

ABSTRACT
Electrochemical CO2 reduction (CO2R) in acidic membrane electrode assemblies (MEAs) represents a promising pathway for sustainable chemical production, but achieving high selectivity, low cell voltage and long-term stability remains challenging. Current approaches using alkali cations can promote selectivity through cationic effects, but relying on H2O as a weak proton donor results in high overpotential and severe precipitation, causing elevated cell voltage and poor operational stability. Here, we introduce NH4+ as a proton-donating cation that simultaneously addresses these challenges in acidic MEAs. As a cation, it electromigrates to the catalyst surface, stabilizing *CO2 intermediates and reducing localized H+ concentration for high selectivity. As a proton donor, it provides superior proton-donating ability compared to H2O when H+ mass transport is limited, which decreases the protonation barrier and reduces CO2R overpotential on CoPc@CNT, resulting in a lower cell voltage. Furthermore, NH4+ effectively donates protons to bicarbonate, promoting its decomposition at significantly lower temperatures compared to KHCO3, thereby enabling easy removal of precipitates through mild heating and maintaining an NH3/NH4+ recirculation system for operational stability. As a result, this approach achieves an average CO2-to-CO selectivity of 86% in acidic MEAs at 100 mA cm−2 and 60°C using CoPc@CNT–NH2 catalyst, with stable performance over 110 h at an average cell voltage of 2.84 V, corresponding to a 40.6% energy efficiency. This strategy advances acidic MEA-based CO2R toward practical implementation by simultaneously achieving high selectivity, low overpotential and stable operation.
Keywords: acidic CO2 reduction, cationic effects, proton-donating effects, NH3/NH4+ recirculation, membrane electrode assemblies
Beyond enhancing selectivity via cation effects, this work employs the proton-donating ability of NH4+ to lower both the protonation barrier of CO2 reduction and the bicarbonate decomposition barrier, achieving high energy efficiency and stability in acidic membrane electrode assemblies.
INTRODUCTION
Electrocatalytic CO2 reduction (CO2R) powered by renewable energy represents a promising strategy for converting CO2 into valuable chemical products, offering a sustainable pathway toward carbon-neutral chemical industry [1–3]. While both alkaline and acidic electrolyzers have been extensively studied for CO2 conversion [4,5], acidic electrolyzers offer unique advantages by preventing CO2 carbonation and eliminating CO2 crossover [6–8], thereby enabling higher single-pass CO2 conversion efficiency and avoiding the purification costs associated with CO2 and O2 mixing [9–11]. However, the implementation of acidic CO2R systems faces critical challenges in achieving both high energy efficiency and long-term stability. High energy efficiency requires the maintenance of high product selectivity while operating at low cell voltage. Addressing these challenges is crucial for realizing the full potential of acidic electrolyzers in industrial-scale sustainable chemical production.
The industrial implementation of CO2R requires electrolyzers that can deliver both high energy efficiency and scalability [12]. Among various configurations, membrane electrode assemblies (MEAs) represent the most promising pathway toward commercialization [13]. By eliminating liquid catholyte and enabling zero-gap operation, MEAs significantly reduce ohmic losses and system complexity, providing a practical route to industrial-scale production. While both cation exchange membranes (CEMs) and anion exchange membranes can be utilized in MEA systems [14,15], CEMs offer a distinctive acid-environment advantage in preventing CO2 carbonation and crossover [16]. However, this acidic environment poses a significant challenge for CO2R selectivity, with a limited selectivity of 37% in pure acidic MEAs [17].
To address the selectivity challenge in acidic MEAs, two main strategies have been developed to modify the local reaction environment. The first strategy employs alkali cations to enhance product selectivity by stabilizing reaction intermediates and reducing local H+ concentration [6,9,11,18,19]. However, the fundamental challenge of bicarbonate precipitation emerges during extended operation [20,21], affecting CO2 transport and electrolyte stability, which limits continuous operation to around 20 h [10,22]. The second strategy utilizes polymer cations to circumvent precipitation issues [16,23–25]; however, the limited density of fixed cations poses an intrinsic ionic conductivity challenge in acidic MEAs. To suppress the hydrogen evolution reaction (HER) and achieve high CO2R selectivity, this strategy requires operation in weak acid or pure water environments, where the significant ohmic losses result in prohibitively high cell voltages (3.6–4.0 V) and low energy efficiency (22%–26%) even at moderate current densities (100 mA cm−2) [16,25], presenting a major barrier to practical implementation.
The progress achieved through alkali cations and polymer cations demonstrates that the cationic effects, including stabilizing reaction intermediates and regulating local pH, are essential for high CO2R selectivity. To make acidic CO2 electrolysis practically viable, we need to maintain this high selectivity while addressing the remaining challenges in cell voltage and stability. The decomposition of bicarbonate requires proton participation, and a more efficient proton source could facilitate bicarbonate decomposition, preventing precipitation and enhancing operational stability. Moreover, our previous mechanistic studies on CoPc@CNT catalyst reveal that protonation is the potential-determining step in the CO2R process [17]. The reliance on water as a proton donor, with its inherently weak proton-donating ability (i.e. a high thermodynamic barrier for proton donation), contributes to a high overpotential. A stronger proton donor could potentially lower the barrier, thereby reducing cell voltage and improving energy efficiency. Therefore, designing a system that can provide efficient ability of proton donating while maintaining the beneficial effects of cationic species becomes crucial for achieving high selectivity, low cell voltage and long-term stability in acidic CO2 electrolysis.
To address these challenges, we propose that a proton-donating cation could provide an ideal solution by combining the beneficial properties of cations with enhanced proton-donating ability (Fig. 1). Our computational and experimental studies identify NH4+ as an optimal candidate that simultaneously fulfills the dual requirements of our design strategy. First, like alkali cations and polymer cations, NH4+ maintains high CO2-to-CO selectivity by stabilizing reaction intermediates and reducing local H+ concentration (Fig. 1a). Second, and uniquely, NH4+ acts as a superior proton donor compared to water under mass transport-limited conditions for H+ (Fig. 1b), effectively lowering the protonation barrier and reducing the CO2R overpotential on CoPc@CNT by 280 mV at 100 mA cm−2. This reduction in cathodic overpotential contributes to lowering of the cell voltage of the MEA system, thereby enhancing energy efficiency. Furthermore, NH4+ proves to be a more efficient proton donor than HCO3−, leading to a significantly lower decomposition temperature for NH4HCO3 relative to KHCO3 (Fig. 1b). This property facilitates the easy removal of NH4HCO3 precipitates via mild heating, allowing NH4+ to function within an NH3/NH4+ recirculation system (Fig. 1c). Consequently, the facile thermal removal of precipitated salts enables continuous NH4+ supply, fundamentally resolving the long-standing salt precipitation issue in acidic MEAs and maintaining consistent performance throughout the reaction. As a result, CoPc@CNT–NH2 catalyst achieves an average CO2-to-CO selectivity of 86% at a current density of 100 mA cm−2 and 60°C in acidic MEAs, with stable performance sustained over 110 h with an average cell voltage of 2.84 V, corresponding to an energy efficiency of 40.6%. To the best of our knowledge, this represents the most stable system among acidic MEAs while maintaining high energy efficiency throughout the CO2R operation. With salt precipitation issues resolved, the system's long-term stability now depends primarily on catalyst stability, suggesting that more stable catalysts would further enhance operational longevity. These findings present a novel strategy for overcoming the inherent challenges of acidic CO2R, offering a sustainable pathway toward carbon-neutral chemical production.
Figure 1.

Schematic of NH4+-mediated CO2R in acidic MEAs. NH4+ enables efficient and stable CO2R with cationic, proton-donating effects, supported by an NH3/NH4+ recirculation system. (a) Cationic effects: NH4+ migrates and accumulates at the electrode surface, stabilizing *CO2 intermediates through dipole–electric field interactions, and suppressing H+ migration due to the competitive migration of NH4+. (b) Proton-donating effects: NH4+ donates protons more readily than water, facilitating the protonation of *CO2 and lowering the CO2R overpotential. Furthermore, NH4+ effectively donates protons to bicarbonate, promoting bicarbonate decomposition. (c) NH3/NH4+ recirculation system: NH4HCO3 precipitates decompose at low temperatures into NH3, which is protonated by H+ in an ‘NH3 adsorption container’ to form NH4+. These NH4+ ions diffuse back to an ‘NH4+ supply container’, ensuring a steady NH4+ supply.
RESULTS AND DISCUSSION
The cationic effects of NH4+ on acidic CO2R
Unlike conventional alkali cations, NH4+ possesses dual functionality as both a cation and a proton donor, making it uniquely suited for acidic CO2R. To understand how NH4+ enhances CO2R selectivity through its cationic properties, we first investigate its effects on the reaction interface using computational calculations.
COMSOL simulations are conducted to assess the effects of NH4+ on localized H+ concentration under reaction conditions. Given that cations significantly accumulate at the electrode surface under the influence of electric field, both spatial constraints and ion size must be considered in the simulations. Therefore, we employ the generalized modified Poisson-Nernst-Planck (GMPNP) model [20,26–28], which accounts for electric field effects, diffusion and reactions to precisely calculate species concentration distributions in the reaction region (Fig. 2a). The simulation details are provided in the Methods and Table S1 of the Supplementary data.
Figure 2.

The cationic effects of NH4+ on acidic CO2R. (a) Schematic of ion transport near the cathode based on the GMPNP model, showing the Stern layer, diffuse layer and diffusion layer, with electromigration and diffusion processes for solvated ions (H+, NH4+ and K+). (b–d) Ion concentration profiles as a function of distance from the cathode for different electrolytes: (b) 0.1 M H2SO4, (c) 0.1 M H2SO4 and 0.5 M (NH4)2SO4, and (d) 0.1 M H2SO4 and 0.5 M K2SO4, simulated under varying current densities. (e) Atomic structure of NH4+-containing electrolyte with CO2 adsorption on CoPc@CNT. (f) Averaged electric field distribution along the z-axis near the catalyst surface under different ion conditions (H2O, K+ and NH4+). (g) Adsorption energy of CO2 on CoPc@CNT in the presence of H2O, K+ and NH4+.
Both the CO2-to-CO conversion and HER similarly affect the electrode interface, as they involve proton consumption at the catalyst surface. The corresponding electrode reactions are as follows:
 |
(1) |
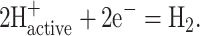 |
(2) |
Here,  refers to the active protons involved in the HER or CO2R processes (Fig. S1). In the case of a pure acidic system, the sources of
refers to the active protons involved in the HER or CO2R processes (Fig. S1). In the case of a pure acidic system, the sources of 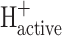 are H+ and H2O. For the NH4+ system,
are H+ and H2O. For the NH4+ system,  originates from H+, NH4+ and H2O. It is important to note that NH4+ and water supply
originates from H+, NH4+ and H2O. It is important to note that NH4+ and water supply  for HER or CO2R, and the resulting NH3 and OH− quickly react with H+ when H+ is sufficient (Fig. S2), converting back to NH4+ and H2O:
for HER or CO2R, and the resulting NH3 and OH− quickly react with H+ when H+ is sufficient (Fig. S2), converting back to NH4+ and H2O:
 |
(3) |
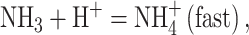 |
(4) |
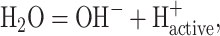 |
(5) |
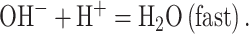 |
(6) |
From an overall reaction perspective, the protons consumed in the electrode reactions all originate from H+. These reactions establish the framework for analyzing species distribution during CO2R. We compare the species distributions in 0.1 M H2SO4 alone and with the addition of 0.5 M (NH4)2SO4, at current densities ranging from 50 to 200 mA cm−2 (Fig. 2b and c). In the pure acidic system (0.1 M H2SO4), a significant accumulation of H+ at the surface occurs under the influence of the electric field, approaching a saturation concentration of 9.50 mol L−1 (Fig. 2b). In the NH4+-containing system, under the influence of the electric field, NH4+ competes with H+ for accumulation at the surface. As the current density increases, the concentration of H+ gradually decreases, while the concentration of NH4+ continues to rise. At the current density of 100 mA cm−2, the surface H+ concentration in the NH4+-containing system is 0.58 M, 6% of that in the pure acidic system. At 200 mA cm−2, it even decreases to 0.12 M, 1.3% of the pure acidic system's concentration.
When comparing the effects of different cations, we found that NH4+ produces species distributions similar to those observed with K+ (Fig. 2d). This can be attributed to two main factors. First, both NH4+ and K+ are cations that compete with H+ for electromigration, which effectively reduces the localized concentration of H+. Second, the comparable hydrated radii and migration rates of NH4+ and K+ result in similar electromigration behaviors. As shown above, NH4+ effectively reduces the localized H+ concentration, thereby inhibiting the conversion of H+ to H2. These findings demonstrate that NH4+ effectively modulates the reaction interface by reducing local H+ concentration, which is crucial for suppressing the competing HER.
Beyond controlling H+ concentration, the accumulated NH4+ at the surface may create a localized electric field that stabilizes key reaction intermediates, particularly those with large dipole moments such as *CO2 [18]. To investigate this effect, we perform density functional theory (DFT) calculations using CoPc@CNT as a model catalyst system. This well-defined single-atom catalyst not only enables accurate computational modeling [29] but also demonstrates high activity under acidic conditions in our previous study [17], making it ideal for studying the role of NH4+ in acidic CO2R. Based on this, we examine the electric field distribution at the interface under various electrolyte conditions (Fig. 2e and f, and Fig. S3). Pure water without cations is used as the blank control, while K+, a well-known cation capable of creating a localized electric field [30,31], serves as the reference. In the presence of pure water, the interfacial electric field remains negligible due to the lack of significant charge separation between water molecules and the CoPc@CNT catalyst. However, upon introducing NH4+ into the electrolyte, a pronounced electric field develops at the interface, with the field strength peaking at z = 15 Å, corresponding to the position of adsorbed *CO2. It is important to note that the electric field strength shown here is z-averaged, and the local cation-induced electric field is significantly stronger in the vicinity of the cation. Thus, we anticipate a much stronger field strength near the active Co site when the cation is close to the Co. Interestingly, we found that the electric field induced by NH4+ closely resembles that generated by K+, suggesting that NH4+ and K+ can induce similar field–dipole interactions. As shown in Fig. 2g, both K+ and NH4+ significantly stabilize the *CO2 intermediate, increasing its binding energy by approximately 0.3 eV. This stabilization is attributed to the large dipole moment of *CO2, which is highly sensitive to the local electric field. The negative field stabilizes the dipole, thus favoring the activation of CO2. Since *H has a minimal dipole moment and is therefore less sensitive to the electric field, our previous work indicates that *H and *CO2 compete with each other in the absence of cations [17]. Consequently, the presence of cations is expected to have a more pronounced promotional effect on CO2R. Integrating the results from COMSOL simulations and DFT calculations, NH4+ electromigrates and accumulates at the catalyst surface, leading to a reduction in localized H+ concentration and stabilization of *CO2 intermediates, which ensures the selectivity for CO2-to-CO conversion.
Collectively, our computational calculations reveal that NH4+ serves two essential functions as a cation: reducing local H+ concentration through competitive electromigration, and stabilizing reaction intermediates via field–dipole interactions. These effects work synergistically to promote CO2R selectivity while suppressing the competing HER, addressing a key challenge in acidic CO2R systems.
The proton-donating effects of NH4+ on acidic CO2R
Having established the role of NH4+ as a cation in modulating the reaction interface, we next investigate its function as a proton donor. While CO2R involves protonation as a key elementary step, the conventional proton source H2O (pKa = 14) presents a high kinetic barrier [32]. NH4+, with a significantly lower pKa of 9.25, could potentially serve as a more efficient proton donor. For our model catalyst CoPc@CNT, where protonation is the potential-determining step [17], this property of NH4+ may substantially reduce the overpotential required for CO2R.
To systematically investigate the proton-donating abilities of NH4+, we first employ rotating disk electrode (RDE) measurements to compare different proton sources under conditions where the mass transfer of H+ is limiting. Linear sweep voltammetry (LSV) measurements during HER are used to analyze the onset potentials at which NH4+ and H2O act as proton sources. Figure 3a shows the LSV results for HER at a rotation speed of 900 r/min in various acidic electrolytes. For the 0.1 M H2SO4 electrolyte, the HER current density increases as the potential becomes more negative, and the LSV curve follows the typical e-exponential behavior described by the Butler–Volmer equation (Fig. S4a) [33]. In contrast, for the electrolyte system with the addition of NH4+ and K+, the LSV curve exhibits distinct differences, with the curve clearly divided into three regions. For the NH4+-containing electrolyte, in the first region (−0.6 to −1.0 V), H+ reduction occurs, but the current density is significantly lower than in the pure acidic system (0.1 M H2SO4). This reduction is attributed to NH4+ lowering the localized H+ concentration, which leads to an elevated local pH, as confirmed by the RDE measurements (Fig. S5). In the second region (−1.0 to −1.2 V), the current density remains nearly constant despite increasingly negative potentials, which is characteristic of mass transport limitation [20]. In this case, the reactant is H+. This phenomenon is not observed under pure acidic conditions (Fig. S4a), indicating that the mass transport limitation of H+ is caused by cations. Higher plateau current densities observed with increased rotation speeds from 900 to 2500 r/min confirm that this potential range is H+ diffusion-limited (Fig. S4b). In the third region (potentials more negative than −1.2 V), the increased current density indicates the involvement of a new proton source in HER. Similarly, LSV curves obtained in an acidic solution containing K+ also display three distinct regions (Fig. 3a and Fig. S4c). The key difference lies in the onset potential of the third region, which corresponds to the potential at which the new proton source begins to participate in HER. In the K+-containing system, water acts as the proton source at −1.5 V (Fig. 3b), whereas in the NH4+ system, HER begins at −1.2 V. The different onset potentials confirm that NH4+, rather than water, serves as the proton source in the NH4+ system (Fig. 3c). The more positive onset potential in the NH4+ system demonstrates that NH4+ is a more effective proton donor than H2O. This characteristic is expected to lower the protonation barrier in the CO2R process, thereby reducing the overpotential and facilitating CO2R.
Figure 3.

The proton-donating effects of NH4+ on acidic CO2R. (a) LSV curves for 0.1 M H2SO4 with 0.5 M (NH4)2SO4 or 0.5 M K2SO4, and for 0.1 M H2SO4 alone, measured using a rotating disk electrode at 900 r/min in an Ar-saturated environment. (b and c) Schematic illustration of ion distribution at the cathode in the presence of (b) K+, where H2O acts as the proton donor, and (c) NH4+, where NH4+ serves as the proton donor. Both K+ and NH4+ hinder the migration of H+ to the electrode surface. (d) Free energy diagrams for CO2R pathways on CoPc@CNT at −1 V vs SHE, comparing H2O and NH4+ as proton donors. (e) Calculated energy span as a function of potential for H2O and NH4+. (f) CO partial current density as a function of potential in 0.1 M H2SO4 electrolytes containing NH4+ or K+. (g) Gibbs free energy diagrams comparing the proton-donating abilities of NH4+ and HCO3−. (h) Thermogravimetric analysis of NH4HCO3 and KHCO3, showing the decomposition temperatures of bicarbonate species. (i) Weight loss profiles of NH4HCO3 at different temperatures.
These experimental results clearly demonstrate the superior proton-donating ability of NH4+ compared to H2O. To understand the thermodynamic basis for this enhancement, we turn to DFT calculations. Our recent work demonstrates that the pH level in proton-coupled electron transfer (PCET) processes can be correlated with the pKa of the proton source [34]. The chemical potential of a coupled proton and electron is determined by the equilibrium with H2 gas at the hydrogen electrode as shown:
 |
(7) |
where  is the Gibbs free energy of a coupled proton–electron pair,
is the Gibbs free energy of a coupled proton–electron pair,  is the Gibbs free energy of H2 in the gas phase at standard state, U is the electrode potential vs standard hydrogen electrode (SHE),
is the Gibbs free energy of H2 in the gas phase at standard state, U is the electrode potential vs standard hydrogen electrode (SHE),  is the Boltzmann constant, T is the absolute temperature, and pKa is the negative log10 of the acid dissociation constant of the relevant acid.
is the Boltzmann constant, T is the absolute temperature, and pKa is the negative log10 of the acid dissociation constant of the relevant acid.
As protons are always reactants in these systems, lowering the pKa of the proton source decreases the overall reaction energy, thereby affecting both the equilibrium and onset potentials. When the H+ mass transfer is limited, water (with a pKa of 14) typically acts as the proton source. However, NH4+ (with a lower pKa of 9.25) can facilitate PCET processes more efficiently. The CO2 conversion to CO on CoPc@CNT proceeds through two key PCET steps: CO2(g) to *COOH, followed by *COOH to CO(g). As shown in Fig. 3d, the first step (CO2 to *COOH) is the potential-determining step, with *COOH being the intermediate with the highest energy level along the reaction pathway. When NH4+ is used as the proton source, there is a significant reduction in the overall energy span (Δ), as follows:
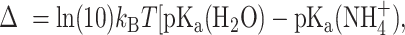 |
(8) |
where  and
and  are pKa values of H2O and NH4+. This is independent of the electrode potential, as depicted in Fig. 3e. Given that the CO2-to-CO reaction involves two PCET steps, the equilibrium potential when NH4+ is used as the proton source is reduced by 2Δ, approximately 0.56 V (Fig. S6). Thus, the pKa of the proton source significantly impacts both the energy span and equilibrium potential for CO2R to CO. A lower pKa results in a reduced energy span, implying slower kinetics according to the Bell–Evans–Polanyi (BEP) relationship when compared to that when water is used as the proton source. Meanwhile, the decrease in equilibrium potential provides a stronger thermodynamic driving force for CO2 conversion to CO when NH4+ is present. Therefore, compared to water, NH4+ offers superior kinetic and thermodynamic advantages, which can effectively reduce the overpotential in CO2R.
are pKa values of H2O and NH4+. This is independent of the electrode potential, as depicted in Fig. 3e. Given that the CO2-to-CO reaction involves two PCET steps, the equilibrium potential when NH4+ is used as the proton source is reduced by 2Δ, approximately 0.56 V (Fig. S6). Thus, the pKa of the proton source significantly impacts both the energy span and equilibrium potential for CO2R to CO. A lower pKa results in a reduced energy span, implying slower kinetics according to the Bell–Evans–Polanyi (BEP) relationship when compared to that when water is used as the proton source. Meanwhile, the decrease in equilibrium potential provides a stronger thermodynamic driving force for CO2 conversion to CO when NH4+ is present. Therefore, compared to water, NH4+ offers superior kinetic and thermodynamic advantages, which can effectively reduce the overpotential in CO2R.
To confirm that NH4+ more effectively promotes the protonation process in CO2R compared to H2O, CO2R activity tests are operated by using a three-electrode flow cell. Unlike two-electrode MEAs, the three-electrode system can accurately evaluate the effect of cations on the cathode potential, minimizing interference from anode-related factors such as gas bubbles and device assembly during testing. Figure 3f and Fig. S7 shows that NH4+-mediated CO2R operates at a more positive reaction potential than K+-mediated CO2R, resulting in a lower overpotential. At a partial current density of 100 mA cm−2 for CO production, the overpotential with NH4+ is up to 280 mV lower than that with K+. In addition, as shown in Fig. S8, the overpotentials for CO2R mediated by Li+ and Na+ are also higher than that of NH4+. These confirm that NH4+, with its superior proton-donating ability compared to H2O, more effectively facilitates the protonation process on CoPc@CNT, thereby significantly reducing the overpotential.
The strong proton-donating ability of NH4+ plays dual beneficial roles in acidic CO2R. First, as demonstrated above, it effectively reduces the reaction overpotential. Second, this same property proves advantageous in addressing another critical challenge in CO2R systems—the formation and removal of bicarbonate precipitates. In both NH4+ and conventional cation systems, the reaction of CO2 with alkaline species (OH− and NH3) leads to bicarbonate precipitation (KHCO3 and NH4HCO3), which can obstruct CO2 transport channels and compromise system stability. The key to removing these precipitates lies in their decomposition mechanism, which fundamentally involves proton transfer processes. Here, the superior proton-donating ability of NH4+ plays a crucial role in facilitating this decomposition. Given that thermal treatment can decompose bicarbonate precipitates, understanding the decomposition mechanisms of KHCO3 and NH4HCO3 is crucial for developing effective mitigation strategies. The decomposition of bicarbonates requires proton transfer processes, as the formation of final products (CO2 and H2O) necessitates the combination of protons with bicarbonate ions. The key difference between KHCO3 and NH4HCO3 lies in their proton transfer pathways during decomposition. The decomposition of KHCO3 requires intermolecular proton transfer between adjacent molecules, while NH4HCO3 can decompose through intramolecular proton transfer:
 |
(9) |
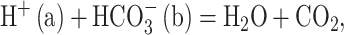 |
(10) |
 |
(11) |
 |
(12) |
Here, a and b represent two adjacent KHCO3 molecules. The energy difference between these decomposition pathways primarily stems from the distinct proton donation barriers of HCO3− and NH4+. In KHCO3, protons must transfer between neighboring molecules, creating a higher energy barrier. In contrast, NH4HCO3 contains its own proton source (NH4+), enabling more efficient intramolecular proton transfer.
DFT calculations indicate that the proton-donation barrier for NH4+ is lower than that for HCO3− (Fig. 3g). This implies that the decomposition temperature of NH4HCO3 is substantially lower than that of KHCO3. Thermogravimetric (TG) analysis further corroborates this finding (Fig. 3h and Fig. S9), with decomposition temperatures of NH4HCO3 and KHCO3 observed at 45°C and 133°C, respectively. Moreover, the decomposition of NH4HCO3 does not yield solid residues, allowing for its complete decomposition. As the temperature increases, the decomposition rate of NH4HCO3 accelerates (Fig. 3i). In contrast, KHCO3 decomposes to form K2CO3, which is more thermally stable and resists further decomposition. This makes it challenging to fully remove KHCO3 through mild heating.
These findings comprehensively demonstrate the dual advantages of NH4+ as a proton donor in acidic CO2R. First, its lower pKa compared to H2O enables more efficient proton transfer, leading to a significant reduction in overpotential (up to 280 mV lower than K+-mediated systems) during CO2R. Second, while both NH4+ and K+ systems face challenges from bicarbonate precipitation, the intramolecular proton transfer ability of NH4HCO3 enables its complete decomposition at mild temperatures (45°C vs 133°C for KHCO3). These unique properties suggest that NH4+-mediated systems can achieve cell voltage through enhanced proton donation and good stability through facile precipitate removal, addressing two key challenges in acidic CO2R.
NH3/NH4+ recirculation system enables stable CO2R in acidic MEA
Based on our understanding of the functions of NH4+ and the thermal decomposition behavior of NH4HCO3, we design an NH3/NH4+ recirculation system to address a critical challenge in MEA operation: maintaining stable NH4+ supply (Fig. 1c and Fig. S10). In MEA devices, NH4+ at the cathode interface is sourced from an ‘NH4+ supply container’, while bicarbonate precipitated at the cathode consumes and decreases its concentration in the supply container. Insufficient NH4+ at the cathode would weaken its crucial roles in stabilizing reaction intermediates, moderating local acidity and providing protons for CO2R. Our system incorporates a mild heating strategy to achieve dynamic equilibrium of NH4+ concentration. The anolyte contains both NH4+ and H+ ions, with NH4+ migrating to the cathode under the electric field to fulfill its multiple functions. When NH4HCO3 precipitation occurs, mild heating facilitates its decomposition into NH3, CO2 and H2O. The gaseous NH3 can be captured by an ‘NH4+ adsorption container’, where it reacts with H+ to regenerate NH4+. These regenerated NH4+ ions can then diffuse through the gas–liquid separation membrane to the ‘NH4+ supply container’. This NH4+/NH3 cycling system maintains sufficient NH4+ concentration at the cathode to sustain both its cationic and proton-donating effects. In contrast, K+-mediated MEAs lack such a self-regulating mechanism. Once K+ is lost to KHCO3 precipitation, it cannot be readily recovered through thermal decomposition and gas-phase transport, leading to continuous depletion of K+ supply and eventual system failure. Therefore, this NH3/NH4+ recirculation system provides a robust foundation for achieving stable CO2R in acidic MEAs.
Before experimentally verifying the stability of NH4+-mediated acidic MEAs, anolyte composition should be first determined, especially for H+ concentration. The H+ concentration in the anolyte plays a critical role in both reaction selectivity and system stability through two mechanisms [10,22]. First, H+ depletion at the cathode interface is necessary for achieving high CO2R selectivity by suppressing the competing HER. Second, H+ helps neutralize the in situ-formed HCO3− in the diffusion layer, preventing undesired salt precipitation. These dual roles of H+ create competing requirements for its concentration. At low H+ concentrations (e.g. 0.1 M), while H+ depletion is readily achieved at low current densities favoring CO2R selectivity, the insufficient H+ cannot effectively neutralize the HCO3− formed from the reaction between NH3 (produced by NH4+ proton donation), CO2 and H2O, leading to salt precipitation. Conversely, higher H+ concentrations (e.g. 0.6 M) better prevent salt precipitation but require impractically high current densities (>150 mA cm−2) to achieve the H+ depletion necessary for high selectivity. To address this trade-off, we fixed the NH4+ concentration at 0.2 M while varying H+ concentrations. SO42− was chosen as the counter anion for its chemical inertness at both electrodes. Selectivity analysis (Fig. 4a and Fig. S11) reveals that intermediate H+ concentrations (0.2 and 0.4 M) achieve the best balance. While both concentrations show comparable CO selectivity at 100 mA cm−2, the higher H+ concentration (0.4 M) demonstrates better stability by more effectively preventing salt precipitation (Fig. 4b). Therefore, we determine the anolyte composition to be 0.1 M (NH4)2SO4 and 0.2 M H2SO4, which balances high selectivity with good stability at our target current density of 100 mA cm−2. Notably, the NH4+/H+ ratio used in the MEA system is significantly lower than that in flow cells, as the absence of flowing catholyte in MEAs minimizes the convective impact on the local pH.
Figure 4.

NH3/NH4+ recirculation system enables stable CO2R in acidic MEA. (a) CO-FE tests at various H+ concentrations with 0.2 M NH4+ in the anolyte on CoPc@CNT. (b) Stability tests with anolytes containing 0.2 M NH4+ and either 0.2 M or 0.4 M H+. (c) CO-FE tests under temperatures from 25 to 60°C, using 0.2 M NH4+ and 0.4 M H+ in the anolyte at 100 mA cm−2. (d) CO-FE tests under CO2 pressures of 1–3 atm at 60°C under identical conditions. (e) Stability tests at 100 mA cm−2, 3 atm and 60°C, comparing catalyst loadings of 1 and 2 mg cm−2. (f) Polarization curves recorded before and after stability testing at 1 mg cm−2 catalyst loading.
The temperature is another key parameter for system performance, as it directly influences the NH4HCO3 decomposition. Figure 4c illustrates that as the temperature increases, stability improves gradually, with stability at 60°C remaining essentially unchanged over a period of 3 h. However, this enhancement in stability comes at the expense of CO selectivity, which decreases from 90% to 72%. This decrease is primarily attributed to the greater influence of temperature on the kinetics of the HER compared to CO2R, with elevated temperatures significantly enhancing HER kinetic activity. To enhance selectivity, increasing the CO2 partial pressure effectively reduces the CO2 adsorption barrier and improves the competitiveness of CO2R relative to HER. Considering that CO2 is typically stored and transported under pressurized conditions (50–100 atm), the use of CO2 at pressures below 10 atm does not require additional energy input. Figure 4d (details in Fig. S12) shows that increasing the pressure from 1 to 3 atm leads to an improvement in CO2R selectivity, increasing from 73% to 80% at a current density of 100 mA cm−2. However, further pressure increases are not pursued, as they have a minimal effect on selectivity while significantly increasing the demands on reactor gas tightness.
Under these conditions, we evaluate the stability of NH4+-mediated MEAs. The system demonstrates stable operation at 100 mA cm−2 for 10 h (Fig. 4e). Interestingly, we observe an increase in CO2R selectivity from 80% to 90% during the initial 10 h, which we attribute to gradual catalyst deactivation. As the number of active catalytic sites decreases, the increased current density per site further reduces local H+ concentration, suppressing HER and enhancing CO2R selectivity. This catalyst deactivation is confirmed by the decreased current density in post-stability cyclic voltammetry measurements (Fig. 4f). Notably, increasing the catalyst loading from 1 to 2 mg cm−2 extends the stable operation period from 10 to 20 h (Fig. 4e and Fig. S13). These results demonstrate that our NH3/NH4+ recirculation strategy effectively solves the salt precipitation challenge in acidic MEAs, with system stability now primarily limited by catalyst deactivation rather than precipitate-related issues. This finding motivates us to focus on catalyst engineering to further improve the stability of acidic CO2R systems.
NH4+-mediated stable CO2R on CoPc@CNT–NH2
Having established an effective NH3/NH4+ recirculation strategy, we next focus on improving catalyst stability to achieve stable operation. The deactivation of CoPc@CNT primarily stems from Co atoms detaching from the phthalocyanine ring during the reaction [35]. Previous studies have shown that CoPc supported on amino-functionalized CNT (CNT–NH2) exhibits enhanced electrochemical stability [36], as the lone-pair electrons from –NH2 groups can coordinate axially with Co atoms, preventing their detachment. Based on this insight, we synthesize the molecularly dispersed catalyst CoPc@CNT–NH2 (Figs S14–S16), and evaluate its performance in acidic MEAs with a catalyst loading of 2 mg cm−2.
To further mitigate the effects of salt precipitation on device performance during prolonged stability testing, we implement an operational protocol consisting of 10-hour reaction cycles followed by 1-hour intermittent pauses. These strategic interruptions serve dual purposes: first, they allow for the maintenance of optimal CO2 humidification conditions through the replenishment of water in the humidification bottle, otherwise dry CO2 would remove water from the cathode region, accelerating salt precipitation; second, they provide sufficient time for the complete decomposition of accumulated NH4HCO3, preventing its precipitation within the system. Based on this, the CoPc@CNT–NH2 catalyst maintains an average CO2-to-CO selectivity of 86% at 100 mA cm−2 for 110 h at an average cell voltage of 2.84 V, corresponding to a 40.6% energy efficiency (Fig. 5a and b, and Fig. S17). To the best of our knowledge, this represents the most stable system among acidic MEAs while maintaining high energy efficiency throughout the CO2R operation (Fig. 5c and Table S2). Furthermore, compared to the most stable neutral MEA systems reported recently [37] (Table S3), our system exhibits clear advantages in both single-pass conversion efficiency (Fig. S18) and cell voltage (Tables S3 and S4), which translate into significantly reduced energy consumption (Fig. S19) and operational costs (Fig. S20).
Figure 5.

NH4+-mediated stable CO2R on CoPc@CNT–NH2. (a and b) Comparison of stability tests for NH4+- and K+-mediated CO2R systems at 0.2 M NH4+/K+ and 0.4 M H+, using CoPc@CNT–NH2 catalyst under conditions of 60°C and 3 atm CO2 at 100 mA cm−2. (c) Comparison of energy efficiency and stability with literature reports. (d) Cross-sectional SEM image and EDS mapping of gas diffusion electrode from the K+-mediated system, showing extensive salt precipitation throughout the electrode structure. (e) Cross-sectional SEM image and EDS mapping of gas diffusion electrode from the NH4+-mediated system, with N signals confined to the catalyst layer, confirming stable operation without salt precipitation.
It should be noted that the system's stability is currently constrained by catalyst degradation rather than salt participation issues. Inductively coupled plasma analysis reveals a significant decrease in Co content in CoPc@CNT–NH2 after electrolysis (Fig. S21 and Table S5). This is further supported by transmission electron microscopy observations showing reduced Co signal intensity (Figs S15 and S22), and X-ray photoelectron spectroscopy results indicating a decline in the Co–N ratio (Fig. S23). Meanwhile, X-ray diffraction analysis shows no evidence of new phase formation (Fig. S24). Together, these characterizations confirm that the primary degradation pathway of CoPc@CNT–NH2 involves the leaching of Co from the phthalocyanine structure. Notably, the Co leaching rate in CoPc@CNT–NH2 is one-fifth of that in CoPc@CNT (Table S5), which correlates well with the slower rate of cell voltage increase observed during operation (Fig. S25). Moreover, the NH4+ concentration remains stable in both the NH3 adsorption container and the NH4+ supply container, and no NH3 leakage is detected (Fig. S26), ruling out the possibility of instability arising from the NH3/NH4+ recirculation system. These findings suggest that further development of more stable catalysts could directly lead to extended device lifetime.
To validate the effectiveness of our NH4+-mediated system, we conduct comparative tests with conventional K+-mediated MEAs under identical conditions (Fig. 5a). Due to the weaker proton-donating ability of H2O compared to NH4+, K+-mediated MEAs exhibit higher CO-Faradaic efficiency (CO-FE), which is consistent with results obtained in flow cell configurations (Fig. S7c). However, its weaker proton-donating ability also leads to a significantly higher cell voltage in the K+-mediated MEA compared to the NH4+-mediated MEAs (Fig. 5b). Moreover, the K+-mediated system shows significant CO-FE fluctuations within the first 4 h (Fig. 5b and Fig. S27), followed by rapid deactivation. This instability manifests through several observable phenomena: a notable decrease in product outlet flow rate, declining flowmeter readings (Fig. S28 and Table S6), and visible salt deposits blocking the gas channel outlet upon post-reaction inspection (Fig. S29). These observations indicate that salt precipitation severely compromises CO2 transport by blocking gas channels. Therefore, while the NH4+ system sacrifices a small degree of selectivity due to its stronger proton-donating ability, it significantly lowers the cell voltage and effectively eliminates the issue of salt precipitation, leading to dramatically improved operational stability. This trade-off is well justified for long-term electrolysis performance.
The contrasting stability of these systems is further evidenced by post-reaction scanning electron microscopy energy-dispersive spectroscopy (SEM-EDS) analysis of the gas diffusion electrodes. In K+-mediated MEAs (Fig. 5d), K+ is detected throughout the entire electrode structure, including the catalyst layer, microporous layer and fiber layer, confirming extensive salt precipitation. In contrast, NH4+-mediated MEAs (Fig. 5e) show nitrogen signals confined to the catalyst layer, likely originating from the catalyst components (CoPc structure and CNT–NH2) and residual nitrogen species. This localized distribution confirms the absence of salt precipitation in our NH4+-mediated system, validating its effectiveness in maintaining stable operation.
These comprehensive results demonstrate the successful development of a stable acidic CO2R MEAs through two key innovations: the NH3/NH4+ recirculation strategy that ensures the stable supply of NH4+, and the modified CoPc@CNT–NH2 catalyst that enables extended operation. The combination achieves stable CO2R performance with over 80% CO selectivity at 100 mA cm−2 for 110 h, representing a significant advancement in acidic MEA systems.
Compared with organic amines, NH4+ salts are among the most readily available and cost-effective proton-donating cations, making them an ideal and practical candidate for fundamental investigation. Therefore, NH4+ is chosen as the primary focus in this work. Meanwhile, common organic amines such as CH3NH3+ and C2H5NH3+ introduce longer carbon chains, which tend to decrease the solubility of bicarbonate salts and increase their decomposition temperature. These properties exacerbate salt precipitation issues and complicate the stability. Addressing such challenges would require precise molecular design and structural tuning, which lie well beyond the scope of the present study. Nevertheless, organic amines offer a unique advantage in their structural tunability. In future studies, we aim to exploit this tunability to improve bicarbonate solubility and lower decomposition temperatures, thereby enabling further reductions in operating temperature.
CONCLUSION
In this work, we demonstrate an effective strategy to achieve both high energy efficiency and stability in acidic CO2R by utilizing NH4+ as a proton-donating cation. Our systematic investigations reveal two distinct roles of NH4+ in promoting CO2R performance. As a cation, NH4+ reduces surface H+ concentration and stabilizes *CO2 intermediates, leading to enhanced CO selectivity, as supported by DFT calculations and COMSOL simulations. As an efficient proton donor, NH4+ offers the advantages over both H2O and HCO3−: it reduces the CO2R overpotential on CoPc@CNT by 280 mV at 100 mA cm−2 through lowered protonation barriers, and enables facile removal of salt precipitation due to the significantly lower decomposition temperature of NH4HCO3 compared to KHCO3. Based on this low-temperature decomposition behavior, an NH3/NH4+ recirculation system is further established to ensure a stable supply of NH₄⁺ during electrolysis. As a result, the NH4+-mediated CoPc@CNT–NH2 system achieves an average CO selectivity of 86% at 100 mA cm−2 with stable performance for 110 h at an average cell voltage of 2.84 V, corresponding to a 40.6% energy efficiency. To the best of our knowledge, this represents the first acidic MEA system that simultaneously achieves high energy efficiency (40.6%) over 110 h, addressing a long-standing challenge in CO2R technology. Notably, the stability of the system is now determined by catalyst stability rather than salt precipitation, indicating that advances in catalyst robustness would extend system lifetime. Beyond the demonstrated high performance of the NH4+-mediated CoPc@CNT–NH2 system achieving stable CO production at acidic MEA, this strategy provides insights for designing next-generation acidic CO2R systems. The dual functionality principle established here could guide the development of other proton-donating species, particularly organic amines with tunable substituents, where the proton-donating ability can be systematically modulated through electronic and steric effects to improve CO2R performance.
Supplementary Material
ACKNOWLEDGEMENTS
Jia Zhu, Xiaojun Wang, Shijia Feng and Hongzhi Zheng acknowledge the micro-fabrication center of the National Laboratory of Solid State Microstructures (NLSSM) for technique support. Philippe Sautet and Dongfang Cheng have been solely supported by Computational resources from the Hoffman2 cluster at the UCLA Institute for Digital Research and Education (IDRE).
Contributor Information
Shijia Feng, National Laboratory of Solid State Microstructures, School of Sustainable Energy and Resources, Jiangsu Key Laboratory of Artificial Functional Materials, Collaborative Innovation Center of Advanced Microstructures, Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, Nanjing 210008, China; College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China.
Ziang Liu, College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China.
Dongfang Cheng, Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Yunfeng Hu, College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China.
Sizhe Chen, College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China.
Xinyuan Zhang, National Laboratory of Solid State Microstructures, School of Sustainable Energy and Resources, Jiangsu Key Laboratory of Artificial Functional Materials, Collaborative Innovation Center of Advanced Microstructures, Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, Nanjing 210008, China.
Jiabao Li, School of Mechanical Engineering, Guangxi University, Nanning 530004, China.
Xiaorui Dong, College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China.
Tianyu Wang, National Laboratory of Solid State Microstructures, School of Sustainable Energy and Resources, Jiangsu Key Laboratory of Artificial Functional Materials, Collaborative Innovation Center of Advanced Microstructures, Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, Nanjing 210008, China.
Ziwei Wang, College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China.
Yulun Wu, Key Laboratory of Mesoscopic Chemistry of Ministry of Education, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China.
Ya Yin, Key Laboratory of Mesoscopic Chemistry of Ministry of Education, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China.
Hongzhi Zheng, National Laboratory of Solid State Microstructures, School of Sustainable Energy and Resources, Jiangsu Key Laboratory of Artificial Functional Materials, Collaborative Innovation Center of Advanced Microstructures, Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, Nanjing 210008, China; College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China.
Philippe Sautet, Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, CA 90095, USA; Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA 90095, USA.
Xiaojun Wang, National Laboratory of Solid State Microstructures, School of Sustainable Energy and Resources, Jiangsu Key Laboratory of Artificial Functional Materials, Collaborative Innovation Center of Advanced Microstructures, Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, Nanjing 210008, China.
Jia Zhu, National Laboratory of Solid State Microstructures, School of Sustainable Energy and Resources, Jiangsu Key Laboratory of Artificial Functional Materials, Collaborative Innovation Center of Advanced Microstructures, Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, Nanjing 210008, China; College of Engineering and Applied Sciences, Nanjing University, Nanjing 210093, China.
FUNDING
Jia Zhu, Xiaojun Wang, Shijia Feng and Hongzhi Zheng acknowledge the support from the National Natural Science Foundation of China (92262305, 92462301, 52525202, 52302225, 52202251 and 52461160296), the ‘GeoX’ Interdisciplinary Project of Frontiers Science Center for Critical Earth Material Cycling (20250106), the New Cornerstone Science Foundation through the XPLORER PRIZE, and the Natural Science Foundation of Jiangsu Province (BK20233001 and BK20243009). Philippe Sautet and Dongfang Cheng have been solely supported by the US National Science Foundation CBET Grant 2103116 and the Audi CO2 Cy Pres Award.
AUTHOR CONTRIBUTIONS
S.F. performed the majority of the synthesis, characterization, electrochemical tests and COMSOL simulations. Z.L. optimized the CO2R stability, performed the energy and cost analysis, and contributed to manuscript revision. D.C. performed DFT calculations, under the guidance of P.S., X.Z., and T.W. optimized the electrochemical performance. J.L. and X.D. were involved in the COMSOL simulations and DFT calculations, respectively. Z.W. conducted the SEM-EDS experiments. Y.W. performed the RDE tests. Y.Y. conducted the TG tests. S.C. and Y.H. contributed to the optimization of the figures presented in the manuscript. S.F., D.C., H.Z., X.W. and J.Z. analyzed the data. S.F. and D.C. drafted the manuscript, while X.W. and J.Z. provided overall direction and oversight for the research.
Conflict of interest statement. None declared.
REFERENCES
- 1. Seh ZW, Kibsgaard J, Dickens CF et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 2017; 355: eaad4998. 10.1126/science.aad4998 [DOI] [PubMed] [Google Scholar]
- 2. Lees EW, Mowbray BAW, Parlane FGL et al. Gas diffusion electrodes and membranes for CO2 reduction electrolysers. Nat Rev Mater 2022;7: 55–64. 10.1038/s41578-021-00356-2 [DOI] [Google Scholar]
- 3. Rabinowitz JA, Kanan MW. The future of low-temperature carbon dioxide electrolysis depends on solving one basic problem. Nat Commun 2020; 11: 5231. 10.1038/s41467-020-19135-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lees EW, Bui JC, Romiluyi O et al. Exploring CO2 reduction and crossover in membrane electrode assemblies. Nat Chem Eng 2024;1: 340–53. 10.1038/s44286-024-00062-0 [DOI] [Google Scholar]
- 5. Sassenburg M, Kelly M, Subramanian S et al. Zero-gap electrochemical CO2 reduction cells: challenges and operational strategies for prevention of salt precipitation. ACS Energy Lett 2023;8: 321–31. 10.1021/acsenergylett.2c01885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li X, Zhang P, Zhang L et al. Confinement of an alkaline environment for electrocatalytic CO2 reduction in acidic electrolytes. Chem Sci 2023; 14: 5602–7. 10.1039/D3SC01040F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ma Z, Yang Z, Lai W et al. CO2 electroreduction to multicarbon products in strongly acidic electrolyte via synergistically modulating the local microenvironment. Nat Commun 2022; 13: 7596. 10.1038/s41467-022-35415-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qiao Y, Lai W, Huang K et al. Engineering the local microenvironment over Bi nanosheets for highly selective electrocatalytic conversion of CO2 to HCOOH in strong acid. ACS Catal 2022; 12: 2357–64. 10.1021/acscatal.1c05135 [DOI] [Google Scholar]
- 9. Huang JE, Li F, Ozden A et al. CO2 electrolysis to multicarbon products in strong acid. Science 2021; 372: 1074–8. 10.1126/science.abg6582 [DOI] [PubMed] [Google Scholar]
- 10. Li H, Li H, Wei P et al. Tailoring acidic microenvironments for carbon-efficient CO2 electrolysis over a Ni-N-C catalyst in a membrane electrode assembly electrolyzer. Energy Environ Sci 2023; 16: 1502–10. 10.1039/D2EE03482D [DOI] [Google Scholar]
- 11. Xie Y, Ou P, Wang X et al. High carbon utilization in CO2 reduction to multi-carbon products in acidic media. Nat Catal 2022;5: 564–70. 10.1038/s41929-022-00788-1 [DOI] [Google Scholar]
- 12. Masel RI, Liu Z, Yang H et al. An industrial perspective on catalysts for low-temperature CO2 electrolysis. Nat Nanotechnol 2021; 16: 118–28. 10.1038/s41565-020-00823-x [DOI] [PubMed] [Google Scholar]
- 13. Yan C, Gao D, Velasco-Vélez J-J et al. Reaction microenvironment control in membrane electrode assemblies for CO2 electrolysis. EES Catal 2024;2: 220–30. 10.1039/D3EY00155E [DOI] [Google Scholar]
- 14. Lee T, Lee Y, Eo J et al. Acidic CO2 electroreduction for high CO2 utilization: catalysts, electrodes, and electrolyzers. Nanoscale 2024; 16: 2235–49. 10.1039/D3NR05480B [DOI] [PubMed] [Google Scholar]
- 15. Yu J, Xiao J, Ma Y et al. Acidic conditions for efficient carbon dioxide electroreduction in flow and MEA cells. Chem Catal 2023;3: 100670. [Google Scholar]
- 16. O'Brien CP, Miao RK, Liu S et al. Single pass CO2 conversion exceeding 85% in the electrosynthesis of multicarbon products via local CO2 regeneration. ACS Energy Lett 2021;6: 2952–9. [Google Scholar]
- 17. Feng S, Wang X, Cheng D et al. Stabilizing *CO2 intermediates at the acidic interface using molecularly dispersed cobalt phthalocyanine as catalysts for CO2 reduction. Angew Chem Int Ed 2024; 63: e202317942. 10.1002/anie.202317942 [DOI] [PubMed] [Google Scholar]
- 18. Gu J, Liu S, Ni W et al. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat Catal 2022;5: 268–76. 10.1038/s41929-022-00761-y [DOI] [Google Scholar]
- 19. Bondue CJ, Graf M, Goyal A et al. Suppression of hydrogen evolution in acidic electrolytes by electrochemical CO2 reduction. J Am Chem Soc 2021; 143: 279–85. 10.1021/jacs.0c10397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qin H-G, Li F-Z, Du Y-F et al. Quantitative understanding of cation effects on the electrochemical reduction of CO2 and H+ in acidic solution. ACS Catal 2023; 13: 916–26. 10.1021/acscatal.2c04875 [DOI] [Google Scholar]
- 21. Bai Q, Xiong L, Zhang Y et al. Salt precipitation and water flooding intrinsic to electrocatalytic CO2 reduction in acidic membrane electrode assemblies: fundamentals and remedies. EES Catal 2024;2: 1228–37. 10.1039/D4EY00170B [DOI] [Google Scholar]
- 22. Pan B, Fan J, Zhang J et al. Close to 90% single-pass conversion efficiency for CO2 electroreduction in an acid-fed membrane electrode assembly. ACS Energy Lett 2022;7: 4224–31. 10.1021/acsenergylett.2c02292 [DOI] [Google Scholar]
- 23. Qin H-G, Du Y-F, Bai Y-Y et al. Surface-immobilized cross-linked cationic polyelectrolyte enables CO2 reduction with metal cation-free acidic electrolyte. Nat Commun 2023; 14: 5640. 10.1038/s41467-023-41396-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fan M, Huang JE, Miao RK et al. Cationic-group-functionalized electrocatalysts enable stable acidic CO2 electrolysis. Nat Catal 2023;6: 763–72. 10.1038/s41929-023-01003-5 [DOI] [Google Scholar]
- 25. Fan J, Pan B, Wu J et al. Immobilized tetraalkylammonium cations enable metal-free CO2 electroreduction in acid and pure water. Angew Chem 2024; 136: e202317828. 10.1002/ange.202317828 [DOI] [PubMed] [Google Scholar]
- 26. Butt NE, Padding JT, Hartkamp R. Size-modified Poisson–Nernst–Planck approach for modeling a local electrode environment in CO2 electrolysis. Sustain Energy Fuels 2023;7: 144–54. 10.1039/D2SE01262F [DOI] [Google Scholar]
- 27. Weng L-C, Bell AT, Weber AZ. Modeling gas-diffusion electrodes for CO2 reduction. Phys Chem Chem Phys 2018; 20: 16973–84. 10.1039/C8CP01319E [DOI] [PubMed] [Google Scholar]
- 28. Bohra D, Chaudhry JH, Burdyny T et al. Modeling the electrical double layer to understand the reaction environment in a CO2 electrocatalytic system. Energy Environ Sci 2019; 12: 3380–9. 10.1039/C9EE02485A [DOI] [Google Scholar]
- 29. Ren X, Zhao J, Li X et al. In-situ spectroscopic probe of the intrinsic structure feature of single-atom center in electrochemical CO/CO2 reduction to methanol. Nat Commun 2023; 14: 3401. 10.1038/s41467-023-39153-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Resasco J, Chen LD, Clark E et al. Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide. J Am Chem Soc 2017; 139: 11277–87. 10.1021/jacs.7b06765 [DOI] [PubMed] [Google Scholar]
- 31. Ringe S, Clark EL, Resasco J et al. Understanding cation effects in electrochemical CO2 reduction. Energy Environ Sci 2019; 12: 3001–14. 10.1039/C9EE01341E [DOI] [Google Scholar]
- 32. Silverstein TP, Heller ST. pKa values in the undergraduate curriculum: what is the real pKa of water? J Chem Educ 2017; 94: 690–5. 10.1021/acs.jchemed.6b00623 [DOI] [Google Scholar]
- 33. Dickinson EJF, Wain AJ. The Butler-Volmer equation in electrochemical theory: origins, value, and practical application. J Electroanal Chem 2020; 872: 114145. 10.1016/j.jelechem.2020.114145 [DOI] [Google Scholar]
- 34. Kowalski RM, Banerjee A, Yue C et al. Electroreduction of captured CO2 on silver catalysts: influence of the capture agent and proton source. J Am Chem Soc 2024; 146: 20728–41. 10.1021/jacs.4c03915 [DOI] [PubMed] [Google Scholar]
- 35. Wu Y, Jiang Z, Lu X et al. Domino electroreduction of CO2 to methanol on a molecular catalyst. Nature 2019; 575: 639–42. 10.1038/s41586-019-1760-8 [DOI] [PubMed] [Google Scholar]
- 36. Li H, Pan Y, Wang Z et al. Coordination engineering of cobalt phthalocyanine by functionalized carbon nanotube for efficient and highly stable carbon dioxide reduction at high current density. Nano Res 2022; 15: 3056–64. 10.1007/s12274-021-3962-2 [DOI] [Google Scholar]
- 37. Hao S, Elgazzar A, Ravi N et al. Improving the operational stability of electrochemical CO2 reduction reaction via salt precipitation understanding and management. Nat Energy 2025; 10: 266–77. 10.1038/s41560-024-01695-4 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.