

Abstract
Mx proteins form a small family of interferon (IFN)-induced GTPases with potent antiviral activity against various negative-strand RNA viruses. To examine the antiviral spectrum of human MxA in homologous cells, we stably transfected HEp-2 cells with a plasmid directing the expression of MxA cDNA. HEp-2 cells are permissive for many viruses and are unable to express endogenous MxA in response to IFN. Experimental infection with various RNA and DNA viruses revealed that MxA-expressing HEp-2 cells were protected not only against influenza virus and vesicular stomatitis virus (VSV) but also against Semliki Forest virus (SFV), a togavirus with a single-stranded RNA genome of positive polarity. In MxA-transfected cells, viral yields were reduced up to 1,700-fold, and the degree of inhibition correlated well with the expression level of MxA. Furthermore, expression of MxA prevented the accumulation of 49S RNA and 26S RNA, indicating that SFV was inhibited early in its replication cycle. Very similar results were obtained with MxA-transfected cells of the human monocytic cell line U937. The results demonstrate that the antiviral spectrum of MxA is not restricted to negative-strand RNA viruses but also includes SFV, which contains an RNA genome of positive polarity. To test whether MxA protein exerts its inhibitory activity against SFV in the absence of viral structural proteins, we took advantage of a recombinant vector based on the SFV replicon. The vector contains only the coding sequence for the viral nonstructural proteins and the bacterial LacZ gene, which was cloned in place of the viral structural genes. Upon transfection of vector-derived recombinant RNA, expression of the β-galactosidase reporter gene was strongly reduced in the presence of MxA. This finding indicates that viral components other than the structural proteins are the target of MxA action.
SFV is a member of the family Togaviridae (genus Alphavirus), a family of mosquito-borne, positive-strand RNA viruses which has a large host range and whose most common complication is encephalitis (17). The virus enters the cells via receptor-mediated endocytosis (22). The uncoating of the nucleocapsids depends on ribosomes which release the capsid proteins from the nucleocapsid and sequester them (35). In contrast to negative-strand RNA viruses, which transport their RNA transcriptase within the virion into the cells, the liberated genomic 49S RNA of Semliki Forest virus (SFV) serves directly as mRNA for the synthesis of the RNA polymerase. For replication, which occurs in the cytoplasm, the parental 49S positive-strand RNA is transcribed into a 49S negative-strand RNA, which in turn serves as a template for either the synthesis of progeny 49S positive-strand genomic RNA or subgenomic 26S mRNA directing the synthesis of structural proteins (15).
SFV infection is strongly impaired in mice following treatment with type I interferon (IFN) (7). IFN also mediates a very potent activity against SFV replication in cell cultures, and the virus is widely used as challenging agent in virus yield reduction assays (20). IFN-α treatment leads to reduced viral protein levels and hinders virus-mediated host shutoff (24). However, little is known about the molecular mechanisms of this antiviral action. The antiviral effect of IFNs is mediated by several IFN-induced proteins which inhibit the multiplication of viruses by distinct mechanisms (for reviews see references 31 and 37). Some members of the Mx protein family were shown to contribute to this antiviral state by inhibiting the multiplication of different negative-strand RNA viruses (5, 6, 8, 16, 23, 27, 33, 39, 43, 44, 46). The Mx proteins form a small group of GTPases (9, 25, 29) and are synthesized under the stringent control of IFN type I (1, 38). The molecular mechanism of Mx action still remains unclear, but the GTPase activity appears to be essential for antiviral function (28). The antiviral properties of Mx proteins differ and are influenced by the intracellular localization of a particular Mx protein (14, 45, 47). Murine Mx1, which accumulates in the nucleus (4, 11), appears to be specific for Orthomyxoviridae (8, 27, 39, 43). The protein interferes with influenza virus replication at the level of primary transcription (18, 19, 26), suggesting an interaction with the viral polymerase complex. Indeed, overexpression of PB2, a subunit of the influenza virus polymerase complex, leads to a partial neutralization of the antiviral effect of Mx1 (12, 41).
The human MxA protein, which accumulates in the cytoplasm, has a broader activity, inhibiting the multiplication of influenza virus, Thogoto virus (Orthomyxoviridae), vesicular stomatitus virus (VSV) (Rhabdoviridae), measles virus (MV), human parainfluenza virus type 3 (Paramyxoviridae), and several members of the family Bunyaviridae (5, 6, 16, 27, 32, 33, 44). In contrast to Mx1, MxA appears to block the multiplication of influenza virus at a poorly defined cytoplasmic step following primary transcription (26). In the case of VSV, MxA inhibits primary transcription (40). For MV, the situation is clearly different. First, the protective effect of MxA against MV was detected only in the human monocytic cell line U937 and in the glioblastoma cell line U87. Furthermore, MxA inhibited the multiplication of MV at the level of either viral RNA synthesis or synthesis of viral glycoproteins, depending on the cell line used (32, 33).
We report here that the antiviral specificity of MxA is extended to SFV, a positive-strand RNA virus. The activity of MxA against SFV appears to be either cell type or species specific, since no inhibitory effect was found in MxA-transfected mouse 3T3 fibroblasts (27). The fact that the accumulation of viral RNAs and proteins was inhibited points to a block occurring early in the replicative cycle. In order to define potential viral targets of MxA, we took advantage of an SFV replicon-based vector coding only for the viral replicase. The viral structural genes are replaced by the bacterial LacZ reporter gene (21). Upon transfection into cells, vector-derived recombinant RNA is amplified by virtue of its self-encoded replicase, and as a consequence large quantities of β-galactosidase (β-Gal) are produced. In MxA-transfected HEp-2 cells but not in mouse 3T3 cells, expression of β-Gal was dramatically reduced. These results demonstrate that the SFV structural proteins are not the target of MxA action and further suggest the involvement of species-specific cellular factors.
MATERIALS AND METHODS
IFNs and IFN treatments.
Recombinant human IFN-α2 (Roferon-A) was obtained from Roche Pharma, Rheinach, Switzerland. Approximately 90%-confluent cell monolayers were treated with 1,000 U of IFN per ml in culture medium for 18 h prior to protein or RNA extraction.
Cells.
Swiss mouse 3T3 cells and human HEp-2 cells were grown in Dulbecco’s modified minimal essential medium containing 10% fetal calf serum. The human monocytic U937 cell line (42) was cultured in RPMI 1640 medium. Stably transfected 3T3, HEp-2, and U937 cells were maintained in culture medium containing 500 μg of G418 per ml.
Viruses.
Stocks of the FPV-B strain (13) of influenza A virus (108 PFU/ml), VSV serotype Indiana (108 PFU/ml), encephalomyocarditis virus (EMCV) (109 PFU/ml), SFV (6.8 × 108 PFU/ml), herpes simplex virus type 1 (HSV-1) (3 × 106 PFU/ml), and mengovirus (2 × 109 PFU/ml) were prepared from supernatants of virus-infected Swiss mouse 3T3 cells.
Transfection.
HEp-2 cells were cotransfected with pSV2-neo (36) and the pHMG-MxA expression vector (27) as previously described (39). Transfected cells were selected in culture medium containing 500 μg of G418 per ml. Resistant clones were examined for MxA expression by indirect immunofluorescence (4). Positive clones were subjected to a second round of subcloning by limiting dilution.
Immunofluorescence analysis.
Cell cultures were prepared as previously described (4). Mouse monoclonal anti-recombinant MxA antibody and polyclonal rabbit anti-SFV C protein serum (27a) were diluted in phosphate-buffered saline (PBS) containing 5% normal goat serum. Rhodamine-conjugated goat anti-rabbit immunoglobulin G and fluorescein-conjugated goat anti-mouse immunoglobulin G (diluted 1:50 in PBS containing 5% normal goat serum; Nordic) were used as secondary antibodies.
Western blot analysis.
The cytoplasmic cell extracts were prepared according to the protocol of a β-Gal enzyme-linked immunosorbent assay (ELISA) kit from Boehringer Mannheim. Briefly, the culture medium was removed, and the cells were washed with precooled PBS. Hypotonic cell lysis buffer (pH 6.5) containing morpholinepropanesulfonic acid (MOPS)-buffered saline and Triton X-100 was added, and the cells were incubated for 30 min at room temperature. To remove the cell debris, the extract was spun in a microcentrifuge at maximum speed. Separation of the sample on sodium dodecyl sulfate (SDS)–10% polyacrylamide gels, protein transfer to nitrocellulose membranes, and immunostaining were done as previously described (1). SFV C protein was detected by immunostaining with a polyclonal rabbit anti-SFV C protein serum. MxA protein was immunostained with a mouse monoclonal anti-MxA (p78) antibody (10). The blot was stained with a horseradish peroxidase-conjugated secondary antibody and then incubated with SuperSignal working solution.
Virus plaque assay.
Cell monolayers in 60-mm-diameter dishes were infected with several hundred PFU of virus. After 90 min at 37°C, unabsorbed virus was removed and overlay agar (culture medium containing 2% fetal calf serum, 20 mM HEPES buffer [pH 7.3], 0.002% DEAE dextran, and 0.4% noble agar) was added. The cultures were incubated for 48 to 72 h at 37°C. To visualize viral plaques, the agar overlay was removed and cells were stained with 1% crystal violet in 20% ethanol.
Virus yield reduction assay.
Confluent cell monolayers were infected as described previously (27). Culture supernatants were collected 24 h postinfection, and virus yields were determined on 3T3 cells by the TCID50 method 24 h postinfection.
RNA extraction and Northern blot analysis.
Total cell RNA was prepared by the acid-guanidinium-phenol-chloroform procedures (3). Northern blot analysis was carried out as described previously (1) with 1.2% agarose gels containing formaldehyde to separate the RNA. As hybridization probes, 32P-labeled cDNA fragments of SFV genomic RNA, human glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and human guanylate-binding protein 1 (GBP1) (2) were used.
SFV eukaryotic expression system.
The eukaryotic expression vector pSFV3-lacZ (21) (GIBCO BRL), coding for the SFV nonstructural proteins (nsP1 to nsP4) and β-Gal protein, was linearized with SpeI. Recombinant RNA was prepared in vitro with SP6-polymerase in the presence of m7G(5′)ppp(5′)G as a capping analog. Transfection of pSFV3-lacZ was carried out according to the protocol of the manufacturer. Briefly, 80%-confluent 35-mm-diameter dishes were transfected with 5 μg of RNA complexed with 10 μl of Lipofectamine (GIBCO BRL). Transfection was carried out for 2 h at 37°C in Opti-MEM medium (GIBCO BRL). Subsequently, Opti-MEM medium was removed and cells were allowed to express RNA for 20 h in complete medium. Cell extracts were prepared, and the amount of protein was measured with Bradford reagent. Expression of β-Gal was measured by quantitative determination of β-Gal in each cell extract (30 μg of protein) by a colorimetric enzyme immunoassay (Boehringer Mannheim). In one experiment, 3 μg of recombinant SFV RNA was cotransfected with 3 μg of pSV2-CAT expression plasmid (Stratagene) under the same conditions. The amount of bacterial enzyme chloramphenicol acetyltransferase type I (CAT) was determined with a CAT ELISA kit (Boehringer Mannheim).
RESULTS
Stable expression of MxA protein.
HEp-2 cells were chosen because they are susceptible to many viruses and do not express endogenous MxA protein. The cells were cotransfected with pHMG-MxA (27) and pSV2-neo (36) and subsequently selected in the presence of G418. Individual colonies were tested for expression by indirect immunofluorescence, and positive clones were subjected to subcloning by limited dilution. MxA accumulated in the cytoplasm of transfected cells and showed a granular staining pattern similar to MxA patterns observed in various human cell lines treated with human IFN-α2. As a second cell line, stably MxA-transfected human monocytic U937 cell clones that had been previously shown to restrict infection of MV, VSV (33), and influenza virus (30) were used. The U937-MxA clonal lines 5 and 6 were also subjected to subcloning by limited dilution. The clonal lines expressed the transgene at levels comparable to the glioblastoma cell line T98G stimulated with human IFN-α2 (data not shown). All subclones expressed MxA in more than 98% of the cells as judged by immunofluorescence analysis.
Inhibition of SFV replication by MxA.
To elucidate the antiviral spectrum of HEp-2-MxA cells, subclones expressing MxA and HEp-2-neo control cells were subjected to viral plaque assays following infection with influenza A fowl plaque virus (Bratislava strain, FPV-B), VSV, SFV, mengovirus, EMCV, and HSV-1. As expected, in the presence of MxA, plaque formation was completely inhibited upon infection with several hundred PFU of influenza virus, and infection with VSV yielded only a few very small plaques (Fig. 1). Surprisingly, we detected no plaque formation with SFV on HEp-2-MxA cells (Fig. 1). By contrast, mengovirus, EMCV, and HSV-1 showed no reduction in the number or size of plaques on HEp-2-MxA cells (data not shown).
FIG. 1.

Inhibition of virus plaque formation by MxA protein expressed in stably transfected human cell lines. Confluent monolayers of HEp-2-neo control cells and the clonal lines HEp-2-MxA 44.12 and 71.2 were infected with several hundred PFU of either influenza A virus (FPV-B), VSV, or SFV. The viruses were allowed to form plaques under soft agar for 46 h (VSV infection) or 70 h (influenza virus and SFV infection).
Inhibition of plaque formation of SFV on HEp2-MxA cells came as a surprise, because mouse 3T3 cells overexpressing mouse Mx1 or human MxA remain sensitive to infection with SFV (27). However, virus yield reduction assays corroborated the finding from the viral plaque assays. To that end, HEp-2-MxA and U937-MxA clonal lines were infected with virus at a multiplicity of infection (MOI) of 0.1 and the tissue culture supernatants were collected 24 h postinfection. HEp-2-neo and U937 cells were used as control cells. SFV yields were reduced between 30- and 700-fold in clonal lines of HEp-2-MxA cells compared to HEp-2-neo cells and between 30- and 1,700-fold in clonal lines of U937-MxA cells compared to U937 cells not expressing MxA (Fig. 2B). Quantitation of MxA signals on Western blots with cytoplasmic cell extracts from the three independent HEp-2-MxA lines and the four U937-MxA lines (Fig. 2A) revealed that there is a good correlation between the amount of MxA and virus inhibition.
FIG. 2.

Expression of MxA protein in stably transfected human HEp-2-MxA and U937-MxA cells leads to inhibition of SFV multiplication. (A) Western blot analysis of cytoplasmic extracts from various HEp-2-MxA and U937-MxA clonal lines. Samples (50 μg of protein per lane) were separated in SDS–10% polyacrylamide gels and subsequently blotted onto nitrocellulose membranes. The blots were immunostained with a monoclonal mouse anti-MxA antibody. The bands were visualized with enhanced chemiluminescence reagents (Amersham). Quantitation of the optical density was carried out with the video documentation system E.A.S.Y (Herolab, Wiesloch, Germany). The relative MxA levels are shown as arbitrary expression units. (B) Reduction of SFV yields by MxA. The different clonal lines were infected with SFV at an MOI of 0.1. Viral yields in culture supernatants 24 h postinfection were determined by the TCID50 method.
In clonal lines of HEp-2-MxA and U937-MxA, the yields for influenza virus were reduced between 100- and 400-fold, and VSV was reduced about 100-fold. The multiplication of EMCV, mengovirus, and HSV-1 was not affected in these cell clones (data not shown), in accordance with the results obtained from the MxA-transfected mouse (27) and human cell lines (30, 32, 33).
Infected cells expressing MxA show strongly reduced levels of SFV C protein.
The remaining virus produced in MxA-expressing human cells could be the result of either a low level of replication in all cells or a breakdown of resistance in a small fraction of cells. To address this question, HEp-2-neo and HEp-2-MxA 71.2 cells were infected with SFV at an MOI of 2. The cells were fixed after 24 h and simultaneously analyzed for expression of MxA and SFV C protein by indirect immunofluorescence analysis (Fig. 3). Only a small fraction of HEp-2-MxA cells (about 1% of the cell population) expressed detectable levels of C protein in the cell cytoplasm (Fig. 3D). C protein was detected only in cells accumulating low levels of MxA (Fig. 3E and F). By contrast, virtually 100% of the HEp-2-neo cells showed high concentrations of the C protein in the cell nucleus and cytoplasm (Fig. 3B). These findings clearly argue in favor of the notion of a few MxA-expressing cells producing large amounts of virus. Very similar results were obtained with MxA-transfected U937 cells (data not shown).
FIG. 3.
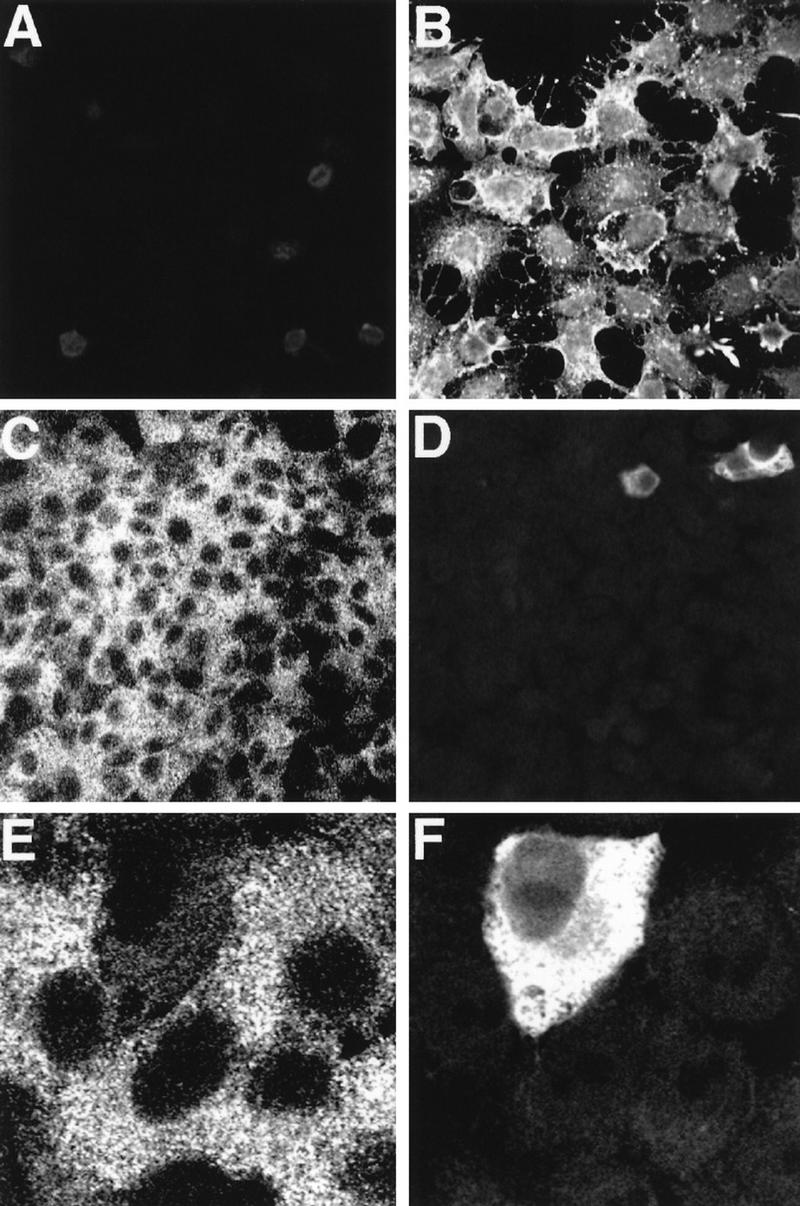
Inhibition of SFV protein synthesis in HEp-2 cells expressing MxA. HEp-2-neo control cells (A and B) and cells of the clonal line HEp-2-MxA 71.2 (C through F) were infected with SFV at an MOI of 2. Cells were fixed 24 h postinfection and subjected to double immunofluorescence analysis with a confocal laser scanning light microscope. Cells were simultaneously immunostained with a mouse monoclonal anti-recombinant MxA antibody (A, C, and E) and a polyclonal rabbit anti-SFV C protein serum (B, D, and F).
The accumulation of C protein in infected cells was further assessed by Western blot analysis. Subconfluent cultures of several independent HEp-2-MxA and U937-MxA clonal lines were infected with SFV at an MOI of 3, and total cell extracts were prepared 7 h postinfection. Extracts (200 μg per lane) of infected cells were analyzed by Western blotting with a rabbit polyclonal antiserum directed against recombinant C protein. In contrast to control cells, HEp-2-MxA cell clones 44.12 and 71.2 showed no detectable accumulation of SFV C protein (Fig. 4A). Similarly, C protein levels were strongly reduced in extracts of cell clones U937-MxA 5.1 and 9 (Fig. 4B).
FIG. 4.

Accumulation of SFV C protein is inhibited in infected cells stably expressing MxA protein. Monolayer cultures of HEp-2-MxA (A) and U937-MxA (B) clonal lines were infected with SFV at an MOI of 3. HEp-2-neo and U937 cells were used as controls. Infected cells were harvested 7 h postinfection. Western blots of whole-cell extracts (200 μg per lane) were immunostained with a polyclonal rabbit anti-SFV C protein antiserum.
SFV mRNA synthesis is inhibited by human MxA protein.
To determine whether MxA inhibited SFV mRNA synthesis, we infected cell monolayers of several independent cell clones stably expressing MxA with SFV at an MOI of 3. Accumulation of viral RNAs was monitored 5 h postinfection by Northern blot analysis with a cDNA probe of the viral genome specific for genomic 49S and subgenomic 26S RNA. Twenty micrograms of total cell RNA was loaded per lane, and the amount was verified by ethidium bromide staining of parallel RNA gels. MxA expression had a strong effect on the accumulation of viral RNA. While HEp-2-neo control cells produced large quantities of 26S and 49S RNA by 5 h postinfection, clonal lines of HEp-2-MxA either lacked detectable levels (HEp-2-MxA 71.2) or contained viral RNAs mounting to 2% (HEp-2-MxA 44.12) and 14% (HEp-2-MxA 34.36) of control cells, respectively (Fig. 5A). The viral RNA levels observed in the different clonal lines reflected the virus yields (Fig. 2B). Similar results were obtained with various clonal lines of U937-MxA where RNA accumulation was at best 1% of control cells (Fig. 5B). The same blots were also hybridized with single-stranded SFV probes of negative or positive polarity. The results were virtually identical to the ones obtained with the double-stranded cDNA probe, indicating that MxA does not discriminate between positive- or negative-strand replication or transcription (data not shown).
FIG. 5.

Reduced accumulation of SFV genomic 49S and subgenomic 26S RNAs in infected cells expressing MxA protein. Monolayer cultures of different clonal lines of HEp-2-MxA (A) and U937-MxA cells (B) were infected with SFV at an MOI of 3, and total cell RNA was prepared 5 h postinfection. HEp-2-neo and U937 cells served as controls. HEp-2 cells were either left untreated or pretreated with IFN-α2 for 18 h prior to infection (A). (C) To examine RNA accumulation throughout the course of infection, the clonal lines HEp-2-MxA 71.2 and HEp-neo were infected with SFV at an MOI of 1 and total cell RNA was prepared 5, 24, 48, and 72 h postinfection (C). The RNA preparations (20 μg per lane) were subjected to Northern blot analysis with radiolabeled genomic cDNA of SFV. Hybridization to a radiolabeled 0.8-kb fragment of human GAPDH cDNA was used as a control (C, lower section). The blots were exposed to X-ray film.
To examine RNA levels at different time points after infection, cell monolayers of HEp-2-neo and HEp-2-MxA cells were infected with SFV at an MOI of 1 and accumulation of viral RNA was examined at 5, 24, 48, and 72 h postinfection (Fig. 5C). In this experiment, large amounts of viral RNA were detected in control cells 24 h postinfection. Since all HEp-2-neo cells had been killed by the virus 48 h postinfection, it was not possible to collect samples after 48 and 72 h. However, most of the HEp-2-MxA cells survived virus infection, and viral RNA was barely visible after 24 and 48 h and no longer detectable after 72 h. To monitor the amount of RNA loaded in each lane, the blot was reprobed with a radiolabeled GAPDH cDNA fragment. In HEp-2 cells, the amount of GAPDH RNA was clearly reduced 24 h postinfection (Fig. 5C). This result is presumably due to the virus-mediated host shutoff. In contrast to the experiments shown in Fig. 5A and B, significant accumulation of viral RNA in HEp-2-neo cells occurred later than 5 h postinfection. This result is explained by the fact that less virus inoculum was used in the later experiment.
HEp-2 cells pretreated with IFN showed no accumulation of viral RNA after infection (Fig. 5A), suggesting that IFN-induced protein(s) other than MxA inhibits the replication of SFV. To exclude the possibility that the observed protection is due in part to the induction of IFN type I in infected HEp-2-MxA cells, we infected HEp-2-neo and HEp-2-MxA cells with SFV at an MOI of 1 and measured the amount of IFN-α present in cell culture supernatants 8, 24, 48, 72, and 168 h postinfection by ELISA (Anawa, Dübendorf, Switzerland). No IFN-α was detected, indicating that the antiviral effect was solely due to MxA. Moreover, probing of a Northern blot parallel to the one shown in Fig. 5C with a radiolabeled cDNA probe of the IFN-inducible human GBP1 gene (2) showed that GBP1 mRNA accumulated in trace amounts 24 h postinfection in HEp-2-neo cells but not in HEp-2-MxA cells. Low levels of GBP1 mRNA also accumulated in HEp-2-MxA cells 48 and 72 h postinfection (data not shown).
Inhibition by MxA protein is independent of SFV structural proteins.
To determine whether MxA interferes with the structural proteins of SFV, we took advantage of the eukaryotic expression system based on the SFV replicon (21). This recombinant viral RNA codes for the SFV nonstructural proteins nsP1 to nsP4 (SFV replicase) but lacks the genetic information for the structural genes, which in the case of plasmid pSFV3-lacZ are replaced by the bacterial LacZ gene (21). Upon transfection into target cells, the viral RNA drives its own replication and capping, resulting in the production of β-Gal.
HEp-2-neo and HEp-2-MxA cells were transfected with recombinant viral RNA synthesized in vitro from the plasmid pSFV3-lacZ. Mx-transfected 3T3 cell clones served as controls, since 3T3-Mx1 and 3T3-MxA cells are sensitive to infection with SFV (27). Twenty-four hours after transfection, the cells were lysed and cytoplasmic extracts were prepared. To monitor transfection efficiency in the various HEp-2 and 3T3 cell lines, a pSV2-CAT expression plasmid was cotransfected with the viral replicon RNA in one experiment. The cell extracts were then simultaneously tested for the production of β-Gal and CAT protein by colorimetric enzyme immunoassays (Fig. 6).
FIG. 6.

Structural proteins are not required for the inhibition of SFV replication by MxA protein. Eighty-percent confluent monolayer cultures of the clonal lines HEp-2-MxA 71.2, 44.12, and 34.36 were cotransfected with 3 μg of in vitro-transcribed RNA from the plasmid pSFV3-lacZ and 3 μg of pSV2-CAT expression plasmid. Clones 3T3-neo, 3T3-MxA, 3T3-Mx1, and HEp-2-neo were used as controls. The recombinant RNA coding for the SFV replicase and the bacterial gene LacZ drives its own replication and capping, resulting in the production of heterologous β-Gal protein. For the quantitative determination of β-Gal and CAT colorimetric enzyme, immunoassays were used.
Figure 6 shows that CAT expression and hence the transfection efficiency varied little (80 to 150% of control values) among the different clonal lines of transfected 3T3 and HEp-2 cells. Similarly, the concentrations of β-Gal did not differ significantly in extracts of 3T3-neo, 3T3-Mx1, and 3T3-MxA cells (Fig. 6). However, in the case of MxA-expressing HEp-2 cells, β-Gal levels were reduced to 2% (HEp-2-MxA 71.2), 7% (HEp-2-MxA 34.36), and 9% (HEp-2-MxA 44.12) of the levels observed in HEp-2-neo cells (approximately 30 ng/mg of protein). The experiment was repeated three times, showing no significant differences from the data presented in Fig. 6.
The data clearly demonstrate that the viral structural proteins are not the target of MxA, since the nonstructural proteins nsP1 to nsP4, which form the SFV transcriptase complex, are the only viral proteins produced in this system. We next tested whether MxA would affect the accumulation of the SFV replicon RNA. To that end, HEp-2-MxA clonal lines and control cells were transfected with pSFV3-lacZ RNA and total cellular RNA was isolated 24 h later. The accumulation of SFV replicon RNA was assayed by Northern blot analysis with radiolabeled cDNA coding for nsP1 to nsP4 (Fig. 7). Although we could detect only a faint signal, the results show that expression of MxA led to a strong reduction of SFV replicon RNA levels (clonal lines 71.2, 44.12, and 34.36) compared to control cells.
FIG. 7.

Accumulation of pSFV3-lacZ replicon RNA is strongly reduced in HEp-2 cells lines expressing MxA protein. Eighty-percent confluent monolayer cultures of the clonal lines HEp-2-MxA 71.2, 44.12, and 34.36 were transfected with 5 μg of in vitro-transcribed RNA from the plasmid pSFV3-lacZ. Clone HEp-2-neo was used as a control. RNA (20 μg per lane) was prepared 24 h after transfection and subjected to Northern blot analysis with a radiolabeled cDNA coding for nsP1 to nsP4. The blots were exposed to X-ray film.
DISCUSSION
The rationale for transfecting human HEp-2 cells with MxA cDNA was to examine the antiviral spectrum of MxA in a homologous cell line that was sensitive to many viruses. Until now, antiviral activity of Mx proteins has been reported only against negative-strand RNA viruses (5, 6, 8, 16, 23, 27, 33, 39, 43, 44, 46). Our experiments now show that in homologous cells, human MxA exerts an intrinsic antiviral activity against SFV, a positive-strand RNA virus. MxA expression led to a drastic reduction of viral protein and RNA synthesis and consequently to reduced viral titers. Moreover, using the SFV RNA replicon expression system, we were able to demonstrate that MxA does not interfere with the structural proteins of SFV.
Human cells expressing MxA were protected from the cytopathic effect of SFV, and most cells not only survived SFV infection but also continued to proliferate. By contrast, the cytopathic killing of control cultures was completed within 24 to 48 h postinfection. Immunofluorescence analysis of SFV-infected cells at different time points postinfection revealed that only a small fraction of approximately 1% (Fig. 3) of MxA-expressing cells showed expression of C protein at detectable levels. These cells showed no or weak expression of MxA. We therefore assume that the infectious virus particles observed in the supernatants of HEp-2-MxA and U937-MxA cells are due to the replication of SFV in a few nonresistant cells. This conclusion is supported by the fact that the virus is completely cleared from these cultures within 2 weeks postinfection (data not shown).
SFV replication is also inhibited in IFN-α2-treated Hep-2 cells, which do not express MxA protein, indicating that MxA is not the only cellular protein that mediates IFN action against this virus (Fig. 5A). Similar observations have been made with VSV in mouse and human cells (27, 46). However, other IFN-induced proteins did not contribute to the protective effect of the transfected MxA, since culture supernatants of SFV-infected HEp-2-MxA cells were completely devoid of IFN-α. This result was confirmed by the finding that mRNA of GBP1, an IFN-inducible gene, was not detectable in RNA preparations of HEp-2-MxA cells harvested 24 h postinfection.
As was the case for all MxA-transfected cell lines tested so far, HEp-2-MxA cells were resistant to infection with VSV and influenza virus. However, preliminary experiments with MV and human parainfluenza virus serotypes 1, 2, and 3 showed no MxA-dependent effect on syncytium formation in transfected HEp-2 cells (data not shown). The fact that the activity of MxA against SFV depended on expression of the protein in human cell lines points to a necessary auxiliary factor which is missing from murine cells. The existence of cellular factors that modulate the activities of Mx proteins has been postulated before (45, 47), and the fact that MV is inhibited only in certain human cell lines, including U937 cells, strongly supports this notion (32, 33). Alternatively, it might well be that the postulated auxiliary factor is present in mouse cells but exhibits a low affinity for the heterologous MxA protein. In this context it will be interesting to see whether Mx2 (46), the murine homolog of MxA, exerts antiviral activity against SFV.
To define the step of the SFV replication cycle which is sensitive to MxA action, we examined the synthesis of viral macromolecules. Viral protein synthesis, subgenomic mRNA transcription, and genome amplification were strongly reduced in MxA-expressing cells, suggesting that MxA interferes with an early step of SFV multiplication. Since the viral particles and the SFV-based RNA replicon enter the cell by a completely different mechanism, it is very unlikely that MxA interferes with normal uptake or uncoating of SFV in the host cell cytoplasm. The observed inhibition of β-Gal expression from the SFV RNA replicon, which lacks the structural genes, by MxA rules out viral structural proteins as targets of MxA action. The only viral proteins expressed by the SFV RNA replicon are the four nonstructural proteins nsP1 to nsP4, which form the SFV transcriptase complex. It is therefore conceivable that the inhibitory function of MxA is associated with either the synthesis or the function of the viral replicase. Possible targets of the MxA activity include synthesis or processing of the nonstructural proteins, replication of viral genomic RNA, and synthesis or capping of viral mRNA. Also, we cannot rule out the possibility that MxA interferes with cellular proteins required for SFV replication.
So far, the only common denominator for the activity of MxA against various negative-strand RNA viruses and SFV is the early inhibition of their replication cycles. For VSV, various members of the bunyavirus family, and MV in U87 glioblastoma cells, the inhibition appears to be at the level of viral RNA synthesis (5, 16, 32, 34, 40). In the case of influenza A virus, which replicates in the nucleus, MxA interferes with a poorly defined stage of replication taking place in the cytoplasm (28). However, MxA has the capacity to inhibit influenza A virus at the level of RNA synthesis as demonstrated by the translocation of MxA to the nucleus by means of a foreign nuclear target signal (45). A further exception appears to be MV in the monocytic cell line U937, where MxA seems to interfere with the synthesis of viral glycoproteins (33). Taken together, these findings suggest that MxA appears able to interfere with different stages of replication. Nevertheless, this conclusion does not preclude the possibility that MxA recognizes a target common to all Mx-sensitive viruses and acts by a single mechanism.
Our results clearly demonstrate that the antiviral activity of MxA is not restricted to negative-strand RNA viruses but includes at least one member of the positive-strand RNA viruses. The availability of SFV in vitro transcription systems and SFV-based RNA replicons offers a unique opportunity to examine the molecular mechanism of the antiviral function of MxA in cell culture and in vitro. It will be interesting to see whether the antiviral spectrum of MxA extends to further positive-strand RNA viruses of the togavirus or the flavivirus family.
ACKNOWLEDGMENTS
We thank E. Mantei and D. Bucher for excellent technical assistance.
This work was supported by grants from the Swiss National Science Foundation and by the Kanton of Zürich.
REFERENCES
- 1.Aebi M, Fäh J, Hurt N, Samuel C E, Thomis D, Bazzigher L, Pavlovic J, Haller O, Staeheli P. cDNA structures and regulation of two interferon-induced human Mx proteins. Mol Cell Biol. 1989;9:5062–5072. doi: 10.1128/mcb.9.11.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng Y S, Patterson C E, Staeheli P. Interferon-induced guanylate-binding proteins lack an N(T)KXD consensus motif and bind GMP in addition to GDP and GTP. Mol Cell Biol. 1991;11:4717–4725. doi: 10.1128/mcb.11.9.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 4.Dreiding P, Staeheli P, Haller O. Interferon-induced protein Mx accumulates in nuclei of mouse cells expressing resistance to influenza viruses. Virology. 1985;140:192–196. doi: 10.1016/0042-6822(85)90460-x. [DOI] [PubMed] [Google Scholar]
- 5.Frese M, Kochs G, Feldmann H, Hertkorn C, Haller O. Inhibition of bunyaviruses, phleboviruses, and hantaviruses by human MxA protein. J Virol. 1996;70:915–923. doi: 10.1128/jvi.70.2.915-923.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frese M, Kochs G, Meier-Dieter U, Siebler J, Haller O. Human MxA protein inhibits tick-borne Thogoto virus but not Dhori virus. J Virol. 1995;69:3904–3909. doi: 10.1128/jvi.69.6.3904-3909.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gresser I. Role of interferon in resistance to viral infection in vivo. In: Vilcek J, De Maeyer E, editors. Interferon 2. New York, N.Y: Elsevier Science Publishers; 1984. pp. 221–247. [Google Scholar]
- 8.Haller O, Frese M, Rost D, Nuttall P A, Kochs G. Tick-borne Thogoto virus infection in mice is inhibited by the orthomyxovirus resistance gene product Mx1. J Virol. 1995;69:2596–2601. doi: 10.1128/jvi.69.4.2596-2601.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horisberger M A. Interferon-induced human protein MxA is a GTPase which binds transiently to cellular proteins. J Virol. 1992;66:4705–4709. doi: 10.1128/jvi.66.8.4705-4709.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horisberger M A, Hochkeppel H K. IFN-alpha induced human 78 kD protein: purification and homologies with the mouse Mx protein, production of monoclonal antibodies, and potentiation effect of IFN-gamma. J Interferon Res. 1987;7:331–343. doi: 10.1089/jir.1987.7.331. [DOI] [PubMed] [Google Scholar]
- 11.Horisberger M A, Staeheli P, Haller O. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc Natl Acad Sci USA. 1983;80:1910–1914. doi: 10.1073/pnas.80.7.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang T, Pavlovic J, Staeheli P, Krystal M. Overexpression of the influenza virus polymerase can titrate out inhibition by the murine Mx1 protein. J Virol. 1992;66:4154–4160. doi: 10.1128/jvi.66.7.4154-4160.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Israel A. Preliminary characterization of the particles from productive and abortive infections of L cells by fowl plague virus. Ann Microbiol. 1979;130B:85–100. [PubMed] [Google Scholar]
- 14.Johannes L, Arnheiter H, Meier E. Switch in antiviral specificity of a GTPase upon translocation from the cytoplasm to the nucleus. J Virol. 1993;67:1653–1657. doi: 10.1128/jvi.67.3.1653-1657.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaariainen L, Takkinen K, Keranen S, Soderlund H. Replication of the genome of alphaviruses. J Cell Sci Suppl. 1987;7:231–250. [PubMed] [Google Scholar]
- 16.Kanerva M, Melen K, Vaheri A, Julkunen I. Inhibition of Puumala and Tula hantaviruses in Vero cells by MxA protein. Virology. 1996;224:55–62. doi: 10.1006/viro.1996.0506. [DOI] [PubMed] [Google Scholar]
- 17.Koblet H. The “merry-go-round”: alphaviruses between vertebrate and invertebrate cells. Adv Virus Res. 1990;38:343–402. doi: 10.1016/s0065-3527(08)60866-0. [DOI] [PubMed] [Google Scholar]
- 18.Krug R M, Shaw M, Broni B, Shapiro G, Haller O. Inhibition of influenza viral mRNA synthesis in cells expressing the interferon-induced Mx gene product. J Virol. 1985;56:201–206. doi: 10.1128/jvi.56.1.201-206.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landis, H., H. P. Hefti, and J. Pavlovic. Mx1 and MxA inhibit influenza virus RNA synthesis in vitro independent of the endonuclease activity of PB2. Submitted for publication.
- 20.Lewis J A. Biological assays for interferons. In: Clemens M J, Gearing A, editors. Lymphokines and interferons. Oxford, England: IRL Press; 1987. pp. 73–88. [Google Scholar]
- 21.Liljestrom P, Garoff H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Bio/Technology. 1991;9:1356–1361. doi: 10.1038/nbt1291-1356. [DOI] [PubMed] [Google Scholar]
- 22.Marsh M, Helenius A. Virus entry into animal cells. Adv Virus Res. 1989;36:107–151. doi: 10.1016/S0065-3527(08)60583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meier E, Kunz G, Haller O, Arnheiter H. Activity of rat Mx proteins against a rhabdovirus. J Virol. 1990;64:6263–6269. doi: 10.1128/jvi.64.12.6263-6269.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munoz A, Carrasco L. Action of human lymphoblastoid interferon on HeLa cells infected with RNA-containing animal viruses. J Gen Virol. 1984;65:377–390. doi: 10.1099/0022-1317-65-2-377. [DOI] [PubMed] [Google Scholar]
- 25.Nakayama M, Nagata K, Kato A, Ishihama A. Interferon-inducible mouse Mx1 protein that confers resistance to influenza virus is a GTPase. J Biol Chem. 1991;266:21404–21408. [PubMed] [Google Scholar]
- 26.Pavlovic J, Haller O, Staeheli P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol. 1992;66:2564–2569. doi: 10.1128/jvi.66.4.2564-2569.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pavlovic J, Zürcher T, Haller O, Staeheli P. Resistance to influenza virus and vesicular stomatitis virus conferred by expression of human MxA protein. J Virol. 1990;64:3370–3375. doi: 10.1128/jvi.64.7.3370-3375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27a.Pavlovic, J., and M. Michel. Unpublished data.
- 28.Pitossi F, Blank A, Schröder A, Schwarz A, Hüssi P, Schwemmle M, Pavlovic J, Staeheli P. A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J Virol. 1993;67:6726–6732. doi: 10.1128/jvi.67.11.6726-6732.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richter M F, Schwemmle M, Herrmann C, Wittinghofer A, Staeheli P. Interferon-induced MxA protein. GTP binding and GTP hydrolysis properties. J Biol Chem. 1995;270:13512–13517. [PubMed] [Google Scholar]
- 30.Ronni T, Sareneva T, Pirhonen J, Julkunen I. Activation of IFN-alpha, IFN-gamma, MxA, and IFN regulatory factor 1 genes in influenza A virus-infected human peripheral blood mononuclear cells. J Immunol. 1995;154:2764–2774. [PubMed] [Google Scholar]
- 31.Samuel C E. Antiviral actions of interferon. Interferon-regulated cellular proteins and their surprisingly selective antiviral activities. Virology. 1991;183:1–11. doi: 10.1016/0042-6822(91)90112-o. [DOI] [PubMed] [Google Scholar]
- 32.Schneider-Schaulies S, Schneider-Schaulies J, Schuster A, Bayer M, Pavlovic J, Ter Meulen V. Cell type-specific MxA-mediated inhibition of measles virus transcription in human brain cells. J Virol. 1994;68:6910–6917. doi: 10.1128/jvi.68.11.6910-6917.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnorr J J, Schneider-Schaulies S, Simon-Jödicke A, Pavlovic J, Horisberger M A, Ter Meulen V. MxA-dependent inhibition of measles virus glycoprotein synthesis in a stably transfected human monocytic cell line. J Virol. 1993;67:4760–4768. doi: 10.1128/jvi.67.8.4760-4768.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwemmle M, Weining K C, Richter M F, Schumacher B, Staeheli P. Vesicular stomatitis virus transcription inhibited by purified MxA protein. Virology. 1995;206:545–554. doi: 10.1016/s0042-6822(95)80071-9. [DOI] [PubMed] [Google Scholar]
- 35.Singh I, Helenius A. Role of ribosomes in Semliki Forest virus nucleocapsid uncoating. J Virol. 1992;66:7049–7058. doi: 10.1128/jvi.66.12.7049-7058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Southern P J, Berg P. Transformation of mammalian cells to antibiotic resistance with a bacterial gene under control of the SV40 early region promoter. J Mol Appl Genet. 1982;1:327–341. [PubMed] [Google Scholar]
- 37.Staeheli P. Interferon-induced proteins and the antiviral state. Adv Virus Res. 1990;38:147–200. doi: 10.1016/s0065-3527(08)60862-3. [DOI] [PubMed] [Google Scholar]
- 38.Staeheli P, Danielson P, Haller O, Sutcliffe J G. Transcriptional activation of the mouse Mx gene by type I interferon. Mol Cell Biol. 1986;6:4770–4774. doi: 10.1128/mcb.6.12.4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell. 1986;44:147–158. doi: 10.1016/0092-8674(86)90493-9. [DOI] [PubMed] [Google Scholar]
- 40.Staeheli P, Pavlovic J. Inhibition of vesicular stomatitis virus mRNA synthesis by human MxA protein. J Virol. 1991;65:4498–4501. doi: 10.1128/jvi.65.8.4498-4501.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stranden A M, Staeheli P, Pavlovic J. Function of the mouse Mx1 protein is inhibited by overexpression of the PB2 protein of influenza virus. Virology. 1993;197:642–651. doi: 10.1006/viro.1993.1639. [DOI] [PubMed] [Google Scholar]
- 42.Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int J Cancer. 1976;17:565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 43.Thimme R, Frese M, Kochs G, Haller O. Mx1 but not MxA confers resistance against tick-borne Dhori virus in mice. Virology. 1995;211:296–301. doi: 10.1006/viro.1995.1404. [DOI] [PubMed] [Google Scholar]
- 44.Zhao H, De B P, Das T, Banerjee A K. Inhibition of human parainfluenza virus-3 replication by interferon and human MxA. Virology. 1996;220:330–338. doi: 10.1006/viro.1996.0321. [DOI] [PubMed] [Google Scholar]
- 45.Zürcher T, Pavlovic J, Staeheli P. Mechanism of human MxA protein action: variants with changed antiviral properties. EMBO J. 1992;11:1657–1661. doi: 10.1002/j.1460-2075.1992.tb05212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zürcher T, Pavlovic J, Staeheli P. Mouse Mx2 protein inhibits vesicular stomatitis virus but not influenza virus. Virology. 1992;187:796–800. doi: 10.1016/0042-6822(92)90481-4. [DOI] [PubMed] [Google Scholar]
- 47.Zürcher T, Pavlovic J, Staeheli P. Nuclear localization of mouse Mx1 protein is necessary for inhibition of influenza virus. J Virol. 1992;66:5059–5066. doi: 10.1128/jvi.66.8.5059-5066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]