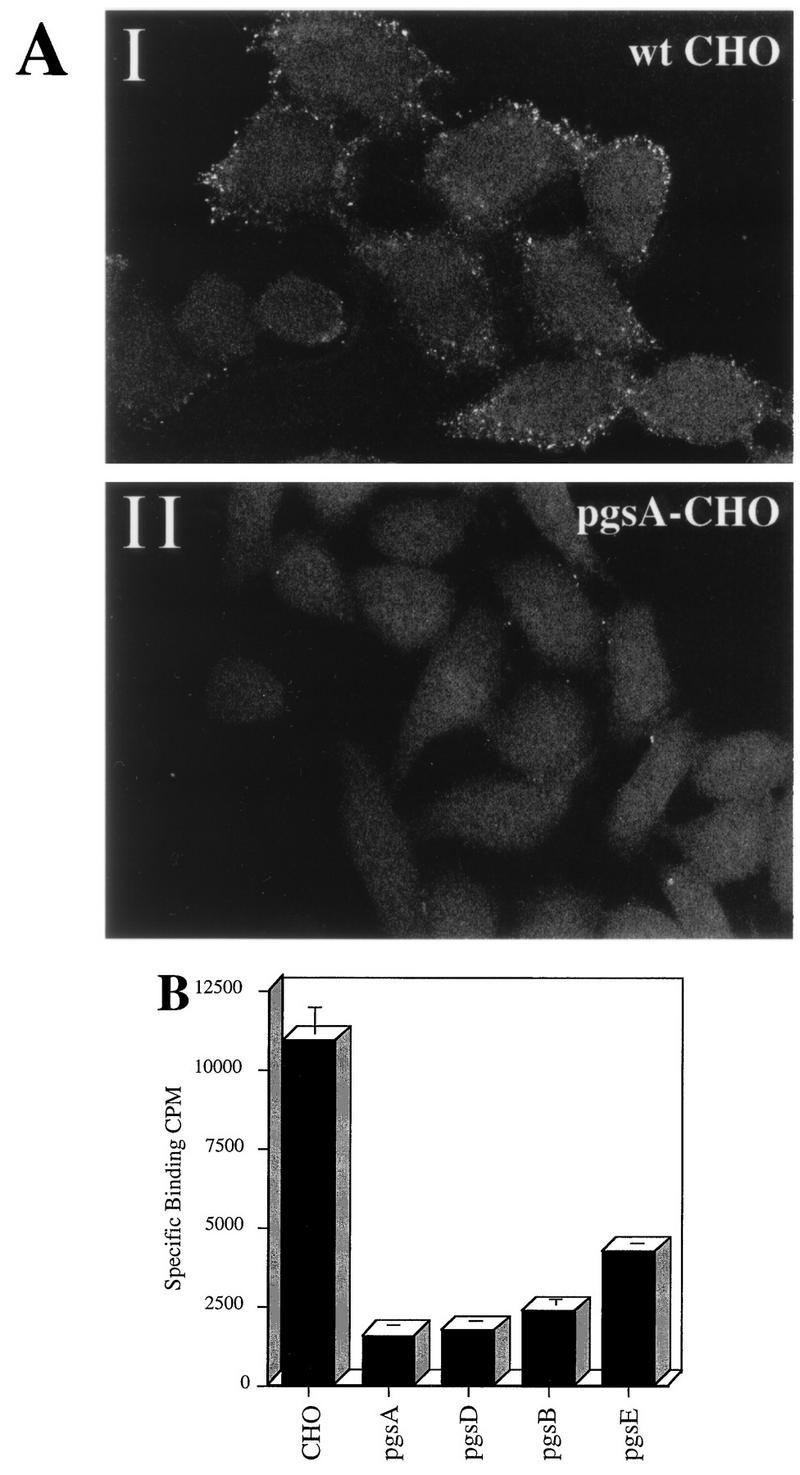
HS proteoglycan serves as a primary attachment receptor for AAV-2. Wild-type CHO-K1 cells and CHO-K1 mutants defective in proteoglycan synthesis were assessed for the ability to bind AAV-2. Cell line pgsA-745 lacks HS and chondroitin sulfate proteoglycans; pgsD-677 lacks HS proteoglycan and produces a threefold excess of chondroitin sulfate proteoglycans; pgsB-618 produces 15% of normal proteoglycans; pgsE-606 produces an undersulfated form of HS proteoglycan and normal levels of chondroitin sulfate proteoglycans. (A) Cy3-labeled AAV-2 was bound to wt CHO cells (I) and the pgsA-745 mutant that lacks proteoglycans (II) as described in Materials and Methods. Images were captured by confocal microscopy. (B) Binding of 3H-AAV to parental and mutant CHO cells. Binding assays were performed at 4°C in Eppendorf tubes. A total of 3 × 105 cells were incubated with 4 × 1011 particles of 3H-AAV for 90 min in HBSB. After thorough washing, cells were pelleted and solubilized, and radioactivity was quantitated as described in Materials and Methods. Nonspecific binding was determined by parallel binding studies done in the presence of a 100-fold excess of unlabeled virus. Data represent the mean specific binding and standard deviation obtained from experiments performed in triplicate.
Although the ability of AAV to bind all mutants was significantly diminished, each mutant still exhibited some binding as assessed by 3H-AAV in the presence of a 100-fold excess of unlabeled virus (Fig. 4B). This finding suggests that AAV may bind to yet unidentified cell surface receptors present in low abundance or that extremely low levels of HS GAG are present on these mutant cells. The pgsA cells have a low level of xylosyltransferase activity, which could result in residual proteoglycan synthesis (16, 17).
Overall, these data indicate that the HS and not chondroitin sulfate moieties of cell surface proteoglycans serve as attachment receptors for AAV. In addition, the diminished ability of AAV to bind pgsE-606 (mutant with undersulfated HS GAGs) suggests that the degree of sulfation of HS is an important factor influencing binding of AAV to HS proteoglycan.
HS proteoglycan mediates AAV infection.
The CHO cell mutants defective in GAG synthesis were also used to provide genetic evidence that HS proteoglycans are necessary for an efficient AAV infection. As with the previous infection experiments, we used an rAAV-LacZ vector that expresses β-galactosidase to assess AAV-2 infectivity as described in Materials and Methods. The lack of cell surface HS proteoglycan significantly impaired the ability of AAV to infect CHO cells (Fig. 5). Compared to AAV transduction of wt cells, there was significant reduction in AAV transduction of pgsA and pgsD cells (Fig. 5A). In addition to lacking HS proteoglycan on the surface, the pgsD cells overproduce chondroitin sulfate proteoglycans. The poor infection of this cell line further demonstrates the specificity of AAV for cell surface heparan and not chondroitin moieties. We observed a 10-fold reduction in AAV transduction of pgsB cells, which produce 85% less proteoglycan than wt cells. It is not clear why the pgsD cells are almost completely refractory to AAV infection (Fig. 5A) when binding to this cell line is similar to that observed with pgsA. pgsD cells that produce excess chondroitin sulfates may bind AAV inefficiently through these substrates. However, this interaction may not lead to a productive infection, an observation seen by others (43). With the exception of the pgsD cell line, the inefficient AAV transduction of the CHO cell mutants clearly paralleled the reduced ability of AAV to bind the cell surface. The above data indicate that infection by AAV is closely coupled to the amount of AAV that can attach to the cell surface and that this attachment is primarily mediated by HS proteoglycans. Finally, the pgsE cells that produce an undersulfated form of HS proteoglycans supported AAV transduction but with 1.4-fold reduction (Fig. 5), consistent with the previously observed level of AAV binding (Fig. 4B).
FIG. 5.

HS proteoglycan mediates AAV infection. (A) AAV infection of wt and mutant CHO cells deficient in proteoglycan synthesis (described in the legend to Fig. 4). rAAV-LacZ virus was incubated with cells at an MOI of 10 for 1 h at 37°C. Cells were harvested 44 h postinfection and assayed for β-galactosidase activity. Data represent the mean and standard deviation of triplicate experiments. (B) UV treatment of wild-type and mutant CHO cells and its effect on rAAV transduction. Cells were treated with UV (45 J/m2) in a UV Stratalinker (Stratagene) prior to infection with rAAV-LacZ as described above. β-Galactosidase activity was measured as described for non-UV-treated cells.
A rate-limiting step in rAAV vector transduction is inefficient synthesis of second-strand copies from virion single-stranded DNA genomes (18, 19, 36). To ensure that the observed differences in AAV infection were a result of inefficient AAV binding and not due to impaired second-strand synthesis, we treated cells with UV, a technique known to overcome inefficiencies at this step (18). When mutant cells were treated with UV to maximize transgene expression, enhancement of rAAV-LacZ transduction was observed (10-fold) but overall AAV infections were similar to those of non-UV-treated cells (Fig. 5B). Taken together, the results obtained with the CHO cell mutants deficient in GAG synthesis provide genetic evidence that HS proteoglycan mediates both attachment and entry of AAV-2.
DISCUSSION
We have used defined mutant CHO cells defective in GAG synthesis and standard biochemical assays to establish that membrane-associated HS proteoglycan serves as a receptor for AAV-2 and mediates both viral attachment to and subsequent infection of target cells. We have shown that binding and infection of cells by AAV is sensitive to (i) competitive inhibition with heparin, a soluble receptor analog, (ii) enzymatic removal of HS but not chondroitin sulfate moieties from the cell surface, and (iii) genetic defects in the cellular pathway for the production of HS. In addition, the use of mutant cell lines provided genetic evidence that HS, and not chondroitin sulfate, proteoglycans are responsible for a productive AAV infection. This is the first report of a role for proteoglycan in cellular attachment of a parvovirus and provides an explanation for the broad host range of AAV.
We have demonstrated that HS proteoglycan serves as a principal attachment receptor for AAV-2. However, additional factors could also participate in AAV host cell attachment. Some viruses can use more than one distinct attachment receptor. For example, HIV uses CD4 as its main attachment receptor but can also attach to glycolipid galactosyl ceramide to mediate infection (10, 55). Further, Ad attachment to target cells can be mediated by αMβ2 integrin as well as CAR (6, 25). Since removal of HS moieties from the cell surface did not completely abolish AAV infectivity and AAV still exhibits some specific binding to cell lines that do not produce HS proteoglycans, it is possible that AAV attachment and infection can also be mediated by an as yet unidentified receptor, albeit inefficiently.
While the inefficient binding and poor infection by AAV in the absence of HS GAGs suggests that HS proteoglycan could mediate both AAV attachment and entry, it remains to be determined whether AAV attachment to HS proteoglycan is sufficient for viral entry. For example, it is well established that the initial interaction of HSV with its host cell is mediated through HS proteoglycans (35, 57, 61) and that another secondary event is responsible for promoting entry (20). Recently, a novel member of the tumor necrosis factor/nerve growth factor receptor family was demonstrated to serve as a mediator of HSV entry (42). In addition, Ad infection is initiated by attachment to CAR (6) followed by subsequent interaction with secondary receptors, identified as αV integrins, that are known to facilitate virus internalization (60). On the other hand, a large percentage of HS proteoglycans are known to undergo endocytosis (26, 63) and could be involved in direct AAV internalization. Such a mechanism of entry has been described for other HS proteoglycan ligands, including basic fibroblast growth factor and lipoprotein lipase (47, 56). AAV may use either or both of these proposed mechanisms of entry. Further, the possibility that a large functional multireceptor complex is required for efficient AAV entry should not be ruled out, since cell surface proteoglycans have been implicated as members of multimeric complexes (7). That is, our results show the HS proteoglycans are required for AAV infection but do not address whether they are in fact sufficient.
The GAG structures can be complex, exhibiting a diversity of disaccharide sequences with heterogeneous sulfation. Some GAG ligands require the presence of a specific sugar sequence for high affinity binding, as is the case for antithrombin, which binds a distinct sequence present in heparin/HS (3, 32). In addition, a recent report identified HS GAGs as a receptor for the pathogenic RNA virus dengue virus. However, the virus appears to require a highly sulfated form of HS GAG in order to be infectious (12). Although we have not identified a specific sugar sequence requirement for AAV binding, our data indicate that AAV requires HS and not chondroitin sulfate moieties. This conclusion is based on the inability of chondroitinase ABC enzymatic digestion to inhibit AAV binding and infection as well as the inability of AAV to appreciably bind and infect mutant CHO cells that lack HS yet have an excess of chondroitin sulfate proteoglycans. HS GAGs consist of repeating disaccharide units composed of alternating glucosamine and hexuronic acid (either glucuronic acid or iduronic acid) monsacharides. The chondroitin sulfate disaccharide units contain a galactosamine monosacharide in place of glucosamine. Chondroitin sulfate B (dermatan sulfate) is the only chondroitin that contains iduronic acid monosacharides that are also found in HS GAGs. The specificity exhibited by AAV for HS moieties demonstrates that AAV prefers an interaction with a glucosamine-hexuronic acid backbone. Further, since excess soluble dermatan sulfate could inhibit AAV binding and infection, AAV may prefer a HS backbone that contains iduronic acid.
Our data may indicate that multiple receptor molecules mediate AAV infection. In cases where the amount of cell-associated HS GAG was reduced, either by enzymatic digestion or in mutant cell lines, the reduction in AAV infection was more sensitive than the reduction of AAV attachment. One possible explanation is that the density of receptors, and thus the increased probability of virus or HS proteoglycan interactions with some other receptor molecule(s), may be an important factor influencing AAV entry. It will be interesting to determine if the amount of cell surface HS proteoglycan influences the ratio of internalized virus to bound virus. Alternatively, another explanation for our results may be that AAV can attach to a subset of surface molecules that are not capable of mediating AAV infection.
Since the degree of HS sulfation affects the amount of AAV that can bind the cell surface, there appears to be an important charge component to the specific AAV-HS interaction. A majority of ligand-proteoglycan interactions are mediated by a cluster of basic amino acids displayed by the ligand and the high density of charge found on sulfated GAGs (27, 29). Until the crystal structure of AAV-2 is determined, we cannot be certain which basic residues are exposed to the virion surface, nor can we address the noncolinear basic amino acids that may be close in space in the intact virion. However, there is a high density of positively charged amino acids within the first 170 residues of the VP1 capsid protein, including three strings of basic amino acids (either K/RX4/5KKR or KX6RKR) that, if exposed on the virion surface, could be involved in an ionic interaction with the cell surface. It is also interesting that viral mutants that map in this region are referred to as low-infectious-particle mutants (44).
Recently, we have demonstrated long-term (1.5-year) gene expression after direct rAAV injection into immunocompetent mouse muscle and brain (37, 62). These data have provided preclinical results suggesting that this viral delivery system may provide an attractive alternative to other vectors. In fact, rAAV has recently been tested in a clinical trial for gene therapy of cystic fibrosis without any signs of toxicity of immune complications (19a). Identification of the AAV receptor should now help facilitate maximum use of this vector with appropriate target cells (bone marrow stem cells, airway epithelia cells, etc.). Fluorescence-activated cell sorting analysis of various human cells has shown a correlation between HS GAGs and virus binding consistent with this report (58a). Identification of the AAV receptor should provide further information concerning primary events involved in AAV infection and future development of this virus as a viral vector.
ACKNOWLEDGMENTS
We thank John Olsen and Raj Batra for helpful discussion; Jeff Bartlett for the gift of Cy3-labeled wt AAV; Ting Qian (Department of Cell Biology and Anatomy, UNC at Chapel Hill) for generously analyzing Cy3-labeled wt AAV bound to CHO cells by confocal microscopy; and Doug McCarty, Charles Yang, and Terry VanDyke for critical reading of the manuscript and helpful suggestions. We further thank the Vector Core Facility at UNC Chapel Hill for rAAV.
This research was supported by NIH grant HL51818.
REFERENCES
- 1.Amicon Division, W. R. Grace Co. Matrex cellufine sulfate. Amicon technical publication 845. Beverly, Mass: Amicon Division, W. R. Grace Co.; 1993. [Google Scholar]
- 2.Atchison R W, Castro B C, Hammond W M. Adeno-associated defective virus particles. Science. 1965;149:754–759. doi: 10.1126/science.149.3685.754. [DOI] [PubMed] [Google Scholar]
- 3.Atha D H, Stephens A W, Rimon A, Rosenberg R D. Sequence variation in heparin octasaccharides with high affinity for antithrombin III. Biochemistry. 1984;23:5801–5812. doi: 10.1021/bi00319a020. [DOI] [PubMed] [Google Scholar]
- 4.Bame K, Esko J D. Undersulfated heparan sulfate in a chinese hamster ovary cell mutant defective in heparan sulfate N-sulfotransferase. J Biol Chem. 1989;264:8059–8069. [PubMed] [Google Scholar]
- 5.Bame K J, Lidholt K, Lindahl U, Esko J D. Biosynthesis of heparan sulfate. J Biol Chem. 1991;266:10287–10293. [PubMed] [Google Scholar]
- 6.Bergelson J M, Cunningham J A, Droguett G, Kurt-Jones E A, Krithivas A, Hong J S, Horwitz M S, Crowell R L, Finberg R W. Isolation of a common receptor for coxsackie B viruses and adenovirus 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 7.Bernfield M, Kokenyesi R, Kato M, Hinkes M T, Gallo R L, Lose E J. Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu Rev Cell Biol. 1992;8:365–393. doi: 10.1146/annurev.cb.08.110192.002053. [DOI] [PubMed] [Google Scholar]
- 8.Berns K I. Parvoviridae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields virology. Vol. 3. Philadelphia, Pa: Lippincott-Raven; 1996. pp. 2173–2197. [Google Scholar]
- 9.Berns K I, Rose J A. Evidence for single stranded adenovirus-associated virus genome. Isolation and separation of complementary single strands. J Virol. 1970;5:693–699. doi: 10.1128/jvi.5.6.693-699.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhat S, Spitalink S L, Gonzalez-Scarano F, Silberg D H. Galactosyl ceramide or a derivative is an essential component of the neural receptor for human immunodeficiency virus type 1 envelope glycoprotein gp120. Proc Natl Acad Sci USA. 1991;88:7131–7134. doi: 10.1073/pnas.88.16.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown K E, Anderson S M, Young N. Erythrocyte P antigen: cellular receptor for B19 parvovirus. Science. 1993;262:114–116. doi: 10.1126/science.8211117. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Maguire T, Hileman R E, Fromm J R, Esko J D, Linhardt R J, Marks R M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat Med. 1997;3:866–871. doi: 10.1038/nm0897-866. [DOI] [PubMed] [Google Scholar]
- 13.Compton T, Nowlin D M, Cooper N R. Initiation of human cytomegleovirus infection requires initial interaction with cell surface heparan sulfate. Virology. 1993;193:834–841. doi: 10.1006/viro.1993.1192. [DOI] [PubMed] [Google Scholar]
- 14.Diertrich C P, Cassaro C M F. Distribution of sulfated mucopolysaccharides in invertebrates. J Biol Chem. 1977;252:2254–2261. [PubMed] [Google Scholar]
- 15.Esko J D, Rostand K S, Weinke J L. Tumor formation dependent on proteoglycan biosynthesis. Science. 1988;241:1092–1095. doi: 10.1126/science.3137658. [DOI] [PubMed] [Google Scholar]
- 16.Esko J D, Stewart T E, Taylor W H. Animal cell mutants defective in glycosaminoglycan synthesis. Proc Natl Acad Sci USA. 1985;82:3197–3201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esko J D, Weinke J L, Taylor W H, Ekborg G, Roden L, Anantharamaiah G, Gawish A. Inhibition of chondroitin and heparan sulfate biosynthesis in chinese hamster ovary cell mutants defective in galactosyltransferase I. J Biol Chem. 1987;262:12189–12195. [PubMed] [Google Scholar]
- 18.Ferrari F K, Samulski T, Shenk T, Samulski R J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 1996;7:3277–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher K J, Gao G P, Weitzman M D, DeMatteo R, Burda J F, Wilson J M. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J Virol. 1996;70:520–532. doi: 10.1128/jvi.70.1.520-532.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.Flotte T. Present at the Cystic Fibrosis Foundation Williamsburg Gene Therapy Conference, June. 1997. [Google Scholar]
- 20.Fuller A O, Santos R E, Spear P G. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J Virol. 1989;63:3435–3443. doi: 10.1128/jvi.63.8.3435-3443.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giraud C, Winocour E, Berns K. Recombinant junctions formed by site-specific integration of adeno-associated virus into an episome. J Virol. 1995;69:6917–6924. doi: 10.1128/jvi.69.11.6917-6924.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giraud C, Winocour E, Berns K I. Site-specific integration by adeno-associated virus is directed by a cellular DNA sequence. Proc Natl Acad Sci USA. 1994;91:10039–10043. doi: 10.1073/pnas.91.21.10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herrler G. The receptor destroying enzyme of influenza C virus is neuraminate-O-acetylesterase. EMBO J. 1985;4:1053–1056. doi: 10.1002/j.1460-2075.1985.tb03809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoggan M D, Blacklow N R, Rowe W P. Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc Natl Acad Sci USA. 1966;55:1457–1471. doi: 10.1073/pnas.55.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang S, Kamata T, Takada Y, Ruggeri Z M, Nemerow G R. Adenovirus interaction with distinct integrins mediates seperate events in cell entry and gene delivery to hematopoietic cells. J Virol. 1996;70:4502–4508. doi: 10.1128/jvi.70.7.4502-4508.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iozzo R V. Turnover of heparin sulfate proteoglycan in human colon carcinoma cells. J Biol Chem. 1987;262:1888–1900. [PubMed] [Google Scholar]
- 27.Jackson R L, Busch S J, Cardin A D. Glycosaminoglycans: molecular propeties, protein interactions, and role in physiological processes. Physiol Rev. 1991;71:481–539. doi: 10.1152/physrev.1991.71.2.481. [DOI] [PubMed] [Google Scholar]
- 28.Jackson T, Ellard F M, Ghazaleh R A, Brookes S M, Blakemore W E, Corteyn A H, Stuart D I, Newman J W I, King A M. Efficient infection of cells in culture by type O foot-and-mouth disease virus requires binding to cell surface heparan sulfate. J Virol. 1996;70:5282–5287. doi: 10.1128/jvi.70.8.5282-5287.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kjellen L, Lindahl U. Proteoglycans: structures and interactions. Annu Rev Biochem. 1991;60:443–475. doi: 10.1146/annurev.bi.60.070191.002303. [DOI] [PubMed] [Google Scholar]
- 30.Kotin R M, Siniscalco M, Samulski R J, Zhu X, Hunter L, Laughlin C A, McLaughlin S, Muzyczka N, Rocchi M, Berns K I. Site-specific integration by adeno-associated virus. Proc Natl Acad Sci USA. 1990;87:2211–2215. doi: 10.1073/pnas.87.6.2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lidholt K, Weinke J L, Kiser C S, Lugemwa F N, Bame K J, Cheifetz S, Massague J, Lindahl U, Esko J D. A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis. Proc Natl Acad Sci USA. 1992;89:2267–2271. doi: 10.1073/pnas.89.6.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindahl U, Backstrom G, Thunberg L, Leder I G. Evidence for a 3-O-sulfated D-glucosamine residue in the anti-thrombin-binding sequence of heparin. Proc Natl Acad Sci USA. 1980;77:6551–6555. doi: 10.1073/pnas.77.11.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linhardt R J. Analysis of glycosaminoglycans with polysaccharide lyases. In: Janssen K, editor. Current protocols in molecular biology. Vol. 3. New York, N.Y: John Wiley & Sons, Inc.; 1994. pp. 17.13.17–17.13.32. [DOI] [PubMed] [Google Scholar]
- 34.Linhardt R J, Cooney C L, Galliher P M. Polysaccharide lyases. Appl Biochem Biotechnol. 1986;12:135–177. doi: 10.1007/BF02798420. [DOI] [PubMed] [Google Scholar]
- 35.Lycke E, Johansson M, Svennerholm B, Lindah U. Binding of herpes simplex virus to cellular heparan sulfate, an initial step in the adsorption process. J Gen Virol. 1991;72:1131–1137. doi: 10.1099/0022-1317-72-5-1131. [DOI] [PubMed] [Google Scholar]
- 36.McCarty D M, Samulski R J. Adeno-associated viral vectors. In: Strauss M, Barranger J, editors. Concepts in gene therapy. Bellin. N.Y: Walter de Gruyter; 1997. pp. 62–78. [Google Scholar]
- 37.McCown T J, Xiao X, Li J, Breese G R, Samulski R J. Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res. 1996;713:99–107. doi: 10.1016/0006-8993(95)01488-8. [DOI] [PubMed] [Google Scholar]
- 38.McKeon C, Samulski R J. NIDDK workshop on AAV vectors: gene transfer into quiescent cells. Hum Gene Ther. 1996;7:1615–1619. doi: 10.1089/hum.1996.7.13-1615. [DOI] [PubMed] [Google Scholar]
- 39.Melnick J L, Mayor H D, Smith K O, Rapp F. Association of 20 millimicron particles with adenoviruses. J Bacteriol. 1965;90:271–274. doi: 10.1128/jb.90.1.271-274.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mettenleiter T C, Zask L, Zukermann F, Sugg N, Kern H, Ben-Porat T. Interaction of glycoprotein gIII with a cellular heparinlike substance mediates adsorption of pseudorabies virus. J Virol. 1990;64:278–286. doi: 10.1128/jvi.64.1.278-286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mizukami H, Brown K E, Young N. Adeno-associated virus type 2 binds to a 150-kilodalton cell membrane glycoprotein. Virology. 1996;217:124–130. doi: 10.1006/viro.1996.0099. [DOI] [PubMed] [Google Scholar]
- 42.Montgomery R I, Warner M S, Lum B J, Spear P G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 43.Murphy-Ulrich J E, Westrick L G, Esko J D, Mosher D F. Altered metabolism of thrombospondin by chinese hamster ovary cells defective in glycosaminoglycan synthesis. J Biol Chem. 1988;263:6400–6406. [PubMed] [Google Scholar]
- 44.Muzyczka N. Use of adeno-associated virus as a general transduction vector for mammalian cells. Curr Top Microbiol Immunol. 1992;158:97–129. doi: 10.1007/978-3-642-75608-5_5. [DOI] [PubMed] [Google Scholar]
- 45.Ponnazhagan S, Wang X, Woody M J, Luo F, Kang L Y, Madahavi L N, Munshi C M, Shang Z Z, Srivasta A. Differential expression in human cells from the p6 promoter of human parvovirus B19 following plasmid transfection and recombinant adeno-associated virus 2 (AAV) infection: human megacaryocytic leukaemia cells are non-permissive for AAV infection. J Gen Virol. 1996;77:1111–1122. doi: 10.1099/0022-1317-77-6-1111. [DOI] [PubMed] [Google Scholar]
- 46.Rogers G N, Herrler G, Paulson J C, Klenk H D. Influenza C viruses uses 9-O-acetyl-N-acetylneuraminic acid as a high affinity receptor determinant for attachment to cells. J Biol Chem. 1986;261:5947–5941. [PubMed] [Google Scholar]
- 47.Roghani M, Moscatelli D. Basic Fibroblast growth factor is internalized through both receptor-mediated and heparin sulfate mediated mechanisms. J Biol Chem. 1992;267:22156–22162. [PubMed] [Google Scholar]
- 47a.Rolling, F., and R. J. Samulski. Unpublished data.
- 48.Rostand K S, Esko J D. Microbial adherence and invasion through proteoglycans. Infect Immun. 1997;65:1–8. doi: 10.1128/iai.65.1.1-8.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruoslahti E. Proteoglycans in cell regulation. J Biol Chem. 1989;264:13369–13372. [PubMed] [Google Scholar]
- 50.Samulski R J. Adeno-associated virus-based vectors for human gene therapy. In: Hui K M, editor. Gene therapy: from laboratory to the clinic. Singapore, Singapore: World Scientific Publishing Co.; 1995. pp. 232–271. [Google Scholar]
- 51.Samulski R J. Adeno-associated virus: integration at a specific chromosomal locus. Curr Opin Genet Dev. 1993;3:74–80. doi: 10.1016/s0959-437x(05)80344-2. [DOI] [PubMed] [Google Scholar]
- 52.Samulski R J. Development of Adeno-associated virus as a vector for invivo gene therapy. In: Houdebine L M, editor. Transgenic animals: generation and use. Chur, Switzerland: Harwood Academic Publishers; 1997. pp. 197–203. [Google Scholar]
- 53.Samulski R J, Chang L-S, Shenk T. Helper-free stocks of recombinant adeno-associated viruses: normal integration does not require viral gene expression. J Virol. 1989;63:3822–3828. doi: 10.1128/jvi.63.9.3822-3828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samulski R J, Zhu X, Xiao X, Brook J D, Housman D E, Epstein N, Hunter L A. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J. 1991;10:3941–3950. doi: 10.1002/j.1460-2075.1991.tb04964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sattentau Q J, Weiss R A. The CD4 antigen: pysiological ligand and HIV receptor. Cell. 1991;52:631–633. doi: 10.1016/0092-8674(88)90397-2. [DOI] [PubMed] [Google Scholar]
- 56.Saxena U, Klein M G, Goldberg I J. Metabolism of endothelial cell-bound lipoprotein lipase. J Biol Chem. 1990;265:12880–12886. [PubMed] [Google Scholar]
- 57.Sheih M, Montgomery R I, Esko J D, Spear P. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J Cell Biol. 1992;116:1273–1281. doi: 10.1083/jcb.116.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Snyder R O, Xiao X, Samulski R J. Production of recombinant adeno-associated virus vectors. In: Dracopoli N, Haines J, Krof B, Moir D, Seidman C, Seichman J, editors. Current protocols in human genetics. New York, N.Y: John Wiley & Sons Ltd.; 1996. pp. 12.1.1–12.2.23. [Google Scholar]
- 58a.Summerford, C., and R. J. Samulski. Unpublished data.
- 59.Walsh C, Johnson M L, Miller J L, Nienhuis A, Samulski R J. Gene therapy for hemaglobinopathy. Mol Cell Biol. 1994;14:289–299. [Google Scholar]
- 60.Wicham T J, Mathias P, Cheresh D A, Nemerow G R. Integrins αVβ3 and αVβ5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309–319. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- 61.WuDunn D, Spear P. Initial interaction of herpes simplex virus with cells is binding to heparin sulfate. J Virol. 1989;63:52–58. doi: 10.1128/jvi.63.1.52-58.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiao X, Li J, Samulski R J. Efficient long term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yanagishita M, Hascall V C. Metabolism of proteoglycans in rat ovarian granulosa cell culture. J Biol Chem. 1984;259:10270–10283. [PubMed] [Google Scholar]