

Abstract
We have previously described a vaccinia virus late transcription factor, VLTF-X, which we found to be present in cells at early and late times in infection. In this study, transcription complementation assays were used to demonstrate that VLTF-X activity is also present in virion extracts and in the cytoplasm of uninfected HeLa cells. Mobility shift assays performed on various VLTF-X preparations revealed that a late promoter DNA-binding activity cochromatographed and cosedimented with VLTF-X activity. Competition experiments demonstrated that this binding was specific for the late promoter region of the probe and that late transcription was dramatically reduced by an oligonucleotide that blocked factor-DNA complex formation but was only minimally affected by an oligonucleotide that did not inhibit complex formation. These results suggest that a cellular factor may participate in vaccinia virus late transcription. These findings also confirm the requirement for VLTF-X and distinguish it from any of the previously described vaccinia virus late transcription factors, which have all been mapped to the viral genome. Finally, these studies also suggest that the biochemical role for VLTF-X may be in late promoter recognition.
Vaccinia virus is a double-stranded DNA virus that replicates in the cytoplasm of infected cells. Its 192,000-bp genome encodes approximately 200 genes, which are temporally regulated and are classified as early, intermediate, and late depending on their time of synthesis relative to the replication of viral DNA. Expression of early genes begins immediately upon entry of the virus into the host cell and requires, among other factors, the virally encoded RNA polymerase and the vaccinia virus early transcription factor, VETF. Biochemical experiments have shown that VETF demonstrates early promoter-specific DNA-binding activity and is responsible for recruiting the RNA polymerase to early promoters (1, 11). The intermediate genes are expressed only after the onset of viral DNA replication and require several factors, in addition to the viral RNA polymerase, for expression. Thus far, none of these factors has been shown to be a sequence-specific DNA binding protein, and it has been suggested that specific DNA binding may be a function of a complex composed of more than one factor (15).
It has previously been shown that vaccinia virus late transcription can be reconstituted in vitro with extracts from virus-infected HeLa cells (21). These extracts have been fractionated by chromatography on phosphocellulose into three crude components eluting at 0.1, 0.3, and 1.0 M NaCl, all of which were necessary for maximal transcription activity (22). A combination of biochemical and genetic experiments has begun to elucidate which proteins, present in these crude fractions, are necessary to reconstitute late transcription. In addition to the multisubunit virally encoded RNA polymerase, three of these factors have been identified as the 17-, 26-, and 30-kDa protein products of the A1L, A2L, and G8R intermediate genes, respectively (5–8, 14, 18, 20). Recently, two additional factors have been identified as being necessary for late transcription in vitro. One of these factors was purified from the phosphocellulose 1.0 M fraction and has been identified as the 36-kDa product of the vaccinia virus H5R gene (9, 10). The other factor, purified from the 0.3 M fraction, was initially referred to as VLTF-2 but was later renamed VLTF-X in order to designate it as a factor which had not yet been mapped (5, 18). VLTF-X was described as necessary for in vitro transcription and present at early times of infection (18).
The A1L, A2L, G8R, and H5R gene products have all been expressed and purified from heterologous systems, and their ability to stimulate vaccinia virus late transcription in vitro has been documented (5, 8–10, 14, 18). Despite this fact, no late transcription-specific biochemical function has been ascribed to any of them. In the present study, we have further purified the factor designated VLTF-X. The data will demonstrate that an activity which complements for VLTF-X is found in uninfected HeLa cell cytoplasmic extracts and virion extracts, defining it as a unique late transcription factor. In addition, VLTF-X copurifies with a late promoter-specific DNA-binding activity, suggesting that it may be the factor which specifically recognizes vaccinia virus late promoters.
MATERIALS AND METHODS
Purification of factors.
VLTF-X was purified from 30 liters of vaccinia virus-infected HeLa cells essentially as previously described (18) over sequential columns of phosphocellulose, heparin agarose, DEAE-cellulose, and hydroxylapatite (see Fig. 1). However, in addition, the pooled active fractions from the hydroxylapatite column were applied to a second phosphocellulose column and eluted with a 0.1 to 0.35 M NaCl gradient. VLTF-X activity eluted from this column between 0.18 and 0.22 M NaCl. Also, flowthrough fractions from the DEAE column which contained the trailing shoulder of VLTF-X activity were concentrated two- to threefold by ultrafiltration and applied to glycerol gradients.
FIG. 1.
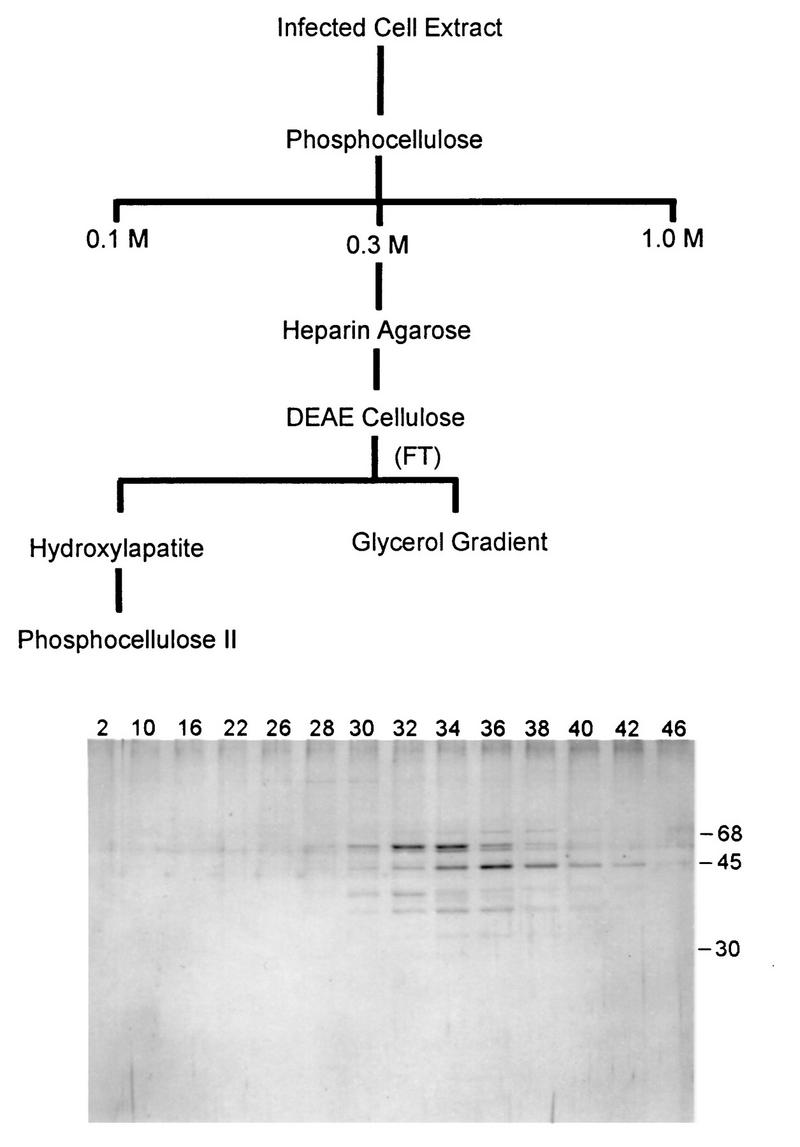
Flow chart of the purification scheme for VLTF-X and a silver-stained polyacrylamide gel of fractions from the phosphocellulose II column. The sizes of molecular-size markers (in kilodaltons) coelectrophoresed with the samples are indicated to the right of the protein gel. This figure and all subsequent figures were processed by using Adobe Photoshop v 3.0 with no enhancements other than brightness and contrast and were printed with a dye sublimation printer.
The A1L and G8R proteins were expressed in a recombinant baculovirus system and purified to the glycerol gradient-pure stage as previously described (18). The A2L protein was also expressed in a recombinant baculovirus system and partially purified to the hydroxylapatite-pure stage as previously described (5). For some of the reactions for which results are shown in Fig. 2 and 4, the A2L protein was expressed as a glutathione S-transferase (GST) fusion protein and purified from Escherichia coli by using glutathione Sepharose. This recombinant fusion protein substitutes for the native protein in transcription reactions (17a). Vaccinia virus RNA polymerase was purified from infected HeLa cells to the glycerol gradient-pure stage as previously described (18).
FIG. 2.

Specific transcription reactions reconstituted with partially purified VLTF-X or virion extracts (VEs). Shown is an autoradiogram of a 4% denaturing polyacrylamide gel in which the radiolabeled RNA products of specific transcription reactions were separated. In lanes 1 to 3 all reaction mixtures contained 3 μl of G8R protein, 2 μl of A1L protein, 4 μl of A2L-GST fusion protein, and 3 μl of the viral RNA polymerase purified from infected HeLa cells (cpol). In addition, the reaction mixture for lane 2 contained 5 μl of hydroxylapatite-purified VLTF-X [VLTF-X (HAP)] and that for lane 3 contained 5 μl of phosphocellulose II-purified VLTF-X [VLTF-X (Pcell II)]. In lanes 4 to 7, 3 μl of G8R protein, 3 μl of A1L protein, and 2.5 μl of A2L protein were present in all reaction mixtures. In addition, the reaction mixtures for lanes 4, 5, and 7 contained 3 μl of viral RNA polymerase purified from infected HeLa cells. Lanes 5 and 6 additionally contained 2.5 μl of a soluble extract (VE) made from sucrose gradient-purified vaccinia virions that was capable of transcribing vaccinia virus early genes (19). Lane 7 additionally contained 5 μl of the flowthrough of two successive DEAE-cellulose columns (DEAE FT) used to purify VETF from the crude virion extracts (19).
FIG. 4.

Electrophoretic mobility shift assays and specific transcription reactions of fractions from the purification of VLTF-X. (Top) Electrophoretic mobility shift assays (MOB SHIFT) with fractions from the hydroxylapatite, phosphocellulose II, and glycerol gradients used to purify VLTF-X (see Fig. 1). Reactions were conducted in 20-μl volumes, which contained approximately 1 ng of a 32P-labeled late promoter-containing fragment as described in Materials and Methods. Proteins used were 2 μl of fractions from the hydroxylapatite column, 4 μl of fractions from the phosphocellulose column, or 7 μl of fractions from the glycerol gradient. The reaction mixtures containing the hydroxylapatite fractions contained 50 ng of poly(dI-dC) · poly(dI-dC) as nonspecific competitor; the reaction mixtures containing the phosphocellulose and glycerol gradient fractions contained 10 ng of poly(dI-dC) · poly(dI-dC) as competitor. Fraction numbers are indicated above the lanes. F indicates the position of free probe. Autoradiograms of the gels are shown. (Bottom) Specific transcription reactions (TX) with fractions from the columns used to purify VLTF-X. Proteins used in transcription reactions were as follows: for the hydroxylapatite column, 1.7 μl of A1L protein, 2 μl of G8R protein, 2 μl of A2L protein, 2.5 μl of RNA polymerase purified from infected cells, and 5 μl of the indicated column fractions (numbers above the lanes); for the phosphocellulose column, 2 μl of A1L protein, 3 μl of G8R protein, 2 μl of A2L protein, 2 μl of RNA polymerase purified from infected cells, and 5 μl of the indicated column fractions; and for the glycerol gradient, 2 μl of A1L protein, 3 μl of G8R protein, 4 μl of A2L-GST fusion protein, 3 μl of RNA polymerase purified from infected cells, and 5 μl of the indicated column fractions. Autoradiograms of the gels are shown as described for Figure 2.
Electrophoretic mobility shift assays.
Electrophoretic mobility shift assays were conducted in a final volume of 20 μl, which contained approximately 1 ng of 32P-labeled target DNA (described below), 2 to 7 μl of column fractions, 20 mM Tris-HCl (pH 7.5), 5% glycerol, 0.1 mM EDTA, 25 μg of bovine serum albumin/ml, 2 mM dithiothreitol, 10 to 35 mM NaCl (depending on the amounts of the protein fractions added), and competitor DNAs as described in the figure legends. The protein fractions were always added to the tubes last. The reaction mixtures were incubated at room temperature for 20 to 30 min, then loaded onto a 4% polyacrylamide gel (acrylamide/bisacrylamide ratio, 80:1) containing 2.5% glycerol and electrophoresed in a buffer containing 6.7 mM Tris-HCl (pH 8), 3.3 mM sodium acetate, and 0.1 mM EDTA for 3 h. The gels were then dried onto Whatman 3M paper and exposed to X-ray film.
Target DNA preparation.
Radiolabeled DNA targets for the mobility shift experiments were prepared by using the plasmid pCFW7, which contains the wild-type promoter and flanking sequences of the vaccinia virus late gene expressing the 11-kDa protein product (F17R) cloned into pUC18 (21). Approximately 1 ng of plasmid DNA was used as the template in PCRs containing PCR buffer II (Perkin-Elmer, Foster City, Calif.); 1.5 mM MgCl2; 0.2 mM deoxynucleotides; 25 ng of M13/pUC forward and reverse primers (Life Technologies, Gaithersburg, Md.), one of which was labeled with [γ-32P]ATP and T4 polynucleotide kinase; and 5 U of AmpliTaq polymerase (Perkin-Elmer). The reaction mixtures were heated to 94°C for 5 min, then cycled for 30 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s, followed by 72°C for 5 min. This created a 273-bp end-labeled fragment which was subsequently gel purified.
Competitor DNA preparation.
Poly(dI-dC) · poly(dI-dC) (Pharmacia Biotech, Piscataway, N.J.) was used as a nonspecific competitor in many reactions. The early promoter-containing oligonucleotide competitor was prepared by annealing the oligonucleotides 5′TAT ATT ACT GAA TTA ATA ATA TAA AAT TCC CAA TCT TGT CAT AAA CA3′ and 5′TGT TTA TGA CAA GAT TGG GAA TTT TAT ATT ATT AAT TCA GTA ATA TA3′ (these sequences are found in the vaccinia virus growth factor (VGF) early gene promoter, which has been used extensively to study VETF-DNA interactions [1]). To form the double-stranded DNA, 40-μl reaction mixtures containing 25 μM each oligonucleotide, 250 mM NaCl, and 10 mM Tris-HCl (pH 8) were placed in a 1.5-ml tube in a beaker of boiling water and the water was allowed to come to room temperature over several hours. The late promoter-containing oligonucleotide competitor was prepared by annealing the oligonucleotides 5′AAG CTT TTT TTT TTT TTT TTT TTT GGC ATA TAA ATA GAC TCG3′ and 5′CGA GTC TAT TTA TAT GAA AAA AAA AAA AAA AAA AAA AAG CTT3′ as described above (these sequences constitute a strong late promoter in vivo [3]). The whole-probe competitor DNA was prepared by using pCFW7 as a template in PCRs with the M13 forward and reverse primers, as described above, except that neither primer was labeled.
Specific transcription reactions.
Specific in vitro transcription assays were conducted under previously described conditions (18, 20, 22). Briefly, protein fractions were incubated with an uncleaved or linearized plasmid containing the late promoter fragment of the gene expressing the 11-kDa protein product fused to 400 bp of DNA lacking G residues in the noncoding strand (17). Reactions were conducted for 30 min at 30°C in a total volume of 50 μl containing 50 mM Tris-HCl (pH 8), 50 mM NaCl, 2 mM dithiothreitol, 0.2 mM EDTA, 2 mM MgCl2, 1 mM ATP, 0.1 mM CTP, 0.02 mM UTP, 5 μCi of [α-32P]UTP (3,000 Ci/mmol), 9% polyvinyl alcohol, 5% glycerol, and approximately 1 μg of DNA template.
RESULTS
Virion extracts complement for VLTF-X activity.
We have previously shown that VLTF-X was absolutely necessary for late transcription in vitro and was present at early and late times in infection but was apparently not present in uninfected-cell extracts or virions (18). Recently, we used an alternate purification scheme for VLTF-X activity that allowed us to identify the A2L protein as a contaminant of our usual preparations of VLTF-X (5). This finding raised the possibility that the previously observed absence of VLTF-X complementation activity in uninfected cells and virion extracts was due to the lack of VLTF-X, the A2L protein, or both. Therefore, VLTF-X was further purified from infected-cell extracts essentially as previously described (18), then passed over an additional phosphocellulose column and eluted with a narrow salt gradient. Figure 1 shows the purification scheme and a silver-stained gel of the fractions from the second phosphocellulose column. While there are bands on the protein gel that correlate with transcription activity, it is not possible to definitively assign any one of them as being VLTF-X. Transcription analysis demonstrated that the fractions from the second phosphocellulose column were free enough of endogenous A2L protein that in vitro transcription could no longer be reconstituted without adding exogenous A2L protein (data not shown).
By using the new VLTF-X preparations and the heterologously expressed A2L protein, the presence of VLTF-X in virion extracts was reevaluated (Fig. 2). Standard transcription assays, employing an uncleaved plasmid containing a late promoter driving a G-less cassette sequence in the absence of added GTP, were used (18, 20, 22). The combination of the A1L, A2L, and G8R proteins and the viral RNA polymerase purified from infected cells did not support transcription alone (Fig. 2, lane 1). However, adding back hydroxylapatite-purified (Fig. 2, lane 2) or phosphocellulose II-purified (Fig. 2, lane 3) VLTF-X reconstituted a high level of transcription. Figure 2, lane 4, is a control for a separate experiment in which the presence of VLTF-X in a virion extract was examined. It has previously been shown that this virion extract will not support late transcription alone (18). As can be seen in Fig. 2, lane 5, this extract from sucrose gradient-purified virions complemented the other components to reconstitute a high level of transcription similar to that seen with purified preparations of VLTF-X. Furthermore, addition of the virion extract abrogated the need for exogenously added infected-cell-purified RNA polymerase (Fig. 2, lane 6), thereby confirming the previous observation that the packaged RNA polymerase works in the late transcription system (18, 19). VLTF-X was additionally observed to be present in the flowthrough of two successive DEAE-cellulose columns used to purify VETF from virions (19) (Fig. 2, lane 7). Further purification of the DEAE flowthrough by phosphocellulose chromatography yielded a late transcription complementation activity eluting in accordance with the chromatographic properties of VLTF-X purified from infected cells (data not shown). Therefore, virion extracts contain an activity that complements for VLTF-X or relieves the need for VLTF-X. This is the only late transcription factor, aside from the viral RNA polymerase, for which virion extracts can substitute.
Detection of VLTF-X-complementing activity in extracts from uninfected HeLa cells.
The above findings suggested that the presence of VLTF-X in uninfected cells should also be reevaluated in the presence of exogenously provided A2L protein. We therefore tested extracts from uninfected cells for their ability to complement for VLTF-X activity. Figure 3, lane 1, shows once again the inability of the A1L, A2L, and G8R proteins and the viral RNA polymerase purified from infected cells to reconstitute transcription. However, adding back fractions from the hydroxylapatite column used to purify VLTF-X from infected cells reconstituted transcription (Fig. 3, lane 2). Surprisingly, adding back a crude cytoplasmic fraction from uninfected HeLa cells, which does not support late transcription alone (data not shown), also reconstituted transcription (Fig. 3, lane 4). It was noticed that the transcript produced by using the uninfected-cell extract appeared to be slightly larger than that produced by using purified VLTF-X from infected cells. To investigate this phenomenon, a linearized template was used, with the result that the transcripts from both systems were the same size (Fig. 3, lanes 3 and 5). Transcription from a crude infected-cell cytoplasmic extract, known to be competent for late transcription, was also examined. Again, there was a size discrepancy between transcription from uncleaved versus linear templates (Fig. 3, lanes 6 and 7). This size difference is perhaps due to the presence of endogenous nucleotides in the unpurified extracts, allowing transcription to proceed beyond the G-less cassette sequences and into the vector. Linearizing the template abrogated this effect. Thus, these experiments demonstrate that cytoplasmic extracts from uninfected HeLa cells contain an activity that substitutes for VLTF-X.
FIG. 3.

Specific transcription reactions reconstituted with VLTF-X purified from infected cells or with uninfected-cell cytoplasmic extracts. The proteins in the reaction mixtures were as follows. Lanes 1 to 5 contained 3 μl of G8R protein, 2 μl of A1L protein, 3 μl of RNA polymerase purified from infected HeLa cells, and 3 μl of A2L protein. Lane 1 contained no additional proteins (−), lanes 2 and 3 contained 3 μl of hydroxylapatite-purified VLTF-X (HAPX), and lanes 4 and 5 contained 3 μl (3 μg) of a cytoplasmic extract made from uninfected HeLa cells (UNIN EXT). Lanes 6 and 7 contained only 5 μl (10 μg) of a cytoplasmic extract made from infected HeLa cells (IN EXT only). Standard transcription reactions were performed in 50 μl with approximately 1 μg of uncleaved template in lanes 1, 2, 4, and 6 or SmaI-linearized template in lanes 3, 5, and 7. Shown is an autoradiogram of the gel as described for Fig. 2.
Copurification of transcription complementation and DNA-binding activities in VLTF-X fractions.
The A1L, A2L, and G8R proteins have all been expressed in heterologous systems and purified. Electrophoretic mobility shift analyses conducted with preparations of these partially purified proteins have not demonstrated that any of them binds to DNA in a sequence-specific manner (unpublished results). In order to determine if VLTF-X binds to DNA, fractions from its purification were tested in specific transcription and standard DNA electrophoretic mobility shift assays (2) using a 32P-labeled 273-bp vaccinia virus late promoter-containing fragment as the target. Specific transcription across the hydroxylapatite column appeared biphasic, peaking in fractions 52 to 68 and again in fractions 76 to 80 (Fig. 4). Transcription experiments with and without exogenously added A2L protein have suggested that the second, smaller peak is probably due to the presence of endogenous A2L protein in these fractions stimulating the trailing edge of VLTF-X activity (4a). Fractions from the hydroxylapatite column were also tested in mobility shift assays. These fractions, which are relatively impure, demonstrated a number of DNA-binding activities; however, there was a strong shift which correlated with the transcription activity of fractions approximately 50 through 70 (Fig. 4). Fractions from the second phosphocellulose column were also tested, and again there was a correlation between band shift and transcription activity in fractions 32 through 36. Similarly, a glycerol gradient used to purify VLTF-X was tested, with the result that the band shift and transcription activities cosedimented at a place in the gradient consistent with the apparent molecular weight of VLTF-X as determined in previous studies (18). Thus, there was a late promoter DNA-binding activity that copurified with transcription activity across the hydroxylapatite and phosphocellulose columns and the glycerol gradient used to purify VLTF-X. It should be noted that this band shift activity occurred in the absence of Mg2+ and nucleotides, although further experiments have demonstrated that the addition of these factors has no apparent effect on the observed shifted bands (data not shown).
Specificity of DNA-binding activity.
The specificity of DNA-binding activity was tested by performing assays in which early or late vaccinia virus promoter-containing DNA fragments or a nonspecific DNA copolymer was allowed to compete with the labeled late promoter probe (Fig. 5). In all assays, the protein factor was added as the last component of the reaction. When a nonspecific DNA [poly(dI-dC) · poly(dI-dC)] was used, the DNA-bound complex formed with the phosphocellulose-purified VLTF-X was partially resistant to a competitor/probe ratio of 20:1 (by weight) but was abrogated at a ratio of 50:1. Additional reactions conducted in the presence of 10 ng of poly(dI-dC) · poly(dI-dC) showed that a late promoter fragment was considerably more effective at reducing late probe binding than an early promoter fragment. The early promoter-containing competitor had no effect on the shifted complexes when present at 10 ng (a 58-fold molar excess over the target) and only partially reduced binding at a 116-fold molar excess. In contrast, the late promoter-containing DNA reduced binding at 2 ng (an approximately 13-fold molar excess over the target) and completely inhibited complex formation at a 66-fold molar excess. The late promoter-containing oligonucleotide is similar to the −30-to-+12 region of the late promoter used as the target sequence (see Materials and Methods for sequences). Unlabeled target DNA almost completely inhibited complex formation at only a fivefold molar excess over the labeled DNA. Thus, despite the similarity in size and overall A/T composition between the early and late oligonucleotides, the late promoter-containing DNA was a more efficient competitor than DNA without a late promoter sequence, suggesting that the observed binding was in the promoter area of the target DNA.
FIG. 5.

Electrophoretic mobility shift assays with phosphocellulose II-purified VLTF-X. Each reaction mixture contained 4 μl of phosphocellulose fraction 35 and the indicated amount of competitor (in nanograms) with the exception of the lane designated C, which contained only the target DNA and no competitor. The competitors are as indicated in the text. Twenty nanograms of early oligonucleotide was approximately a 116-fold molar excess over the target DNA, and 20 ng of late oligonucleotide was approximately a 132-fold molar excess over the target DNA. The positions of free (F) and bound (B) probes are indicated.
DNA-binding activities in crude cell extracts.
Having found a VLTF-X complementing activity in uninfected-cell extracts and a DNA-binding activity that copurified with VLTF-X, it was important to determine if there was a similar DNA-binding activity in extracts from uninfected cells. Preliminary experiments demonstrated that crude cytoplasmic preparations of both infected and uninfected HeLa cells had such an activity that was resistant to over 1,000 ng of poly(dI-dC) · poly(dI-dC) (data not shown). Both cytoplasmic and whole-cell extracts from several different preparations of uninfected HeLa cells demonstrated this activity. Fractionation of infected and uninfected cytoplasmic extracts over phosphocellulose in step fractions demonstrated that this activity predominated in the 0.3 M NaCl fraction in both (data not shown). Figure 6 shows an early oligonucleotide and late oligonucleotide competition experiment performed with crude cytoplasmic extracts from infected or uninfected cells similar to that of Fig. 5 for the purified phosphocellulose fractions of VLTF-X. The DNA-binding activities of both types of extracts behaved similarly, being relatively unaffected by an approximately 3,000-fold molar excess of early oligonucleotide but completely eliminated by a 3,000-fold molar excess of late oligonucleotide. Thus, although these cruder and more concentrated extracts required a quantitatively higher amount of competitor than did the purified fractions of VLTF-X, they behaved qualitatively similarly and were indistinguishable from each other.
FIG. 6.

Electrophoretic mobility shift assays with cytoplasmic extracts from infected or uninfected cells. Each reaction mixture contained 2 μg of protein from cytoplasmic extracts made from infected or uninfected HeLa cells, 500 ng of poly(dI-dC) · poly(dI-dC), and early or late oligonucleotide competitor in the amounts indicated below the lanes (in nanograms). The lanes designated with a minus sign show control reactions lacking cytoplasmic extract. The positions of free (F) and bound (B) probes are indicated.
Inhibition of transcription with oligonucleotides.
In order to independently confirm that the promoter DNA-protein complex seen in the mobility shift experiments is the interaction relevant to transcription, the abilities of the early and late promoter-containing oligonucleotides to inhibit specific transcription were investigated. In these experiments, a cytoplasmic extract from infected cells (used in the experiments of Fig. 3 and 6) was preincubated either with 1,000 ng of poly(dI-dC) · poly(dI-dC) alone or with the early or late promoter-containing oligonucleotides and was then added to standard transcription reactions. The level of transcription when poly(dI-dC) · poly(dI-dC) alone was added to the reactions was 24% that of a control reaction (Fig. 7, lane 2). The early promoter-containing oligonucleotide did not reduce transcription further when present at 1,620 ng (approximately a 108-fold molar excess over template DNA) and reduced transcription to 27% of that with poly(dI-dC) · poly(dI-dC) alone when present at 3,240 ng (Fig. 7, lanes 5 and 6, respectively). In contrast, when the late promoter-containing oligonucleotide was present at 1,620 ng (a 115-fold molar excess over template DNA), transcription was 28% of that with poly(dI-dC) · poly(dI-dC) alone, and when it was present at 3,240 ng, transcription was reduced to a level that was below background. Therefore, specific transcription responded to competition with oligonucleotides in a manner analogous to that in the mobility shift assays, strengthening the argument made by copurification that the protein(s) participating in the mobility shift and transcription assays is the same.
FIG. 7.

Specific transcription reactions performed in the presence of competitors. Standard transcription reactions were performed with an uncleaved template and 4 μl (8 μg) of infected-cell cytoplasmic extract. In the reactions of lanes 2 through 6, the extract was preincubated for 10 min at room temperature with 1,000 ng of poly(dI-dC) · poly(dI-dC) only (lane 2), 1,000 ng of poly(dI-dC) · poly(dI-dC) and 1,620 (lane 3) or 3,240 (lane 4) ng of late promoter-containing oligonucleotide, or 1,000 ng of poly(dI-dC) · poly(dI-dC) and 1,620 (lane 5) or 3,260 (lane 6) ng of early promoter-containing oligonucleotide. The extract in lane 1 (control) was preincubated without competitors. Subsequent to the preincubation, the other components of the transcription reaction were added and the reactions were incubated at 30°C for 20 min. Shown is an autoradiogram of the gel as described for Fig. 2. For quantitation, small areas of the dried gel containing the transcription products and, as blanks, similarly sized areas of the gel directly above the transcripts, were excised and counted in a scintillation counter. For each lane, the value obtained from the blank was subtracted from the value obtained from the gel slice containing the transcription product, resulting in the number used as the value for specific transcription.
DISCUSSION
Further purification of the transcription factor previously designated VLTF-X and heterologous expression and purification of the 26-kDa vaccinia virus late transcription factor have led to a number of new observations. One of the most unanticipated results of the studies presented here is the finding that uninfected-cell cytoplasmic extracts complemented for VLTF-X activity. The simplest explanation for these findings is that a cellular factor is needed to support vaccinia virus late transcription. However, until the factor from uninfected cells is further purified and identified, alternative explanations for these results must be considered. It is possible that VLTF-X purified from infected cells and the factor which complements from uninfected cells are different. In this case, uninfected-cell extracts would be providing an analogous activity or providing an activity which abrogates the need for VLTF-X.
This is not the first account of a cellular factor potentially participating in vaccinia virus transcription, as it has previously been reported that a factor needed for vaccinia virus intermediate gene transcription, VITF-2, was present in nuclear extracts of uninfected cells (16). VITF-2 activity was found in a number of different cell lines, but not in rabbit kidney cells nonpermissive for vaccinia virus K1L mutants or in Trichoplusia ni insect cells. However, VLTF-X does not appear to be the same as VITF-2 because it is found in the cytoplasm of uninfected cells, it has different chromatographic and sedimentation properties from those reported for VITF-2 (16), and VITF-2 preparations apparently do not bind to DNA in electrophoretic mobility shift assays (15).
These results also identify VLTF-X as an activity separate from the other, previously identified late transcription factors. In particular, there has been some question as to the separate identities of VLTF-X and the 36-kDa product of the vaccinia virus H5R open reading frame. In this study, we have found that VLTF-X elutes from phosphocellulose at 0.2 M NaCl, a salt concentration at which the H5R protein would be expected to bind to this resin (9, 10). Also, previous experiments have shown that H5R activity is not found in uninfected cells or purified virions (10). Thus, it would seem that VLTF-X is not the product of the H5R gene. Future experiments to definitively determine whether uninfected cells are the exclusive source of this factor should further help to clarify this issue.
The present study has also demonstrated that virion extracts abrogate the need for exogenously added VLTF-X. Crude extracts from virions complemented for VLTF-X activity, and this activity had the same chromatographic profile over DEAE-cellulose and phosphocellulose as that for VLTF-X purified from infected cells. As for the complementing activity in uninfected cells, the mechanism of this complementation is currently unknown. Virion extracts may actually contain this same protein, or they may contain an analogous factor or one which alleviates the need for VLTF-X. However, if VLTF-X is a cellular factor, this evidence would suggest that at least a portion of a cellular factor needed for late transcription is packaged into virions. The proposed packaging of a cellular protein in poxvirus virions is not unprecedented, as it has previously been shown that a large subunit of cellular RNA polymerase II, as well as several other unidentified cellular proteins, appears to be packaged in a rabbitpox virus system (13).
In addition to unexpectedly finding VLTF-X activity in uninfected cells and virions, these studies demonstrated that a late promoter-specific DNA-binding activity copurified with VLTF-X. We have also found in extracts from uninfected cells a late promoter-specific DNA-binding activity which thus far is indistinguishable from the activity found in infected-cell extracts. This result is in accordance with the finding that uninfected-cell extracts complement for VLTF-X activity.
Competition experiments demonstrated that an oligonucleotide containing a vaccinia virus late promoter sequence was much more effective in blocking the observed DNA-protein interaction than an oligonucleotide containing an early promoter. In concert with these results, it was found that late transcription was dramatically reduced by preincubation of a transcription-competent extract with the late promoter-containing oligonucleotide, but not with an early promoter-containing oligonucleotide. While it will not be possible to prove directly that VLTF-X binds to DNA until it is cloned and heterologously expressed, the results presented here suggest that this factor may be the one which specifically recognizes late promoters. The proposed viral DNA-binding activity of VLTF-X provides an explanation for its apparent packaging into viral particles. Such a mechanism has recently been proposed for the targeting of VETF to virions (12).
Previous studies have shown that mutations in the TAAAT sequence common to all vaccinia virus late promoters dramatically reduce the level of late transcription in vivo (3, 4). In preliminary studies, we have found that a mutation in this TAAAT sequence partially reduces the ability of VLTF-X to bind to the DNA (unpublished results). Further analysis is needed to determine precisely the areas of DNA-protein interaction and to correlate these results with the findings of experiments performed by others to examine sequences critical for late promoter function.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant AI-31220 from the National Institute of Allergy and Infectious Diseases and by departmental funds from the Medical University of South Carolina and the Armed Forces Institute of Pathology.
We thank Stewart Shuman for helpful discussions and critical reading of the manuscript.
REFERENCES
- 1.Broyles S S, Li J, Moss B. Promoter DNA contacts made by the vaccinia virus early transcription factor. J Biol Chem. 1991;263:15539–15544. [PubMed] [Google Scholar]
- 2.Chodosh L A. Mobility shift DNA-binding assay using gel electrophoresis. In: Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley and Sons; 1996. pp. 12.2.1–12.2.10. [Google Scholar]
- 3.Davison A J, Moss B. Structure of vaccinia virus late promoters. J Mol Biol. 1989;210:771–784. doi: 10.1016/0022-2836(89)90108-3. [DOI] [PubMed] [Google Scholar]
- 4.Hänggi M, Bannwarth W, Stunnenberg H G. Conserved TAAAT motif in vaccinia virus late promoters: overlapping TATA box and site of transcription initiation. EMBO J. 1986;5:1071–1076. doi: 10.1002/j.1460-2075.1986.tb04324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Hubbs, A. E. Unpublished data.
- 5.Hubbs A E, Wright C F. The A2L intermediate gene product is required for in vitro transcription from a vaccinia virus late promoter. J Virol. 1996;70:327–331. doi: 10.1128/jvi.70.1.327-331.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keck J G, Baldick C J, Moss B. Role of DNA replication in vaccinia virus gene expression: a naked template is required for transcription of three late trans-activator genes. Cell. 1990;61:801–809. doi: 10.1016/0092-8674(90)90190-p. [DOI] [PubMed] [Google Scholar]
- 7.Keck J G, Feigenbaum F, Moss B. Mutational analysis of a predicted zinc-binding motif in the 26-kilodalton protein encoded by the vaccinia virus A2L gene: correlation of zinc binding with late transcriptional transactivation activity. J Virol. 1993;67:5749–5753. doi: 10.1128/jvi.67.10.5749-5753.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keck J G, Kovacs G R, Moss B. Overexpression, purification, and late transcription factor activity of the 17-kilodalton protein encoded by the vaccinia virus A1L gene. J Virol. 1993;67:5740–5748. doi: 10.1128/jvi.67.10.5740-5748.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovacs G R, Moss B. The vaccinia virus H5R gene encodes late gene transcription factor 4: purification, cloning, and overexpression. J Virol. 1996;70:6796–6802. doi: 10.1128/jvi.70.10.6796-6802.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacs G R, Rosales R, Keck J G, Moss B. Modification of the cascade model for regulation of vaccinia virus gene expression: purification of a prereplicative, late-stage-specific transcription factor. J Virol. 1994;68:3443–3447. doi: 10.1128/jvi.68.5.3443-3447.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Broyles S. Recruitment of vaccinia virus RNA polymerase to early gene promoters by the viral early transcription factor. J Biol Chem. 1993;268:2273–2280. [PubMed] [Google Scholar]
- 12.Li J, Pennington M J, Broyles S S. Temperature-sensitive mutations in the gene encoding the small subunit of the vaccinia virus early transcription factor impair promoter binding, transcription activation, and packaging of multiple virion components. J Virol. 1994;68:2605–2614. doi: 10.1128/jvi.68.4.2605-2614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrison D K, Moyer R W. Detection of a subunit of cellular pol II within highly purified preparations of RNA polymerase isolated from rabbit poxvirus virions. Cell. 1986;44:587–596. doi: 10.1016/0092-8674(86)90268-0. [DOI] [PubMed] [Google Scholar]
- 14.Passarelli A L, Kovacs G R, Moss B. Transcription of a vaccinia virus late promoter template: requirement for the product of the A2L intermediate-stage gene. J Virol. 1996;70:4444–4450. doi: 10.1128/jvi.70.7.4444-4450.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosales R, Harris N, Ahn B-Y, Moss B. Purification and identification of a vaccinia virus-encoded intermediate stage promoter-specific transcription factor that has homology to eukaryotic transcription factor SII (TFIIS) and an additional role as a viral RNA polymerase subunit. J Biol Chem. 1994;269:14260–14267. [PubMed] [Google Scholar]
- 16.Rosales R, Sutter G, Moss B. A cellular factor is required for transcription of vaccinia viral intermediate-stage genes. Proc Natl Acad Sci USA. 1994;91:3794–3798. doi: 10.1073/pnas.91.9.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sawadogo M, Roeder R. Factors involved in specific transcription initiation by the human RNA polymerase II system. J Biol Chem. 1985;259:5321–5326. [PubMed] [Google Scholar]
- 17a.Wright, C. F. Unpublished data.
- 18.Wright C F, Coroneos A M. Purification of the late transcription system of vaccinia virus: identification of a novel transcription factor. J Virol. 1993;67:7264–7270. doi: 10.1128/jvi.67.12.7264-7270.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright C F, Coroneos A M. The H4 subunit of vaccinia virus RNA polymerase is not required for transcription initiation at a viral late promoter. J Virol. 1995;69:2602–2604. doi: 10.1128/jvi.69.4.2602-2604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wright C F, Keck J G, Tsai M M, Moss B. A transcription factor for expression of vaccinia virus late genes is encoded by an intermediate gene. J Virol. 1991;65:3715–3720. doi: 10.1128/jvi.65.7.3715-3720.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wright C F, Moss B. In vitro synthesis of vaccinia virus late mRNA containing a 5′ poly(A) leader sequence. Proc Natl Acad Sci USA. 1987;84:8883–8887. doi: 10.1073/pnas.84.24.8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright C F, Moss B. Identification of factors specific for transcription of the late class of vaccinia virus genes. J Virol. 1989;63:4224–4233. doi: 10.1128/jvi.63.10.4224-4233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]