

Abstract
The precise modulation of physicochemical and ADME properties is critical in drug discovery. We report a unified, mild, and broadly applicable metallaphotoredox-enabled protocol for the N-alkylation of sulfoximines, a versatile S(VI) motif gaining prominence as a bioisostere. This method overcomes limitations of previous N-substitution approaches by accommodating diverse alkyl sources, including alcohols, alkyl bromides, and carboxylic acids, under visible-light irradiation. The strategy features high functional group tolerance and offers efficient access to both primary and secondary N-alkyl sulfoximines. Its utility is showcased through the late-stage functionalization of pharmacologically active compounds, natural product derivatives, and short peptides, enabling rapid access to novel analogues with enhanced structural diversity. Furthermore, we demonstrate the strategic coupling of bicyclo[1.1.1]pentyl moieties with sulfoximines to achieve a synergistic bioisosteric design. A preliminary drug optimization campaign, exemplified by an atuveciclib analogue, highlights the platform’s potential for improving key ADME properties such as lipophilicity and cellular permeability, underscoring its value for lead diversification and refinement in medicinal chemistry.
Keywords: Catalysis, Photochemistry, Photoredox, S(VI)-compounds, Bioisosteres, N-Alkylation, ADME Properties
Graphical Abstract

Introduction.
In modern drug discovery, medicinal chemists routinely encounter major challenges in both hit identification and lead optimization. Key objectives include enhancing binding affinity and selectivity, mitigating toxicity, and improving physicochemical as well as ADME (absorption, distribution, metabolism, excretion) properties.[1,2] One powerful strategy to address these challenges is the use of bioisosteres, i.e. functional groups or frameworks that retain the biological activity of known motifs while offering improved therapeutic profiles.[3–5] This concept, known as bioisosteric replacement or scaffold hopping,[6–8] allows for the design of structurally distinct compounds through the strategic substitution of established motifs, often leading to enhanced potency, selectivity, and drug-like characteristics.
Sulfur(VI) functional groups, particularly sulfonyl and sulfonamide derivatives, are among the most common scaffolds used in modern drug design.[9–11] These motifs are present in over 150 FDA-approved drugs and are widely applied to treat a range of diseases due to their robust pharmacological properties and synthetic accessibility.[12] However, drawbacks such as limited lipophilicity, potential allergic reactions, risks of hepatotoxicity, and development of resistance motivate the search for alternative structures with improved profiles.[13–15]
Sulfoximines, the monoaza analogues of sulfones, have recently emerged as attractive alternatives in this context.[16–19] Their favorable features include adjustable polarity, unique hydrogen-bonding behavior, and the opportunity to introduce additional substituents at the nitrogen center.[20–22] These properties enable the fine-tuning of physicochemical characteristics and pharmacokinetics, including solubility, permeability, and protein binding. In the past decade, sulfoximines have seen increased adoption in medicinal chemistry, with several compounds advancing into clinical trials (Figure 1).[19,23,24] This trend has been supported by greater synthetic accessibility and a deeper understanding of their behavior in biological systems. Nevertheless, the vast majority of reported sulfoximine-containing structures remain unsubstituted at nitrogen, limiting their ability to serve as modular bioisosteres of sulfones or sulfonamides.
Figure 1.
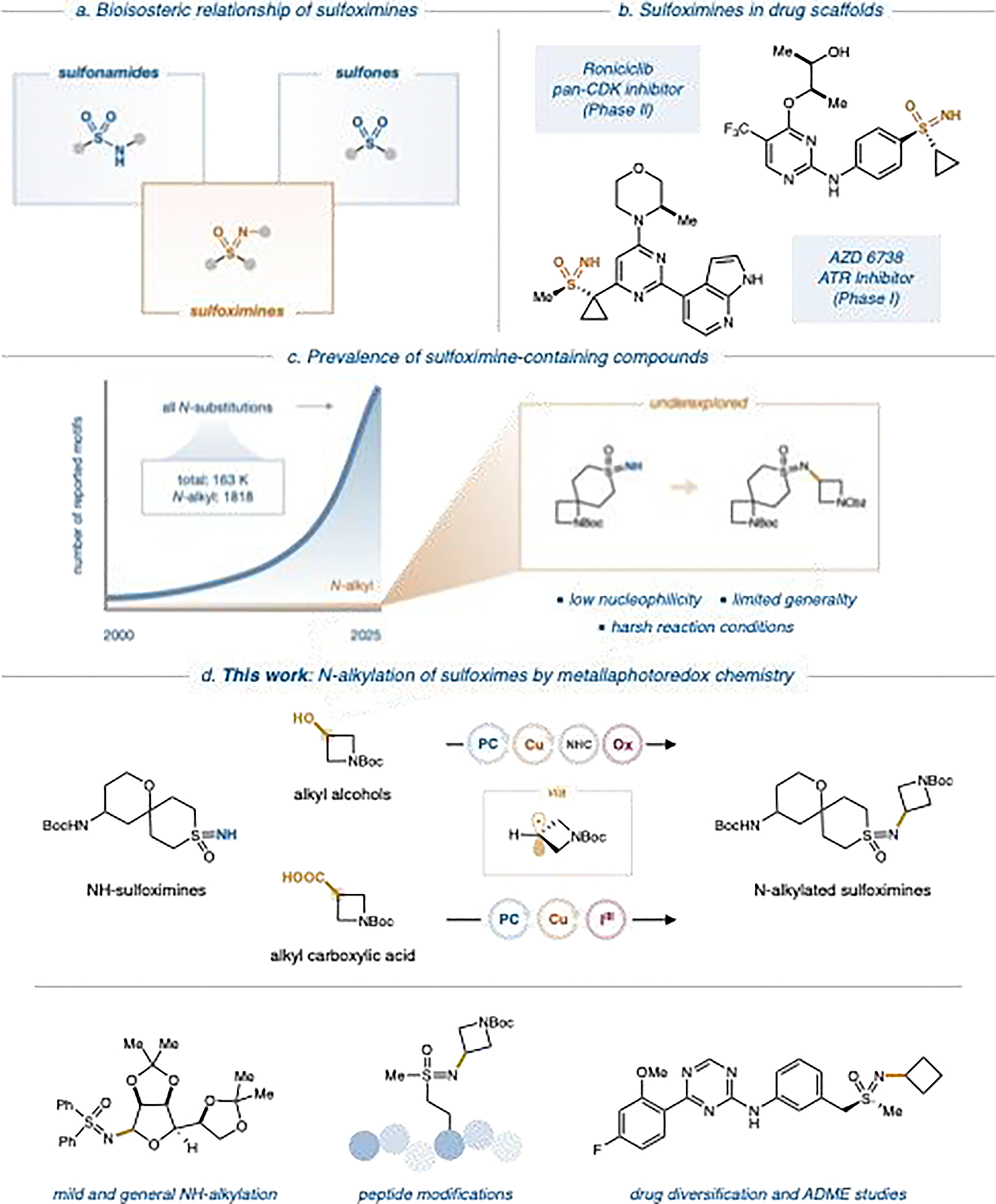
N-Alkylation of sulfoximines.
Substitution at the sulfoximine nitrogen has the potential to modulate pharmacological and metabolic profiles significantly, but practical methods to achieve such modifications remain scarce. Current methods for N-substitution—particularly alkylation—are often hindered by the poor nucleophilicity of the imine-like nitrogen, necessitating harsh reagents or conditions that restrict functional group compatibility.[25–28] The development of mild and general strategies for N-alkyl sulfoximine synthesis remains a key unmet challenge.
Previous studies have shown that both iron and copper catalysis can facilitate N-alkylation via radical intermediates thermally generated from peroxides;[29–32] however, these methods are hindered by safety concerns and substrate scope limitations. More recently, metallaphotoredox catalysis has emerged as a general approach for forming carbon–heteroatom bonds under mild conditions.[33] This technology relies on visible-light activation to generate reactive radical intermediates and has enabled a range of previously challenging transformations. Copper-based systems are particularly well suited for challenging coupling reactions, as they bypass oxidative addition and promote efficient reductive elimination.[34–37]
Building on our previous work using copper and photoredox catalysis to achieve alcohol-based alkylation of nitrogen heterocycles,[38] we sought to develop a unified strategy for the N-alkylation of sulfoximines. Our objective was to establish a modular protocol that could accommodate a wide array of radical precursors, including both alcohols and carboxylic acids, converging on a common bond-forming process that involves alkyl radical capture by an intermediate Cu(II) species. This approach would allow for orthogonal reactivity and broad late-stage functionalization potential.
We have developed strategies to generate alkyl radical intermediates in situ from either alcohol or carboxylic acid precursors. Under this paradigm, alcohols are activated with benzoxazolium-based reagents under photoredox conditions to form alkyl radicals.[39] Carboxylic acids are converted to the corresponding radicals via decarboxylation using a hypervalent iodine reagent previously reported in our laboratory.[40] We demonstrate herein that this orthogonal reagent strategy enables the direct and selective N-alkylation of a wide range of sulfoximines, providing a general platform for the construction of valuable building blocks with potential applications in drug discovery and development.
Both activation pathways converge on a common mechanistic sequence for carbon–nitrogen bond formation. A proposed mechanism for the deoxygenative pathway is outlined in Figure 2. First, alcohol 1 reacts with the NHC salt under mildly basic conditions, forming NHC-alcohol adduct I. Subsequently, the employed photocatalyst (PC) is excited by visible, blue light yielding a sufficiently long-lived exited triplet state (PC*) to participate in bimolecular electron transfer. Using 4CzIPN as the photocatalyst (E1/2red [PC*/PC•−] = +1.43 V vs. saturated calomel electrode (SCE) in MeCN),[41] the excited-state can be readily quenched by I (E1/2red = 1.0 V vs. SCE in MeCN)[39] yielding the radical anion of the photocatalyst (PC•−) and a radical cation of I, which can undergo a deprotonation/β-scission sequence affording the desired alkyl radical II and inert carbamate byproduct III. The resulting radical II is intercepted by an in situ formed copper(II)–sulfoximine complex IV to form a putative copper(III) intermediate V. Facile reductive elimination from this species furnishes the cross-coupled product 3, thereby opening a coordination site for another sulfoximine molecule (2). The resulting copper(I) complex VI is reoxidized to copper(II) by an external oxidant, which additionally allows for the turnover of the photocatalytic cycle regenerating 4CzIPN from the radical anion (E1/2red [PC/PC•−] = −1.24 V vs. SCE in MeCN).[41]
Figure 2.

Plausible mechanism for the deoxygenative N-alkylation of sulfoximines; Cbz, benzyloxycarbonyl; PC, photocatalyst; Ox, oxidant.
Results and Discussion
We began our studies by optimizing the reaction conditions for the alcohol substrate class, using S,S-diphenyl sulfoximine (1) and Cbz-protected 4-hydroxypiperidine (2) as model substrates (Table 1). Under optimized conditions, 1 was combined with the NHC adduct of 2 in methyl tert-butyl ether (MTBE), with 4CzIPN as the photocatalyst, copper(II) acetate with 1,10-phenanthroline as the copper catalyst, 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG) as base, and iodosomesitylene (MesIO) as oxidant in acetonitrile. Irradiation with blue light emitting diodes at 450 nm for eight hours afforded the desired carbon–nitrogen cross-coupled product 3 in 74% isolated yield on 0.5 mmol scale. Control experiments supported the proposed mechanism, as product formation beyond trace amounts was only observed when the photocatalyst, copper catalyst, base, and oxidant were all present under light irradiation. Omitting either the ligand or the external oxidant significantly reduced yields; similar reductions were observed when the reaction was conducted under air. Using 1,1,3,3-tetramethylguanidine (TMG) as a weaker base alternative to BTMG led to slightly diminished yields (see Supporting Information for optimization and control experiments).
Table 1.
Control experiment for deoxygenative N-alkylation of sulfoximines.

| ||
|---|---|---|
| entry | deviation | yielda |
| 1 | none | 79% (74%b) |
| 2 | no light | <1% |
| 3 | no photocatalyst | <1% |
| 4 | no copper catalyst | <1% |
| 5 | no ligand | 6% |
| 6 | no base | <1% |
| 7 | TMG as base (2.0 equiv.) | 61% |
| 8 | no oxidant (N2 atmosphere) | <1% |
| 9 | air as oxidant (18G needle) | 8% |
Performed on a 0.05 mmol scale. The alcohol activation was stirred for 30 min at room temperature before being transferred to the reaction mixture, resulting in 0.05 M reaction in 1:1 MeCN/MTBE. Irradiation for 8 h with 450 nm blue LEDs in the integrated photoreactor. Yields given as 1H-NMR assay yields against 1,3,5-trimethoxybenzene as internal standard.
Isolated yield on 0.5 mmol scale.
Abbreviations: Ac, acetyl; BTMG, 2-tert-butyl-1,1,3,3-tetramethylguanidine; Cbz, benzyloxycarbonyl; Mes, mesityl; MTBE: methyl tert-butyl ether; TMG, 1,1,3,3-tetramethylguanidine; Ph, phenyl; phen, phenanthroline pyr, pyridine. See the Supporting Information (SI) for further details.
With these optimal conditions in hand, we set out to explore the applicability of this transformation to a variety of differently substituted alcohols and sulfoximines (Table 2). Owing to the versatility of the benzoxazolium reagent, a broad range of alcohols featuring diverse functional groups could be coupled to S,S-diphenylsulfoximine as the parent sulfoximine. Primary alcohols derived from chiral amino acids (4 and 5) provided the desired N-alkylated sulfoximines in high to excellent yields (67% and 86%). Moreover, sterically encumbered β-quaternary alcohols—challenging substrates in traditional cross-coupling reactions—proved competent coupling partners (6, 60%), as did alcohols containing acid-labile acetal protecting groups, such as solketal 7 (58%).
Table 2.
Scope of metallaphotoredox-enabled deoxygenative N-alkylation of sulfoximines.a

|
Performed on a 0.5 mmol scale with sulfoximine (1.0 equiv.), alcohol (2.0 equiv), NHC-H (2.2 equiv), pyridine (2.2 equiv), 4CzIPN (5 mol%), Cu(OAc)2 (15 mol%), 1,10-phenanthroline (17.5 mol%), BTMG (2 equiv), MesIO (2 equiv.) in MeCN/MTBE (0.05 M) in the integrated photoreactor (450 nm, 75% light intensity for 8 h).
Performed on a 0.5 mmol scale with sulfoximine (1.0 equiv.), bromide (2.0 equiv), AdNHSi(TMS)3 (2.2 equiv.) 4CzIPN (2 mol%), Cu(OAc)2 (25 mol%), 1,10-phenanthroline (28 mol%), TMG (2 equiv) in MeCN (0.05 M) under air in the integrated photoreactor (450 nm, 10% light intensity for 12 h).
Use of TMG (2.0 equiv.) instead of BTMG and neocuproine (17.5 mol%) instead of 1,10-phenanthroline.
Starting material was present as a diastereomeric mixture of 1:1. See the Supporting Information for experimental details.
There are few reported methods for installing secondary alkyl residues at the sulfoximine nitrogen, and existing approaches often rely on specific alkyl scaffolds or harsh conditions, limiting their generality and efficiency.[31,42,43] We were therefore pleased to find that our system enables straightforward and efficient coupling of various secondary alkyl fragments with the parent sulfoximine. Seven- (8), six- (3, 9, 10), five- (11), and four-membered rings (12) were well tolerated, undergoing substitution at different ring positions in moderate to high yields (52–74%). Additionally, non-cyclic secondary alcohols delivered the product in high yield (13, 75%).
The cyclopropyl moiety, due to the nature of its orbitals, is typically incompatible with SN1 or SN2 conditions, necessitating alternative activation strategies. Applying our methodology to cyclopropanol led to unsatisfactory conversion (<30%) presumably due to the low β-scission rate of the corresponding NHC-adduct upon single-electron oxidation and deprotonation (<104 M−1s−1). As a more practical entry point for this motif, we turned to bromocyclopropane. Building on prior work from our group employing adamantylaminosilane (AdNHSi(TMS)3) as a potent halogen atom abstraction (XAT) reagent,[37,44] following oxidation and an aza-Brooks rearrangement, we reoptimized our protocol for alkyl halide substrates (see Supporting Information for proposed mechanism). This approach furnished N-cyclopropyl sulfoximine 14 in 73% yield, showcasing the complementary nature of our platform and its compatibility with diverse radical precursors and activation modes.
With a robust method for engaging a variety of alkyl fragments, we next explored the diversity of sulfoximine coupling partners as a means to further enhance molecular complexity. Our strategy allows coupling of enantiomerically pure sulfoximines with achiral alcohols with complete stereoretention, delivering product 15 in 81% yield and >99% ee. Furthermore, the introduction of a chiral alkyl fragment enabled construction of product 16 with three absolute stereogenic centers in a single step with high diastereoselectivity (58%, d.r. >17.5:1), exemplifying the power of this modular approach to generate stereochemical complexity.
We next evaluated arene–alkyl sulfoximines and found that a wide array of (hetero)aryl groups at the sulfoximine scaffold were tolerated, including phenyl (17 and 18), pyridyl (19), and pyrazinyl (20) substituents, all delivering products in moderate to high yields (48–83%). The methodology was also applicable to various alkyl sulfoximines, including benzylic variants (21, 71%). For an electron-deficient S(VI)–CF3 derivative, changing the base to TMG and the ligand to neocuproine, afforded the desired product 22 in 57% yield. Cyclic sulfoximines also demonstrated high reactivity, including substrates derived from dibenzothiophene (23, 69%), tetrahydrothiophene (24, 66%), 1,4-oxathiane (25, 50%), thiomorpholine (26, 63%), thiaazaspiro[3.5]nonane (27, 82%). Encouragingly, the successful coupling of fully S,S-alkyl-substituted sulfoximines was also extended to acyclic systems (28–30), with yields ranging from 55% to 67%.
A further advantage of this system is its tolerance to potentially reactive aryl halides (9 and 26), which remain intact under the reaction conditions due to the high kinetic barrier for oxidative addition to copper complexes. This feature enables the use of aryl halides as strategic handles for downstream diversification, greatly expanding the accessible chemical space.
We next aimed to apply our N-functionalization platform to more complex alcohols and sulfoximines, particularly those derived from naturally occurring biomolecules and scaffolds frequently found in pharmacologically active compounds (Table 3). The broad availability of alcohols, combined with the straightforward synthesis of sulfoximines via oxidation/nitrene transfer from thioethers,[45] enabled access to products from structurally diverse and complex starting materials. A range of differently substituted hydroxypiperidines, including N-heterobenzoylated (31 and 32), N-heteroarylated (33), and N-sulfonylated (34) alcohols were effectively employed as alkylating agents for sulfoximines bearing various substitution patterns, affording the desired products in high to very high yields (59–81%).
Table 3.
Scope of complex drug-like substrates.a

|
Isolated yields are reported. See Supporting Information for experimental details.
Starting material was present as a mixture of diastereoisomers.
Given the growing emphasis on incorporating C(sp3)-rich molecular frameworks into pharmaceutical candidates as a strategy correlated with clinical success,[46] we were especially pleased to obtain spirocyclic sulfoximines 35 and 36, featuring an exceptionally high degree of C(sp3) hybridization within their carbon skeleton in high yields (55% and 63%, respectively). Additionally, anti-androgen-derived 37 and an avanafil analog 38 were efficiently synthesized from the corresponding alcohols (61% and 54%, respectively).
The selective derivatization of glucosides, as well as the incorporation of bioisosteric elements into these structures, is of particular interest in drug discovery.[47] Functionalization of glucosides often proves challenging due to the difficulty of activating sugar-derived alcohols and the fragile nature of acetal protecting groups.[48] However, the mild activation and radical generation enabled by the NHC-based reagent allows for the direct conversion of native sugar alcohols into alkyl radicals suitable for coupling.
To demonstrate this utility, we applied the method to an unnatural nucleoside (39), galactopyranose (40), and mannofuranose (41). In each case, the corresponding alkylated sulfoximines were obtained in high to very high yields (57–81%) with excellent diastereoselectivity (d.r. = 13:1 for 39 and d.r. > 20:1 for 41). Owing to the mild conditions of this protocol, this represents the first example of direct conversion of glucosides into their S(VI)-derivatives, offering valuable synthetic handles for future structure–activity relationship (SAR) studies.
Late-stage functionalization of complex molecules is a powerful strategy in drug discovery, enabling efficient structural diversification and optimization of physicochemical properties.[49,50] To highlight the utility of our method in this context, we applied it to the direct alkylation of drug-like and bioactive compounds. This approach offers a modular and broadly applicable route to libraries of novel alkylated sulfoximines without the need for de novo synthesis, allowing installation of the sulfoximine moiety at advanced stages of synthesis.
We applied our reaction platform to a selection of pharmacologically active compounds and their derivatives bearing alcohol groups, benchmarking its applicability for late-stage installation of sulfoximine bioisosteres (Table 4). To our delight, the sulfoximine group was readily incorporated into molecules derived from nateglinide (42), lonazolac (43), and linagliptin (45), affording the corresponding products in moderate to high yields (49–66%). Coupling with androsterone furnished sulfoximine 44 in 61% yield with a 1:1 diastereomeric ratio, providing access to both diastereomers for evaluation of potentially distinct biological activity or pharmacokinetic profiles. In addition, differently substituted complex sulfoximines were effective in the late-stage functionalization of alcohols derived from alogliptin (46) and metoprolol (47), delivering the desired products in high yields (66% and 63%, respectively), further underscoring the versatility of this methodology in connecting diverse and structurally complex building blocks.
Table 4.
Use of drug molecules and bioactive substrates.a

|
Isolated yields are reported. See Supporting Information for experimental details.
This applicability was further demonstrated using racemic atuveciclib, a CDK inhibitor, as the sulfoximine coupling partner.[51] By employing our optimized conditions with bromocyclopropane and AdNHSi(TMS)3, we obtained the corresponding N-cyclopropanated atuveciclib derivative 48 in 56% yield. This result highlights the practicality of the alkylation strategy, as the parent compound can be directly engaged in the transformation without the need for extensive prior modification.
Apart from small molecules, the late-stage modification of peptides represents a powerful strategy in medicinal chemistry, enabling the precise and efficient diversification of complex peptide scaffolds without the need for lengthy de novo synthesis.[52] By facilitating streamlined access to analogues, such methods significantly enhance the speed and precision of peptide-based drug development. To this end, we synthesized a tripeptide model substrate containing a Met–Phe–Leu sequence and subjected it to our established oxidation protocol for converting thioethers into sulfoximines. Exposure of the resulting NH-sulfoximine peptide (MSO–Phe–Leu) to our alkylation procedure enabled the installation of an azetidine moiety in synthetically useful yield (49, 49%, d.r. = 1:1). Notably, this transformation would not have been feasible using traditional substitution techniques or previously reported metal-catalyzed methods. We envision that this methodology could prove useful in peptide library synthesis by converting native methionine residues into unnatural amino acids in a straightforward and versatile manner.
We further applied our protocol to the synthesis of a lapatinib analogue (50), which was obtained in high yield (63%). In this case, compound 50 represents a direct bioisostere of the parent lapatinib, where the sulfone moiety, characterized by two hydrogen bond acceptors, was replaced by an alkylated sulfoximine that mimics these key acceptor features. Importantly, both bioisosteric compounds can be accessed from a common thioether precursor, thereby streamlining synthetic accessibility and enabling a late-stage divergence in the overall synthetic strategy.
Building on the successful use of alcohols and bromocyclopropane as alkylating agents for N-alkylation of sulfoximines, we sought to expand the molecular toolbox of viable starting materials. Carboxylic acids, among the most abundant and versatile functional groups in medicinal chemistry, serve as attractive functionalization handles due to their wide availability and commercial accessibility.[53] In parallel, bicyclo[1.1.1]pentanes (BCPs) have gained prominence as valuable bioisosteres for para-substituted phenyl rings and alkynes, offering superior physicochemical and pharmacokinetic properties.[54–56] Their rigid, saturated, and three-dimensional structure improves metabolic stability and lowers lipophilicity, while preserving essential spatial and electronic attributes of the original motifs.
In line with our group’s long-standing interest in designing new methods to access rich bioisosteric chemical space,[57–59] we set out to incorporate BCP motifs into our reaction protocol. This approach would allow for the convergence of two bioisosteric elements in a single transformation, providing entry to synergistic bioisosteric chemical space that may serve as a replacement for aryl sulfones or sulfonamides (Table 5). By fine-tuning our reaction conditions and modifying the redox linchpins—including pre-activating the carboxylic acids using (diacetoxyiodo)mesitylene [MesI(OAc)2] yielding diacetate 51, [Ir(dF(Me)ppy)2(dtbbpy)]PF6 as the photocatalyst, and Cu(acac)2 as the copper source—we successfully coupled a variety of differently substituted sulfoximines with BCP-carboxylic acids (52–55) in moderate to excellent yields (55–93%; see Supporting Information for details).
Table 5.
Decarboxylative amination of bicyclo[1.1.1]pentanes (BCPs).a

|
Performed on a 0.4 mmol scale with corresponding iodonium carboxylate (1.0 equiv.), sulfoximine (2.0 equiv), Ir[dF(Me)ppy]2(4,4′-dtbbpy)PF6 (2 mol%), Cu(acac)2 (50 mol%), BTTP (1 equiv) in 1,4-dioxane (0.33 M) in the integrated photoreactor (450 nm, 25% light intensity for 1 h); BTTP: tert-butylimino-tri(pyrrolidino)phosphorane. See Supporting Information for experimental details.
A mechanistic hypothesis for this transformation is presented in Figure 3. In contrast to the deoxygenative approach, no external oxidant is required as the system is based on a redox-neutral catalytic cycle. In situ formed Cu(I) complex VI (E1/2red [CuII(BPhen)2/CuI(BPhen)2] = + 0.08 V vs. SCE in DMF, BPhen = 4,7-diphenyl-1,10-phenanthroline)[60] possesses the ability of reductively quenching the exited state of the Ir-based photocatalyst (E1/2red [*IrIII/IrII] = +0.99 V vs. SCE in MeCN)[61] yielding the reduced Ir(II) photocatalyst as well as the required Cu(II)-sulfoximine complex IV. For the decarboxylative approach, we propose that iodomesitylene dicarboxylate 51, generated from carboxylic acid 56 with iodomesitylene diacetate [MesI(OAc)2], would undergo facile reduction by the Ir(II) species (E1/2red [IrIII/IrII] = −1.41 V vs. SCE in MeCN)[61]; Ep [51/51•−] = −1.14 V vs. SCE in MeCN)[40] leading to the formation of a carboxyl radical as well as the regeneration of the ground-state Ir(III) photocatalyst. Upon subsequent decarboxylation the desired BCP-radical IX is obtained. In line with the mechanism for the deoxygenative approach, the desired putative Cu(III) intermediate X is obtained upon coordination of radical IX to Cu(II) complex IV, which subsequently undergoes reductive elimination, furnishing the desired N-BCP sulfoximine 52.
Figure 3.
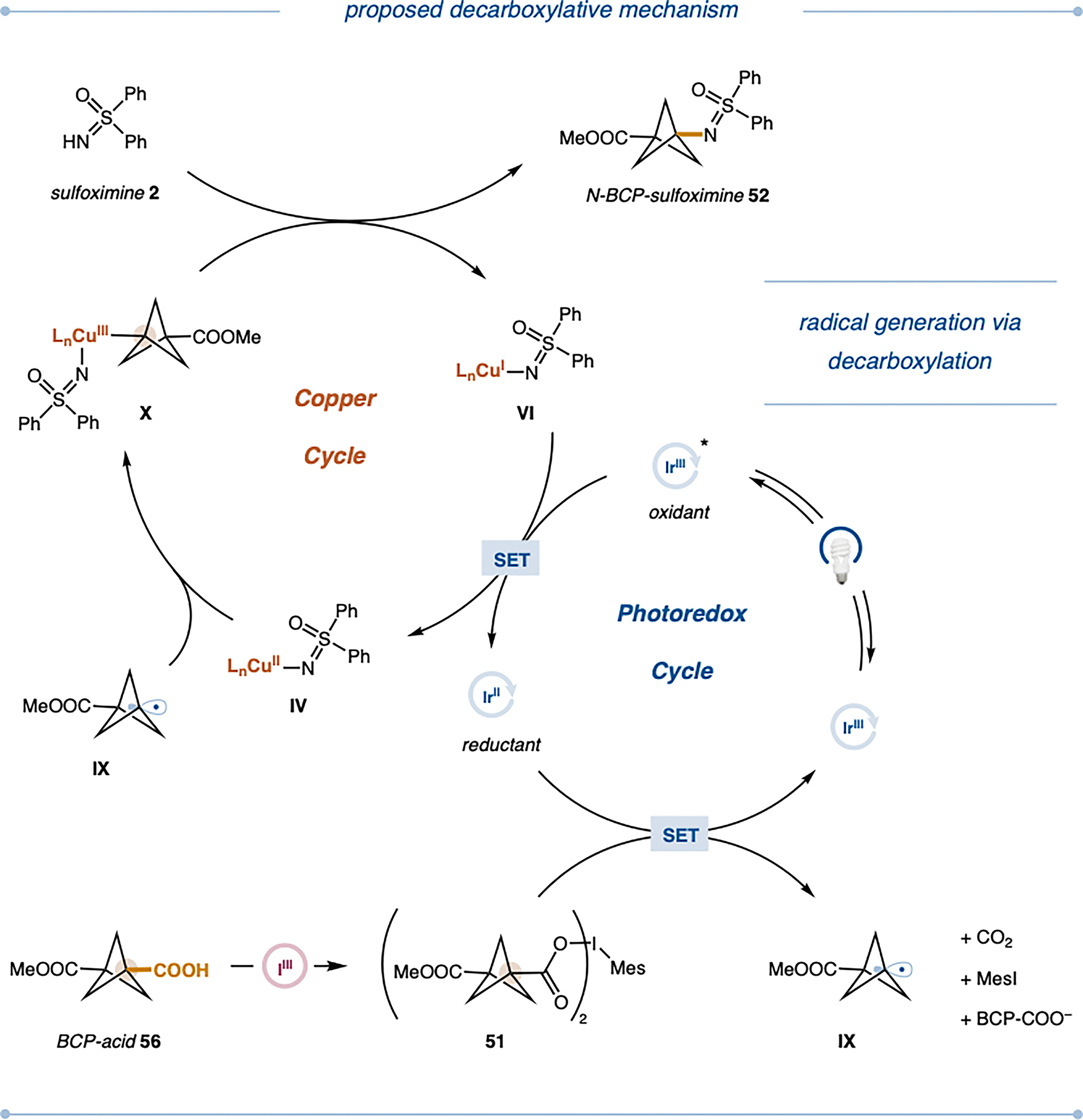
Plausible mechanism for the decarboxylative N-alkylation of sulfoximines; PC, photocatalyst; IIII: (diacetoxyiodo)benzene.
Finally, we evaluated the potential of our method in a small-scale drug optimization campaign as a blueprint for applications in drug discovery. Sulfoximines have gained prominence in medicinal chemistry due to their unique combination of physicochemical and pharmacokinetic advantages. Compared to traditional functional groups such as sulfones and sulfonamides, sulfoximines offer enhanced metabolic stability, reduced off-target effects, and lowered risk of undesired drug-drug interactions. Their tunable hydrogen bonding capacity often translates into improved aqueous solubility and oral bioavailability. The structural diversity of sulfoximines, supported by their three-dimensional, chiral architecture, enables precise tuning of molecular properties such as permeability and plasma protein binding, key determinants of in vivo clearance (Figure 2). As a result, sulfoximines are valuable functional groups for optimizing ADME profiles in drug development. To evaluate this potential, we selected atuveciclib, an established CDK inhibitor,[51] to assess whether the introduction of an alkyl group at the nitrogen atom of the sulfoximine would lead to improved pharmacological properties. Based on computational docking studies,[57] we selected the alkylated derivative 57 as a suitable candidate for our endeavor. Accordingly, we applied our NHC-based protocol using cyclobutanol as the alkylating reagent, affording 55 directly from atuveciclib in 53% yield with full retention of stereochemical configuration.
Compound 57 was subsequently evaluated in HeLa human cervical carcinoma cells (CCL-2) to assess its cellular potency relative to the parent compound (Figure 4). Gratifyingly, the IC50 value (0.7 μM) was comparable to atuveciclib (0.3 μM), indicating that N-alkylation does not compromise cellular activity. Encouraged by this result, we next examined whether N-alkylation improved the compound’s physicochemical properties in vivo. An increase in logD was observed, suggesting enhanced lipophilicity, which may contribute to improved membrane permeability.[62] In parallel, kinetic solubility showed a notable improvement compared to atuveciclib (Figure 4). Together, these properties are expected to increase the fraction absorbed (Fa) in vivo, potentially resulting in higher oral bioavailability. To further assess its ADME profile, we measured the permeability of compound 57 in Madin-Darby canine kidney (MDCK) cells, a model for epithelial absorption. Compound 57 exhibited enhanced permeability compared to the parent compound, suggesting improved oral bioavailability or tissue distribution. Together, these results underscore the potential of N-alkylated sulfoximines as valuable motifs for modulating both physicochemical and pharmacokinetic properties in drug development.
Figure 4.

Tunability of pharmacological properties by bioisosteric replacement.
Conclusion
In summary, we present an efficient and broadly applicable protocol for the N-alkylation of sulfoximines using aliphatic alcohols, alkyl bromides, and carboxylic acids as versatile alkyl sources. This unified strategy accommodates a diverse substrate scope, enabling the coupling of both primary and secondary alcohols—including complex, drug-like examples—with a wide range of structurally distinct sulfoximines. The method demonstrates exceptional functional group tolerance and broad applicability, offering direct access to N-alkyl sulfoximines from readily available starting materials.
The power of this approach in late-stage functionalization was exemplified by the successful modification of pharmacologically active molecules, natural product derivatives, and short peptides. These results highlight the potential for rapidly generating analogues with enhanced structural diversity and complexity. The platform was further expanded to include the modular synthesis of N-substituted sulfoximines bearing bicyclo[1.1.1]pentyl or cyclopropyl motifs, using carboxylic acids and bromocyclopropane as alkyl radical precursors, respectively.
We further applied this methodology in a preliminary drug optimization study, showcasing its value for structure–activity relationship exploration. A sulfoximine analogue of atuveciclib, synthesized in one step, demonstrated improved physicochemical and ADME properties, underlining the potential of this platform to streamline lead diversification and refinement. We anticipate that this reaction will be a valuable addition to the medicinal chemistry toolbox, facilitating the discovery of novel sulfoximine scaffolds and their strategic incorporation as bioisosteric replacements in drug development.
Supplementary Material
Supporting Information. Detailed experimental procedures; optimization data; additional scope examples; characterization data of new compounds; biological studies; NMR spectra for new compounds.
ACKNOWLEDGMENT
The authors thank L. Harstad and A. Pace for helpful discussions, Rebecca Lambert for assistance in preparing this manuscript and B. Kennedy (Lotus Separations) for assistance with compound purification and chiral separation.
Funding Sources
Research reported in this work was supported in part by the National Institute for General Medical Sciences of the National Institutes of Health (R35GM134897), in part by the Office of Naval Research (N000142112138), the Princeton Catalysis Initiative, and kind gifts from Merck, Pfizer, Janssen, Bristol Myers Squibb, Genentech, Novartis and Genmab. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institutes of Health or the Office of Naval Research. J.G. thanks the Deutsche Akademie der Naturforscher Leopoldina (LPDS 2023–03) for financial support.
ABBREVIATIONS
- 4CzIPN
1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene
- Ac
acetyl
- ADME
absorption, distribution, metabolism, excretion
- AdNHSi(TMS)3
adamantylaminosilane
- BCP
bicyclo[1.1.1]pentane
- BTTP
tert-butylimino-tri(pyrrolidino)phosphorane
- BTMG
2-tert-butyl-1,1,3,3-tetramethylguanidine
- Cbz
benzyloxycarbonyl
- CDK
cyclin-dependent kinase
- d.r.
diastereomeric ratio
- ee
enantiomeric excess
- MDCK
Madin-Darby Canine Kidney (cells)
- Mes
mesityl
- MesIO
iodosomesitylene
- SAR
structure–activity relationship
- TMG
1,1,3,3-tetramethylguanidine
- XAT
halogen atom abstraction
Footnotes
The authors declare the following competing financial interest(s): D.W.C.M. declares a competing financial interest with respect to the integrated photoreactor. A provisional U.S. patent has been filed by D.W.C.M. and J.G. based in part on this work, 63/736,215. The remaining authors declare no competing interests.
REFERENCES
- (1).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Vitro Models Sel. Dev. Candidates 1997, 23, 3–25. [Google Scholar]
- (2).Shultz MD Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62, 1701–1714. [DOI] [PubMed] [Google Scholar]
- (3).Hamada Y; Kiso Y The Application of Bioisosteres in Drug Design for Novel Drug Discovery: Focusing on Acid Protease Inhibitors. Expert Opin. Drug Discov. 2012, 7, 903–922. [DOI] [PubMed] [Google Scholar]
- (4).Meanwell NA Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [DOI] [PubMed] [Google Scholar]
- (5).Meanwell NA Applications of Bioisosteres in the Design of Biologically Active Compounds. J. Agric. Food Chem. 2023, 71, 18087–18122. [DOI] [PubMed] [Google Scholar]
- (6).Acharya A; Yadav M; Nagpure M; Kumaresan S; Guchhait SK Molecular Medicinal Insights into Scaffold Hopping-Based Drug Discovery Success. Drug Discov. Today 2024, 29, 103845. [DOI] [PubMed] [Google Scholar]
- (7).Sun H; Tawa G; Wallqvist A Classification of Scaffold-Hopping Approaches. Drug Discov. Today 2012, 17, 310–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Callis TB; Garrett TR; Montgomery AP; Danon JJ; Kassiou M Recent Scaffold Hopping Applications in Central Nervous System Drug Discovery. J. Med. Chem. 2022, 65, 13483–13504. [DOI] [PubMed] [Google Scholar]
- (9).Zhao C; Rakesh KP; Ravidar L; Fang W-Y; Qin H-L Pharmaceutical and Medicinal Significance of Sulfur (SVI)-Containing Motifs for Drug Discovery: A Critical Review. Eur. J. Med. Chem. 2019, 162, 679–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mustafa M; Winum J-Y The Importance of Sulfur-Containing Motifs in Drug Design and Discovery. Expert Opin. Drug Discov. 2022, 17, 501–512. [DOI] [PubMed] [Google Scholar]
- (11).Scott KA; Njardarson JT Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. In Sulfur Chemistry; Jiang X, Ed.; Springer International Publishing: Cham, 2019; pp 1–34. [Google Scholar]
- (12).Kitamura S; Zheng Q; Woehl JL; Solania A; Chen E; Dillon N; Hull MV; Kotaniguchi M; Cappiello JR; Kitamura S; Nizet V; Sharpless KB; Wolan DW Sulfur(VI) Fluoride Exchange (SuFEx)-Enabled High-Throughput Medicinal Chemistry. J. Am. Chem. Soc. 2020, 142, 10899–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ovung A; Bhattacharyya J Sulfonamide Drugs: Structure, Antibacterial Property, Toxicity, and Biophysical Interactions. Biophys. Rev. 2021, 13, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Di L; Kerns EH Profiling Drug-like Properties in Discovery Research. Curr. Opin. Chem. Biol. 2003, 7, 402–408. [DOI] [PubMed] [Google Scholar]
- (15).Serrano-Arias B; Araya-Zúñiga A; Waterhouse-Garbanzo J; Rojas-Barrantes Z; Arguedas-Chacón S; Zavaleta-Monestel E A Comprehensive Review of Sulfonamide Hypersensitivity: Implications for Clinical Practice. Clin. Rev. Allergy Immunol. 2023, 65, 433–442. [DOI] [PubMed] [Google Scholar]
- (16).Lücking U New Opportunities for the Utilization of the Sulfoximine Group in Medicinal Chemistry from the Drug Designer’s Perspective. Chem. Eur. J. 2022, 28, e202201993. [DOI] [PubMed] [Google Scholar]
- (17).Lücking U Sulfoximines: A Neglected Opportunity in Medicinal Chemistry. Angew. Chem. Int. Ed. 2013, 52, 9399–9408. [Google Scholar]
- (18).Han Y; Xing K; Zhang J; Tong T; Shi Y; Cao H; Yu H; Zhang Y; Liu D; Zhao L Application of Sulfoximines in Medicinal Chemistry from 2013 to 2020. Eur. J. Med. Chem. 2021, 209, 112885. [DOI] [PubMed] [Google Scholar]
- (19).Mäder P; Kattner L Sulfoximines as Rising Stars in Modern Drug Discovery? Current Status and Perspective on an Emerging Functional Group in Medicinal Chemistry. J. Med. Chem. 2020, 63, 14243–14275. [DOI] [PubMed] [Google Scholar]
- (20).Anselmi E; Montigny B; Lõkov M; Kesküla C; Soosaar JE; Tshepelevitsh S; Billard T; Magnier E; Leito I Focus on Physico-Chemical Properties of Sulfoximines: Acidity, Basicity and Lipophilicity. Chem. Eur. J. 2025, 31, e202402329. [DOI] [PubMed] [Google Scholar]
- (21).Sirvent JA; Lücking U Novel Pieces for the Emerging Picture of Sulfoximines in Drug Discovery: Synthesis and Evaluation of Sulfoximine Analogues of Marketed Drugs and Advanced Clinical Candidates. ChemMedChem 2017, 12, 487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Frings M; Bolm C; Blum A; Gnamm C Sulfoximines from a Medicinal Chemist’s Perspective: Physicochemical and in Vitro Parameters Relevant for Drug Discovery. Eur. J. Med. Chem. 2017, 126, 225–245. [DOI] [PubMed] [Google Scholar]
- (23).Reck M; Horn L; Novello S; Barlesi F; Albert I; Juhász E; Kowalski D; Robinet G; Cadranel J; Bidoli P; Chung J; Fritsch A; Drews U; Wagner A; Govindan R Phase II Study of Roniciclib in Combination with Cisplatin/Etoposide or Carboplatin/Etoposide as First-Line Therapy in Patients with Extensive-Disease Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 701–711. [DOI] [PubMed] [Google Scholar]
- (24).Kwon M; Kim G; Kim R; Kim K-T; Kim ST; Smith S; Mortimer PGS; Hong JY; Loembé A-B; Irurzun-Arana I; Koulai L; Kim K-M; Kang WK; Dean E; Park W-Y; Lee J Phase II Study of Ceralasertib (AZD6738) in Combination with Durvalumab in Patients with Advanced Gastric Cancer. J. Immunother. Cancer 2022, 10, e005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ghosh P; Ganguly B; Das S N−H and C−H Functionalization of Sulfoximine: Recent Advancement and Prospects. Asian J. Org. Chem. 2020, 9, 2035–2082. [Google Scholar]
- (26).Hendriks CMM; Bohmann RA; Bohlem M; Bolm C N-Alkylations of NH-Sulfoximines and NH-Sulfondiimines with Alkyl Halides Mediated by Potassium Hydroxide in Dimethyl Sulfoxide. Adv. Synth. Catal. 2014, 356, 1847–1852. [Google Scholar]
- (27).Dodd CJ; Schultz DC; Li J; Lindsley CW; Bender AM Alkylation of NH-Sulfoximines under Mitsunobu-Type Conditions. Org. Biomol. Chem. 2023, 21, 5181–5184. [DOI] [PubMed] [Google Scholar]
- (28).Wang H; Frings M; Bolm C Halocyclizations of Unsaturated Sulfoximines. Org. Lett. 2016, 18, 2431–2434. [DOI] [PubMed] [Google Scholar]
- (29).Shi P; Tu Y; Ma D; Bolm C Copper-Catalyzed N-Alkylations of NH-Sulfoximines Under Visible Light. Adv. Synth. Catal. 2023, 365, 1613–1617. [Google Scholar]
- (30).Zhu H; Teng F; Pan C; Cheng J; Yu J-T Radical N-Arylation/Alkylation of Sulfoximines. Tetrahedron Lett. 2016, 57, 2372–2374. [Google Scholar]
- (31).Cheng Y; Dong W; Wang L; Parthasarathy K; Bolm C Iron-Catalyzed Hetero-Cross-Dehydrogenative Coupling Reactions of Sulfoximines with Diarylmethanes: A New Route to N-Alkylated Sulfoximines. Org. Lett. 2014, 16, 2000–2002. [DOI] [PubMed] [Google Scholar]
- (32).Teng F; Sun S; Jiang Y; Yu J-T; Cheng J Copper-Catalyzed Oxidative C(Sp3)–H/N–H Coupling of Sulfoximines and Amides with Simple Alkanes via a Radical Process. Chem. Commun. 2015, 51, 5902–5905. [Google Scholar]
- (33).Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Großkopf J; Gopatta C; Martin RT; Haseloer A; MacMillan DWC Generalizing Arene C–H Alkylations by Radical–Radical Cross-Coupling. Nature 2025, 641, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Le C; Chen TQ; Liang T; Zhang P; MacMillan DWC A Radical Approach to the Copper Oxidative Addition Problem: Trifluoromethylation of Bromoarenes. Science 2018, 360, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lavagnino MN; Liang T; MacMillan DWC HARC as an Open-Shell Strategy to Bypass Oxidative Addition in Ullmann–Goldberg Couplings. Proc. Natl. Acad. Sci. 2020, 117, 21058–21064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dow NW; Cabré A; MacMillan DW C. A General N-Alkylation Platform via Copper Metallaphotoredox and Silyl Radical Activation of Alkyl Halides. Chem 2021, 7, 1827–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Carson WPI; Tsymbal AV; Pipal RW; Edwards GA; Martinelli JR; Cabré A; MacMillan DWC Free-Radical Deoxygenative Amination of Alcohols via Copper Metallaphotoredox Catalysis. J. Am. Chem. Soc. 2024, 146, 15681–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dong Z; MacMillan DWC Metallaphotoredox-Enabled Deoxygenative Arylation of Alcohols. Nature 2021, 598, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Liang Y; Zhang X; MacMillan DWC Decarboxylative Sp3 C–N Coupling via Dual Copper and Photoredox Catalysis. Nature 2018, 559, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Speckmeier E; Fischer TG; Zeitler K A Toolbox Approach To Construct Broadly Applicable Metal-Free Catalysts for Photoredox Chemistry: Deliberate Tuning of Redox Potentials and Importance of Halogens in Donor–Acceptor Cyanoarenes. J. Am. Chem. Soc. 2018, 140, 15353–15365. [DOI] [PubMed] [Google Scholar]
- (42).Chen Y; Chen Q; Zhang S; Feng K; Kong X; Chen X; Li W; Cao Z-Y I2-Catalyzed Benzylation of NH-Sulfoximines with Diarylmethanes and Alkylarenes. Org. Biomol. Chem. 2025, 23, 2111–2114. [DOI] [PubMed] [Google Scholar]
- (43).Kong X; Tian Y; Chen X; Chen Y; Wang W Electrochemical Oxidative C(Sp3)–H/N–H Coupling of Diarylmethanes with Sulfoximines or Benzophenone Imine. J. Org. Chem. 2021, 86, 13610–13617. [DOI] [PubMed] [Google Scholar]
- (44).Sakai HA; Liu W; Le C. “Chip”; MacMillan DWC Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc. 2020, 142, 11691–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Andresini M; Tota A; Degennaro L; Bull JA; Luisi R Synthesis and Transformations of NH-Sulfoximines. Chem. Eur. J. 2021, 27, 17293–17321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- (47).Jiang H; Qin X; Wang Q; Xu Q; Wang J; Wu Y; Chen W; Wang C; Zhang T; Xing D; Zhang R Application of Carbohydrates in Approved Small Molecule Drugs: A Review. Eur. J. Med. Chem. 2021, 223, 113633. [DOI] [PubMed] [Google Scholar]
- (48).Li X; Zhu J Glycosylation via Transition-Metal Catalysis: Challenges and Opportunities. Eur. J. Org. Chem. 2016, 2016, 4724–4767. [Google Scholar]
- (49).Guillemard L; Kaplaneris N; Ackermann L; Johansson MJ Late-Stage C–H Functionalization Offers New Opportunities in Drug Discovery. Nat. Rev. Chem. 2021, 5, 522–545. [DOI] [PubMed] [Google Scholar]
- (50).Castellino NJ; Montgomery AP; Danon JJ; Kassiou M Late-Stage Functionalization for Improving Drug-like Molecular Properties. Chem. Rev. 2023, 123, 8127–8153. [DOI] [PubMed] [Google Scholar]
- (51).Lücking U; Scholz A; Lienau P; Siemeister G; Kosemund D; Bohlmann R; Briem H; Terebesi I; Meyer K; Prelle K; Denner K; Bömer U; Schäfer M; Eis K; Valencia R; Ince S; von Nussbaum F; Mumberg D; Ziegelbauer K; Klebl B; Choidas A; Nussbaumer P; Baumann M; Schultz-Fademrecht C; Rühter G; Eickhoff J; Brands M Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChem 2017, 12, 1776–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Boto A; González CC; Hernández D; Romero-Estudillo I; Saavedra CJ Site-Selective Modification of Peptide Backbones. Org. Chem. Front. 2021, 8, 6720–6759. [Google Scholar]
- (53).Beil SB; Chen TQ; Intermaggio NE; MacMillan DWC Carboxylic Acids as Adaptive Functional Groups in Metallaphotoredox Catalysis. Acc. Chem. Res. 2022, 55, 3481–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Fang Z; X. Qian; Lu Xuehe; Wan Nianfeng; Yang Wu-Lin. The Application of Bicyclo[1.1.1]Pentane as a Bioisostere of the Phenyl Ring in Pharmaceutical Chemistry. Synthesis 2024, 57, 1171–1179. [Google Scholar]
- (55).Subbaiah MAM; Meanwell NA Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug Design. J. Med. Chem. 2021, 64, 14046–14128. [DOI] [PubMed] [Google Scholar]
- (56).Bär RM; Langer L; Nieger M; Bräse S Bicyclo[1.1.1]Pentyl Sulfoximines: Synthesis and Functionalizations. Adv. Synth. Catal. 2020, 362, 1356–1361. [Google Scholar]
- (57).Wiesenfeldt MP; Rossi-Ashton JA; Perry IB; Diesel J; Garry OL; Bartels F; Coote SC; Ma X; Yeung CS; Bennett DJ; MacMillan DWC General Access to Cubanes as Benzene Bioisosteres. Nature 2023, 618, 513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Garry OL; Heilmann M; Chen J; Liang Y; Zhang X; Ma X; Yeung CS; Bennett DJ; MacMillan DWC Rapid Access to 2-Substituted Bicyclo[1.1.1]Pentanes. J. Am. Chem. Soc. 2023, 145, 3092–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zhang X; Smith RT; Le C; McCarver SJ; Shireman BT; Carruthers NI; MacMillan DWC Copper-Mediated Synthesis of Drug-like Bicyclopentanes. Nature 2020, 580, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Sanna G; Pilo MI; Zoroddu MA; Seeber R; Mosca S Electrochemical and Spectroelectrochemical Study of Copper Complexes with 1,10-Phenanthrolines. Inorg. Chim. Acta 1993, 208, 153–158. [Google Scholar]
- (61).Ladouceur S; Fortin D; Zysman-Colman E Enhanced Luminescent Iridium(III) Complexes Bearing Aryltriazole Cyclometallated Ligands. Inorg. Chem. 2011, 50, 11514–11526. [DOI] [PubMed] [Google Scholar]
- (62).Goldberg FW; Kettle JG; Xiong J; Lin D General Synthetic Strategies towards N-Alkyl Sulfoximine Building Blocks for Medicinal Chemistry and the Use of Dimethylsulfoximine as a Versatile Precursor. Tetrahedron 2014, 70, 6613–6622. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.