

Abstract
We have studied 18 participants in phase I/II clinical trials of recombinant gp120 (rgp120) subunit vaccines (MN and SF-2) who became infected with human immunodeficiency virus type 1 (HIV-1) during the course of the trials. Of the 18 individuals, 2 had received a placebo vaccine, 9 had been immunized with MN rgp120, and seven had been immunized with SF-2 rgp120. Thirteen of the 18 infected vaccinees had received three or four immunizations prior to becoming infected. Of these, two were placebo recipients, six had received MN rgp120, and five had received SF-2 rgp120. Only 1 of the 11 rgp120 recipients who had multiple immunizations failed to develop a strong immunoglobulin G antibody response to the immunogen. However, the antibody response to rgp120 was transient, typically having a half-life of 40 to 60 days. No significant neutralizing activity against the infecting strain was detected in any of the infected individuals at any time prior to infection. Antibody titers in subjects infected despite vaccination and in noninfected subjects were not significantly different. Envelope-specific cytotoxic T-lymphocyte responses measured after infection were infrequent and weak in the nine vaccinees who were tested. HIV-1 was isolated successfully from all 18 individuals. Sixteen of these strains had a non-syncytium-inducing (NSI) phenotype, while two had a syncytium-inducing (SI) phenotype. NSI strains used the CCR5 coreceptor to enter CD4+ cells, while an SI strain from one of the vaccinees also used CXCR4. Viruses isolated from the blood of rgp120 vaccinees were indistinguishable from viruses isolated from control individuals in terms of their inherent sensitivity to neutralization by specific monoclonal antibodies and their replication rates in vitro. Furthermore, genetic sequencing of the env genes of strains infecting the vaccinees did not reveal any features that clearly distinguished these viruses from contemporary clade B viruses circulating in the United States. Thus, despite rigorous genetic analyses, using various breakdowns of the data sets, we could find no evidence that rgp120 vaccination exerted selection pressure on the infecting HIV-1 strains. The viral burdens in the infected rgp120 vaccine recipients were also determined, and they were found to be not significantly different from those in cohorts of placebo-vaccinated and nonvaccinated individuals. In summary, we conclude that vaccination with rgp120 has had, to date, no obvious beneficial or adverse effects on the individuals we have studied.
The need for a vaccine that is effective against human immunodeficiency virus type 1 (HIV-1) remains urgent, since the virus continues to spread worldwide (60, 155). Many candidate HIV-1 vaccines have been developed during the past decade, and several have entered clinical trials in the United States and elsewhere (14, 58). To date no efficacy trial has been conducted, and the suitability of current candidates for such trials in one or more parts of the world is a matter of vigorous debate (22, 94, 95, 106, 155). The type of vaccine that has progressed the furthest toward widespread use in humans is based on recombinant forms of the HIV-1 envelope glycoproteins (rgp120 and rgp160) (14, 58).
The first rgp120 and rgp160 immunogens were based on the sequences of HIV-1 LAI and HIV-1 SF-2, two of the earliest-isolated viruses (8, 11, 75, 133). MN rgp120 superseded LAI rgp120 in product development because the latter strain was noted to have an atypical V3 loop sequence, and at that time it was believed that V3-directed antibodies were of paramount importance for protection against HIV-1 (90). MN and SF-2 rgp120 proteins of high quality and purity, expressed in mammalian cell lines, have now been tested extensively in animals and humans; they are immunogenic and are generally regarded as safe (6, 37, 49, 56, 68, 133). Immunization with LAI Env-based immunogens has protected slightly over half of test chimpanzees from intravenous challenge with HIV-1 LAI, generally under optimal conditions (7, 8, 15, 49, 53, 143). Furthermore, both MN and SF-2 rgp120 or rgp160 have protected some chimpanzees from intravenous challenge with HIV-1 SF-2 (10, 37, 54). The significance of the chimpanzee experiments for protection of humans against HIV-1 is uncertain, principally because HIV-1 replicates only to low (LAI) or very low (SF-2) levels in chimpanzees and these strains do not cause disease in these animals. Moreover, HIV-1 LAI and HIV-1 MN are neutralization-sensitive, T-cell line-adapted (TCLA) viruses, and HIV-1 SF-2, even when grown in primary cells, is an unusual strain which is extremely sensitive to neutralization by certain reagents in vitro (101, 115, 146). These strains contrast with most primary isolates of HIV-1, which are generally relatively resistant to neutralization (98, 110, 148, 156). Furthermore, TCLA strains like LAI, MN, and SF-2 all use CXCR4 as an entry cofactor (34), whereas most strains of HIV-1 that are transmitted sexually use the CCR5 coreceptor (29, 34, 65). In contrast to the chimpanzee data, rgp120 immunogens derived from SIVmac were unable to protect rhesus macaques from intravenous challenge with the autologous, virulent virus (28). Also, immunization of cats or ponies with rgp120 immunogens derived from the feline immunodeficiency virus or the equine infectious anemia virus, respectively, failed to induce protective immunity but instead caused enhancement of disease when the animals were later challenged with virulent virus (66, 135, 151). The ambiguous and contradictory experiences with subunit vaccines in animal models therefore mandate that human trials of these immunogens be carefully analyzed, particularly with respect to individuals who become infected despite vaccination.
Two rgp120 proteins produced by Genentech, Inc., and Chiron/Biocine, Inc., based on the sequences of the clade B strains MN and SF-2, respectively, are currently under evaluation in phase I-II clinical trials conducted in the United States by the AIDS Vaccine Evaluation Group (AVEG), a contractor for the National Institutes of Health. These vaccines induce antibodies capable of neutralizing the TCLA strains of virus from which the vaccine is derived but not heterologous primary viruses (6, 9, 10, 37, 56–59, 69, 96, 98, 99, 156). The ability of these immunogens to induce cellular immunity, particularly cytotoxic T lymphocytes (CTL), is limited (5, 32, 58, 59). Partly due to these factors, efficacy trials of these products have not been carried out. Eighteen of 596 trial participants in the phase I/II trials of these proteins have become HIV-1 infected, indicating that the rgp120 vaccines gave less than complete protection from infection in the trial cohort as a whole. Here we describe analyses of the immunological responses induced in the HIV-1-infected rgp120 vaccine recipients before and after infection, we provide information on the amount of plasma HIV-1 virion-associated RNA in each infected trial participant, and we report on the env gene sequences, phenotypes, and in vitro growth characteristics of the infecting HIV-1 strains. Further information on these individuals and the clinical aspects of this study are provided elsewhere (57, 99, 127). Independent studies of two other individuals who became infected after immunization with SF-2 or MN rgp120 have been described previously (69, 100). The purpose of these studies was not to determine formally the efficacy of the rgp120 vaccines but to acquire information that could guide the design of future generations of HIV-1 vaccines.
MATERIALS AND METHODS
The Correlates of HIV-1 Immune Protection (CHIP) network.
The laboratories in the CHIP consortium include those responsible for the following: confirming the infection status and monitoring the plasma viral burden (R. Connor and D. Ho), ascertaining the phenotype of the isolated viruses (E. Fenamore and R. Connor), determining the DNA sequence of the virus directly from blood (K. Kunstman and S. Wolinsky) and after isolation in vitro (F. Gao and B. Hahn), defining the humoral (A. Trkola and J. Moore) and mucosal (J. Mestecky and S. Jackson) antibody responses, determining the HIV-1-specific cell-mediated immune response (B. Walker and S. Kalams), analyzing the DNA sequences and maintaining the central database (D. McDonald, A. Neumann, and B. Korber), and evaluating epidemiological relationships (S. Vermund).
Study and control cohorts.
Clinical trials of MN (Genentech Inc., South San Francisco, Calif.) and SF-2 (Chiron/Biocine Inc., Emeryville, Calif.) rgp120s are being conducted by the AVEG. By definition, individuals receiving either rgp120 immunogen or a placebo are considered vaccinees. Clinical samples were collected as described previously (56, 57, 99). During the trials, participants were monitored for HIV-1 infection by Western blot assay at 1- to 6-month intervals. Contemporary and archival serum and/or plasma samples from any individual suspected to be HIV-1 infected were sent in a blinded fashion to the central repository of the CHIP consortium at the Aaron Diamond AIDS Research Center for a variety of tests, including determination of virion-associated HIV-1 RNA levels in plasma. After the quantitative RNA data were reported to the central database at Los Alamos National Laboratory, the samples were made available for other studies in several of the consortium laboratories. Information on the specific time and use of antiretroviral therapy for the four infected vaccinees with known intercurrent treatment was available.
Control cohorts consisting of nonvaccinated individuals with symptomatic and asymptomatic primary infections were established to screen for the significance of potential observed differences in viral genotype. Control cohorts were also used to compare plasma viremia in vaccinated and nonvaccinated individuals. Case controls were selected and matched by age (plus or minus 5 years), gender, presumed risk of exposure (male homosexual, heterosexual, or intravenous drug user), and the year of primary infection. Controls were not matched for geographic location; however, geographic location within the United States generally does not correspond to clustering patterns in viral phylogenetic analyses (76). One phylogenetic cluster of viral sequences from two infected vaccine cases and two controls was observed in this study. For these four cases, we requested information concerning the city where the study subject was residing (see Results).
Control samples were obtained from nonvaccinated individuals among (i) men enrolled in the Multicenter AIDS Cohort Study (MACS), a natural history study of HIV-1 infection of homosexual men from Baltimore, Pittsburgh, Chicago, and Los Angeles; (ii) men and women enrolled in the Baltimore-based ALIVE Study, a natural history study of HIV-1 infection by intravenous drug use; (iii) men and women enrolled in the San Francisco component of the HIV-1 Vaccine Efficacy Trials Network (HIVNET), a vaccine preparedness cohort; and (iv) men and women enrolled in the Chicago-based National Institute of Drug Abuse-sponsored study to assess risk factors for infection due to intravenous drug use.
Clinical material.
Blood obtained from the infected vaccinees at each AVEG study site was collected in tubes containing acid citrate-dextrose anticoagulant and directly shipped to the Aaron Diamond AIDS Research Center. Plasma and peripheral blood mononuclear cells (PBMC) were separated by Ficoll-Hypaque discontinuous density gradient centrifugation and used for HIV-1 isolation and for quantifying the plasma viral burden. Blood samples from the control subjects were processed by the laboratory associated with each cohort study, and the stored samples from each of the respective repositories were accessed. Parotid saliva was collected by placing Schaffer cups over Stenson’s duct, as described previously (87, 134). Pre-ejaculate and ejaculate (semen), vaginal washings, and cervical, rectal, and nasal secretions were collected as described in detail in a manual for collection of human external secretions (120).
Anti-rgp120 antibody titers.
The antigen capture enzyme-linked immunosorbent assay (ELISA) used has been described previously (105, 112). Briefly, ELISA plates (Immulon II; Dynatech Inc., Chantilly, Va.) wells were coated with 100 μl of a 5-μg/ml solution of sheep antibody D7324 (antibody D6205, lot D017-G; International Enzymes Inc., Fullbrook, Calif.). After the plate wells were washed, MN or SF-2 rgp120 was added at 20 or 800 ng/ml, respectively, in Tris-buffered saline (TBS) containing 10% fetal calf serum (FCS). Control wells lacked gp120. The solution concentrations of MN and SF-2 rgp120 yield an equal amount of D7324-bound gp120, as determined by the binding of CD4-immunoglobulin G (CD4-IgG) (Genentech, Inc.) (18).
Vaccinee sera were titrated in 3.3-fold serial dilutions over the range 1:300 to 1:100,000 in TBS containing 2% nonfat milk powder and 20% sheep serum. Human IgG bound to gp120 was detected with alkaline phosphatase-conjugated goat anti-human IgG (Accurate Chemicals, Inc.) and the AMPAK ELISA Amplification System (Dako Diagnostics, Inc.) (105, 112). In some experiments, gp120 was denatured by boiling the sample in the presence of 1% sodium dodecyl sulfate and 50 mM dithiothreitol followed by alkylation of sulfhydryl groups with 100 mM iodoacetamide at 4°C and then diluting the sample 200-fold in TBS containing 10% FCS (109). Each experiment was conducted with a longitudinal set of sera from one individual. Reference control titrations of CD4-IgG (0.003 to 0.1 μg/ml) and of HIV-1-positive serum from a long-term survivor (17) (1:1,000 to 1:300,000 dilutions) were also included. Absorbance (optical density at 492 nm [OD492]) values derived from wells lacking gp120 were subtracted from the OD492 values derived from the gp120-containing wells at the same serum dilution to correct for assay background; this was usually significant only at serum dilutions of 1:300 and 1:1,000. Midpoint titers were calculated from the titration curves by using a computer program (a version of this program created by Aaron Halpern is available through anonymous file transfer protocol at ftp://atlas.lanl.gov/progs/AbTiter).
ELISA for measurement of antigen-specific antibodies in mucosal samples.
To determine the level of antigen-specific antibodies in mucosal samples, ELISA plates (Dynatech) were coated with MN rgp120 (Genentech, Inc.) at a concentration of 1 μg/ml overnight at 4°C. The plates were washed and blocked with 5% FCS (Mediatech, Inc., Herndon, Va.) in phosphate-buffered saline (PBS). Dilutions of external secretions were added to the plates and incubated overnight at 4°C. After being washed with PBS, the plates were developed with biotinylated F(ab)2 fragments of goat anti-human IgG or IgA antibodies. After another washing with PBS, they were subjected to consecutive incubations with avidin-peroxidase (Sigma, St. Louis, Mo.) and 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS; Sigma). The levels of IgG and IgA anti-gp120 antibodies were measured against standard curves obtained by using solutions with known amounts of polyclonal secretory (colostral) IgA or serum IgG (Moni-Trol ES Chemistry Control; Baxter, Stone Mountain, Ga.). Serum from an individual with high levels of IgG and IgA anti-HIV-1 antibodies was used as a positive control. Negative controls consisted of external secretions and sera of noninfected individuals. The cutoff (nonspecific background binding) was set at 100 ng of IgA or IgG per ml. The levels of total IgG and IgA isotypes were determined by a capture ELISA (86, 104, 120).
Virus isolation and culture.
PBMC were isolated from each vaccine recipient by standard methods as described above. The cells were serially diluted fivefold (1 × 106 to approximately 3 × 102) and cocultivated with 2 × 106 phytohemagglutinin (PHA)-stimulated normal donor PBMC as previously described (17). Culture supernatants were monitored for HIV-1 p24 antigen production on days 7, 14, and 21 by a commercial enzyme immunoassay (Abbott Laboratories, North Chicago, Ill.). A culture was considered positive if the p24 value was above a cutoff value of 30 pg/ml. The virus present in the supernatant of the first positive well was propagated by a single passage (14 to 21 days) in PHA-activated PBMC and titered on PBMC to determine the 50% tissue culture infective dose (TCID50). All virus stocks were aliquoted and stored at −80°C until further use. Viruses were successfully isolated from all infected vaccinees by this method. In one case (C16), it was necessary to deplete CD8+ T cells from the patient’s PBMC prior to coculture. CD8+ T-cell depletion was performed with immunomagnetic beads, following the protocol of the manufacturer (Dynal, Inc.).
Phenotype determination.
To assess the syncytium-inducing (SI) properties of HIV-1 strains from the vaccine recipients, MT-2 cells (5 × 105) were inoculated with 100 TCID50 of each isolate. The cells were washed after 24 h and monitored visually by light microscopy for the appearance of cell fusion on days 4, 7, and 10 after inoculation. Samples of the culture supernatants were also assayed for the presence of HIV-1 p24 antigen. Isolates which scored positively for both syncytia and p24 were considered SI, while those that were negative for both were considered non-SI (NSI). The growth kinetics of a subset of isolates were determined by inoculation of 100 TCID50 into cultures of PHA-activated PBMC. The cells were washed after 24 h, and samples of the culture supernatants were collected and assayed for HIV-1 p24 antigen production over time.
HIV-1 coreceptor use.
To assess coreceptor use by a subset of isolates from the infected vaccinees, three NSI viruses (C07, C08, and C13) and one SI virus (C20) were tested for their ability to infect HOS.CD4 cells expressing either CCR1, CCR2b, CCR3, CCR4, CCR5, or CXCR4 (30). NSI isolates from three of the putative transmitters to the infected vaccinees (pC05, pC18, and pC21) were also evaluated, as well as three NSI viruses obtained from nonvaccinated individuals during the acute phase of infection (AD60, AD74, and AD75) (12, 107). The various HOS.CD4 lines (104 cells/well) were incubated with 103 TCID50 of each isolate in a final volume of 1.0 ml of Dulbecco modified Eagle medium containing 10% FCS, antibiotics, and 1.0 μg of puromycin per ml for 24 h at 37°C and then washed three times with fresh medium. Samples of the culture supernatants were assayed for HIV-1 p24 antigen on days 0, 4, 7, 10, and 14.
Virus neutralization.
HIV-1 neutralization was assessed with an assay based on mitogen-stimulated PBMC as target cells and p24 antigen detection as a measure of virus output (17, 144). Briefly, serum or plasma samples from the infected vaccinees was diluted 1:8 in RPMI 1640 medium with 10% FCS, incubated with 100 TCID50 of the autologous HIV-1 isolate, and added to PHA-stimulated normal donor PBMC. Following overnight incubation at 37°C, the cells were washed extensively to remove any residual serum or plasma antibodies that could interfere with the p24 antigen ELISA. Control cultures were established in duplicate by infecting cells with the autologous virus in the absence of serum or plasma. On day 7 after infection, samples of the culture supernatants were taken and assayed for HIV-1 p24 antigen. Percent neutralization was calculated by dividing the amount of p24 antigen production in the test well by the average production in the duplicate control wells. Multiple, sequential serum samples from each of the infected vaccine recipients were tested against an autologous isolate obtained at the earliest time point after HIV-1 infection had occurred.
MAbs.
Monoclonal antibodies (MAbs) 2G12 and 2F5 were donated by H. Katinger (Polymun Scientific Inc., Vienna, Austria) (22, 23, 108, 116, 132, 133, 145), MAb IgG1b12 was provided by D. Burton (Scripps Research Institute, San Diego, Calif.) (16), and the CD4-IgG2 molecule was a gift from P. Maddon (Progenics Pharmaceuticals Inc., Tarrytown, N.Y.) (1). MAb 447/52-D was purchased from Cellular Products Inc. (Buffalo, N.Y.) (55). Murine MAb B13 was a gift from G. Lewis (Institute of Human Virology, Baltimore, Md.) (111).
Cell lines.
Epstein-Barr virus-transformed B-lymphoblastoid cell lines (B-LCL) were established from PBMC obtained from each of the 18 individuals, as described previously (149). The transformed B-LCL were maintained in RPMI 1640 medium containing 20% FCS supplemented with 2 mM l-glutamine, 50 U of penicillin per ml, and 10 mM HEPES.
Limiting-dilution assays of CTL precursors.
Limiting-dilution precursor frequency analysis was used to determine the relative strength of the postinfection CTL response against HIV-1 proteins, as described previously (83, 84). PBMC were cultured at 250 to 16,000 cells per well in 24 replicate wells of a 96-well microtiter plate. Gamma-irradiated (30 Gy) PBMC (2.5 × 104) from an HIV-1-seronegative donor were added to each well with 0.1 mg of the anti-CD3 MAb 12F6 per ml. Ten to 14 days later, the cells were split and assayed for cytotoxicity on 51Cr-labeled autologous B-LCL infected with recombinant vaccinia viruses expressing HIV-1 Gag (vABT141) (Therion Biologics, Cambridge, Mass.), reverse transcriptase (vCF21), Nef, or Env proteins. The Env proteins were derived from the LAI (vPE11 and vABT299), MN (vMN462), RF (vRF222), or SF2 (Vpe11) strain (74, 84, 142, 149). The fraction of nonresponding wells was determined as the number of wells in which the 51Cr released did not exceed the mean plus three standard deviations of the spontaneously released 51Cr of the 24 control wells divided by the number of assayed wells. Activated-cell frequency was estimated by the maximum-likelihood method (19, 31).
HIV-1 RNA assay.
HIV-1 virion-associated RNA in the serum or plasma of infected vaccine recipients was measured by the Amplicor HIV-1 Monitor assay (Roche Molecular Systems). Briefly, total RNA was extracted from 200 μl of either serum or plasma collected in acid citrate-dextrose to which a standard, HIV-1-unrelated RNA preparation of known copy number was added. A 142-bp sequence of the HIV-1 gag gene was reverse transcribed and, in the same reaction, amplified by PCR for 30 cycles. The amplified products were serially diluted fivefold and hybridized to oligonucleotide probes coated on the wells of a microtiter plate. Following hybridization, the plates were washed extensively and the bound products were detected with an avidin-horseradish peroxidase conjugate. The HIV-1 RNA copy number was calculated on the basis of the ratio of the absorbance (OD450) reading from the bound HIV-1 products relative to that from the internal-standard RNA. The results are expressed as HIV-1 RNA copies per milliliter of serum or plasma. The results of HIV-1 RNA quantitation for samples from the nonvaccinated control subjects did not show a log-normal distribution, possibly due to the differences in sample collection procedures between the different cohorts. Therefore, nonparametric statistics were used to compare the RNA values of the infected vaccinee cases to those of the controls.
DNA sequencing.
env genes were amplified directly from uncultured PBMC DNA (gp120) and from cultured PBMC DNA (gp160) from each individual and then cloned and sequenced as described previously (153). The gp120 sequences from PBMC DNA taken directly from the study subject were determined at Northwestern University by methods described previously (153, 154), and the gp160 sequences from primary-culture DNA were determined at the University of Alabama as described elsewhere (51). The use of these two methods, in two different laboratories, ensured the integrity of both the PCR products and the viral isolates (81, 91).
Viral sequence analysis.
Comparisons were based on various breakdowns of the available PCR product sequence data sets. Initial sequence alignments were generated by using a hidden Markov model (HMMER, version 1.8; http://genome.wustl.edu/eddy/HMMER/main.html) (36, 117). Both DNA and protein alignments were hand edited by using the multiple alignment sequence editor (39). Simple sequence similarity comparisons were made by using the multiple alignment sequence editor after removing positions in the alignment at which gaps had been inserted to maintain the alignment. These were calculated as Hamming distances, or (1 − s) × 100, where s is the fraction of shared sites in two aligned nucleotide sequences (80).
Neighbor-joining phylogenetic reconstructions with bootstrap resampling (41, 56) were generated with the PHYLIP programs dnadist, dnaboot, and neighbor, with a Kimura two-parameter distance matrix which had a transition-to-transversion ratio of 1.3 (Phylogeny Inference Package, version 3.5c; http://evolution.genetics.washington.edu/phylip.html) (42, 43, 44, 154). Bootstrap resampling was done with 100 replicates (63). Qualitatively similar trees were generated with the program FastDNAml, version 1.0 (http://central.pasteur.fr:80/docs/doc-gensoft/fastDNAml), with three randomizations of input order but using only one sequence per person (40, 124).
To screen for potential contamination of product DNA (88), all PCR-amplified viral sequences included in the study were cross-compared through phylogenetic analysis, by screening within- and between-subject Hamming distances, and by comparing them with sequences in the viral subsection of GenBank by using BLAST (2). Evidence of potential problems is the near identity of viral sequences to those of laboratory strains or to those derived from epidemiologically unlinked individuals that are distinct from other viral sequences obtained from the putative source (81, 91).
To search for signature patterns that might be indicative of distinct characteristics among the viral sequences from the infected vaccinees, we first created sequence sets comprised of a single sequence per person from the infected vaccine recipients and from the control group. The MN and SF2 rgp120 vaccine recipients were considered separately for this analysis, with only those who had three or more vaccinations prior to infection being included. The control set of viral sequences was derived from nonvaccinated, asymptomatic individuals with a documented infection, matched for time of primary infection, route of transmission, and relative risk of infection. All of gp120 was scanned for potentially interesting amino acid signatures. The program VESPA was used to search for differences in the most common amino acid in a given position between the infected vaccine cases and controls (77, 79). A minimum frequency change of 0.5 in the most common amino acid was required for selection as a signature site. Power tests for sample sizes of 7 for the query data set and 31 for the background data set indicate that differences of 0.5 can yield a P value of <0.01 with Fisher’s exact test. The program ENTROPY was used to determine if there was greater variation in the sequences of the viruses from the infected vaccinees for any position in the alignment relative to the control sequences (80). Potential asparagine (N)-linked glycosylation sites were considered as both unlinked amino acids and as units, where an N of an N-X-T/S sequon was distinguished from other asparagine residues (93, 118, 119).
The program MotifScan (available through anonymous ftp at ftp://atlas.lanl.gov/progs/Motifscan) was developed to examine variations in short contiguous amino acid motifs relative to the vaccine sequence. Amino acid sequence stretches between 4 and 8 amino acids long were tested for identity or variation relative to the vaccine strain. A sliding window was used so that each possible motif in gp120 was examined. The numbers of identities and variants were tallied for each motif, and a one-sided Fisher’s exact test was run to rank the motifs according to distinctiveness, relative to the vaccine strain, of the vaccine recipients compared to controls. Motifs which had a P value of 0.05 or less were considered distinctive. Gaps in the sequence alignment were considered as characters; all other unusual characters (signifying frameshifts, stop codons, etc.) disqualify the sequence motif in which they are embedded from consideration. To assess the statistical significance of the most-distinctive motifs identified, a Monte Carlo randomization approach was used (80). The infected vaccine recipient and control viral sequence sets were combined into a single pooled set and then randomly partitioned into two data sets of the same respective sizes as the original vaccine recipient and control sequence sets. MotifScan was then run on each of 100 randomized sets to determine the background level of distinctive motifs, i.e., what is typically observed by chance alone.
Nucleotide sequence accession numbers.
Viral sequences generated for this study have been submitted to GenBank under accession no. U84792 through U84887.
RESULTS
Immunization and infection profiles.
The 401 study group comprised all individuals who became infected during phase I-II trials of subunit or composite vaccines conducted by the AVEG. Enrollment in the 401 study began in January 1994 and ended in June 1995. In this paper, we report exclusively on 18 individuals who participated in trials of rgp120 (MN or SF-2) and yet became infected with HIV-1 despite intensive counseling to avoid high-risk behaviors (57, 60, 127). Several other recipients of different immunogens have become HIV-1 infected, and they are described elsewhere (57, 69, 100).
A summary of the trial cohorts is provided in Table 1; note that most (16 of 20) of the 401 group was derived from the 201 trial involving individuals considered to be at relatively high risk for HIV-1 infection. In the 201 trial, 7 of 128 individuals receiving MN rgp120 became infected, as did 6 of 129 receiving SF-2 rgp120 and 2 of 39 placebo recipients (57, 99, 127). The remaining three trial participants were considered to be at relatively low risk for infection at the time of enrollment in the original trial. Of the 18 infected vaccinees, 9 received MN rgp120, 7 received SF-2 rgp120, and 2 received a placebo (Table 1). However, five individuals (two MN and three SF-2 rgp120 recipients) became infected before receiving their third and fourth immunizations (Fig. 1 and 2). Although we present information on these cases (C04, C05, C06, C08, and C09), it is questionable whether they represent true failures of the candidate vaccines to protect against HIV-1 infection. Regardless of whether the five individuals infected after only a partial vaccination course are included, the distribution of infected vaccinees among the MN rgp120, SF-2 rgp120, and placebo groups broadly reflects the composition of the trial cohorts (Table 1).
TABLE 1.
Distribution of infected vaccinees among immunogen groupsa
| Immunogen | Trial participants
|
Infected participants
|
Infected participants receiving three or four immunizations
|
|||
|---|---|---|---|---|---|---|
| n | % of total | n | % of total | n | % of total | |
| All trials (high and low risk) | ||||||
| MN gp120 | 276 | 46 | 9 | 3.3 | 7 | 2.5 |
| SF2 gp120 | 234 | 39 | 7 | 3.0 | 4 | 1.7 |
| Placebo | 86 | 14 | 2 | 2.3 | 2 | 2.3 |
| Total | 596 | 100 | 18 | 3.0 | 13 | 2.2 |
| 201 trial (high-risk volunteers) | ||||||
| MN gp120 | 128 | 43 | 7 | 5.5 | 6 | 4.6 |
| SF2 gp120 | 129 | 44 | 6 | 4.7 | 4 | 3.1 |
| Placebo | 39 | 13 | 2 | 5.1 | 2 | 5.1 |
| Total | 296 | 100 | 15 | 5.1 | 12 | 4.1 |
The compositions of the trial cohorts in which the infected vaccinees participated are listed. The 201 trial was in a high-risk group; 15 of the 18 HIV-1 infections occurred among the 201 participants. Also recorded is the number and proportion of infected vaccine recipients who received more than three immunizations with MN or SF2 rgp120 or a placebo.
FIG. 1.

Temporal relationship between entry into the vaccine trial, receipt of immunizations, and the period during which HIV-1 infection occurred. The date of entry (first immunization) into one of the vaccine trials is designated as day 0, and all time points represent days elapsed from that date. Arrows indicate days on which immunization occurred, and hatched bars correspond to the interval between the last negative and first positive HIV-1 RNA PCR result, based on measurement of viral RNA in plasma or serum. The cases are arranged in order of the time between commencement of immunization and the time of HIV-1 infection.
FIG. 2.
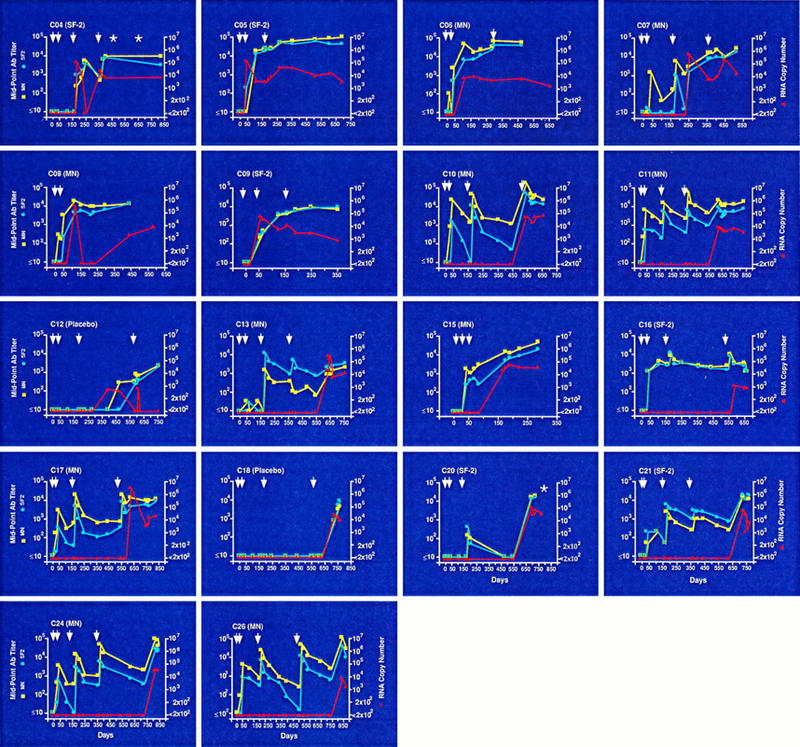
Longitudinal profiles of plasma viral burden, and antibody responses to MN and SF-2 rgp120, for the 18 infected vaccinees. Each panel represents one of the 18 infected vaccinees; the corresponding case number and respective immunogen are shown in the upper left-hand corner of each profile. The red triangles represent the level of plasma HIV-1 virion-associated RNA in copies per milliliter, the blue circles represent the midpoint antibody titers to SF-2 rgp120, and the yellow squares represent midpoint antibody titers to MN rgp120. Day 0 designates the date of entry into one of the vaccine trials, and all time points represent days elapsed from that point. The days on which immunizations were received are indicated by arrows. Four individuals (C04, C15, C20, and C26) received antiretroviral therapy following HIV-1 infection while still participating in a vaccine trial. The day(s) on which therapy was initiated is indicated by an asterisk. For two cases (C15 and C26), treatment was initiated after the last time point in which viral RNA levels and antibody responses were tested. C04 was treated with dideoxyinosine (beginning day 498) and AZT (beginning on day 658); C15 was treated with dideoxycytosine (beginning on day 490); C20 was treated with AZT (beginning on day 811); and C26 was treated with AZT, 3TC, and Retonavir (beginning on day 976).
The temporal relationship between entry into the trial, receipt of immunizations, and the period during which HIV-1 infection occurred is summarized for the 18 cases in Fig. 1. The date of entry into one of the vaccine trials (first immunization) is designated as day zero, and all time points represent days elapsed from that date. The cases are arranged in order of the time between the commencement of immunization and the time of HIV-1 infection (which occurred during the period represented by hatching in Fig. 1). No obvious clustering of HIV-1 infection was associated with the times of immunization, indicating that the pattern of infection was random with respect to time.
Longitudinal profiles of the 18 individual cases, including antibody responses to both MN and SF-2 rgp120, viral burden (plasma HIV-1 virion-associated RNA), the times at which immunizations were received, and the identity of the immunogen, are presented in Fig. 2. Of the individuals who were vaccinated at least three times prior to infection, C12 and C18 received a placebo; C13, C16, C20, and C21 received SF-2 rgp120; and C07, C10, C11, C15, C17, C24, and C26 were recipients of MN rgp120. The specific time and use of antiretroviral therapy are also indicated for the four treated participants (C04, C15, C20, and C26).
Antibody responses. (i) gp120 binding antibodies.
To gain an understanding of the immune response of each individual to MN or SF-2 rgp120 proteins, we measured serum anti-gp120 IgG midpoint titers to both proteins by using an antigen capture ELISA. Every serum sample was titrated against both rgp120s under conditions in which a control HIV-1-positive serum specimen and the CD4-IgG molecule bound equivalently to each rgp120 (data not shown). Thus, antibody titers to the two rgp120 proteins may be compared within and between each longitudinal profile (Fig. 2). Considering only those 11 individuals who received three or more immunizations with rgp120 before becoming infected, several general observations may be made.
The first immunization usually elicited a weak, often undetectable IgG antibody response. Subsequent booster immunizations caused rapid increases in anti-gp120 titers in most individuals; in only one case (C20, an SF-2 rgp120 recipient) was there an almost complete failure to develop an antibody response to the immunogen. In contrast, several individuals developed a very strong anti-rgp120 response to each booster immunization, exemplified by MN rgp120 recipients C11, C24, and C26, whose peak anti-MN rgp120 midpoint titers approached those found in long-term nonprogressors (Table 2) (12). However, the antibody response to rgp120 was transient. Measurements of the exponential decay of anti-gp120 antibodies in several of the best-responding individuals showed that the half-lives were typically about 40 to 60 days, although there was some variation among individuals and among the responses to repeat booster immunizations in the same individual (data not shown). Anti-rgp120 responses sustained between booster immunizations were noted in most individuals, but these were relatively low (titers of 1:1,000 to 1:5,000). Samples from vaccinated, uninfected individuals were not available for this analysis. However, an independent analysis confirmed that antibody titers in subjects infected despite vaccination were not significantly different from those in noninfected subjects from the same original study (57). Thus, it is highly improbable that those individuals who became infected with HIV-1 were clustered among the poorer responders to vaccination. Consistent with this, the peak anti-immunogen rgp120 titers in the infected vaccine recipients were distributed over a 200-fold range (Table 2).
TABLE 2.
Peak vaccine-induced midpoint anti-gp120 titersa
| Individual or group | gp120 immunogen | Titer
|
Autologous titer/heterologous titer | |
|---|---|---|---|---|
| Anti-MN gp120 | Anti-SF2 gp120 | |||
| C07 | MN | 6,060 | 1,150 | 5.3 |
| C10 | MN | 36,600 | 7,610 | 4.8 |
| C11 | MN | 54,100 | 7,570 | 7.1 |
| C15 | MN | 2,930 | 265 | 11.1 |
| C17 | MN | 21,200 | 8,290 | 2.6 |
| C24 | MN | 48,700 | 6,270 | 7.8 |
| C26 | MN | 49,750 | 13,340 | 3.7 |
| C13 | SF-2 | 1,630 | 12,920 | 7.9 |
| C16 | SF-2 | 11,500 | 9,110 | 0.8 |
| C20 | SF-2 | 105 | 320 | 3.0 |
| C21 | SF-2 | 1,280 | 3,270 | 2.6 |
| Mean ratio | 5.2 | |||
| Seven long-term survivors | 94,000b | 69,000b | 1.4 | |
| Nine rapid progressors | 89,000b | 62,000b | 1.4 | |
| PC04 | 100,000 | 76,000 | 1.3 | |
| PC05 | 325,000 | 240,000 | 1.4 | |
Peak midpoint preinfection anti-gp120 titers are recorded for the 12 MN and SF-2 rgp120 vaccinees who became infected after receiving at least three rgp120 immunizations. Also shown are the mean anti-gp120 titers measured in groups of long-term survivors and rapid progressors of HIV-1 infection (12, 17) and the anti-gp120 titers in two putative transmitters of HIV-1 infection to two of the vaccinees near the time of transmission.
Mean value.
Some individuals became infected fairly soon after receiving booster immunizations, when their antibody titers were close to maximum—for example, C17 (an MN rgp120 recipient) and C16 (an SF-2 rgp120 recipient) (Fig. 2). Other individuals became infected after their antibody responses had decayed significantly from the last booster peak—for example, C24 (an MN rgp120 recipient) and C21 (an SF-2 rgp120 recipient). It was notable that HIV-1 infection was associated with a very rapid increase in anti-gp120 antibody titers in several individuals (C10, C11, C13, C21, C24, and C26). We presume that this phenomenon represents an anamnestic response to the antigens expressed by the infecting virus strain. Other individuals developed an antibody response to the infecting strain more gradually (Fig. 2).
In all but one vaccinee (C16, an SF-2 rgp120 recipient), there was a significantly stronger (on average, 5.0-fold) response to the rgp120 used for immunization than to the heterologous rgp120 (i.e., anti-MN rgp120 titers were usually higher than anti-SF-2 rgp120 titers in MN rgp120 recipients, and vice versa) (Table 2). In contrast, the anti-MN and anti-SF-2 rgp120 titers in two unrelated cohorts of HIV-1-infected people were very similar, the mean ratio of the antibody titers to the two proteins being 1.4. The same ratio of anti-rgp120 titers (1.3 to 1.4) was found in sera from two individuals, pC04 and pC05, who were the putative transmitters of HIV-1 to individuals C04 and C05, respectively (Table 2). Furthermore, the anti-rgp120 titers in the vaccinees postinfection were less dependent on the test rgp120 than they were prior to infection (Fig. 2). One interpretation of these data is that although both MN and SF-2 rgp120s were cloned from clade B strains, a significant fraction of the antibody response to these proteins is to type-restricted epitopes, probably within the variable loops. In contrast, in HIV-1-infected people, the infection-induced anti-gp120 response is probably more broadly directed, leading to increased cross-reactivity with both rgp120 proteins. Alternatively, infection of the vaccinees by HIV-1 strains genetically equidistant from both MN and SF-2 (see below) might account for the broadening of antibody binding. However, it is clear that some cross-reactive anti-rgp120 binding antibodies were raised in response to both MN and SF-2 rgp120 immunizations (Table 2).
Sera from humans immunized with certain rgp120 (LAI) immunogens preferentially recognize denatured forms of the rgp120 molecule (109, 147, 148). Furthermore, some MAbs raised against rgp120 or rgp160 in rodents also show an abnormally strong reactivity with denatured rgp120 (111). In contrast, sera from HIV-1-infected humans contain antibodies more reactive with correctly folded (e.g., CD4 binding competent) rgp120 than with denatured rgp120 (105, 107, 109, 147). To determine the quality of the anti-rgp120 response in the MN and SF-2 rgp120 vaccine recipients, we measured the relative reactivities of selected high-titer preinfection sera with correctly folded and denatured forms of MN and SF-2 rgp120s. As controls, we showed that CD4-IgG was unable to bind to denatured gp120 whereas MAb B13 bound much more strongly to denatured gp120 (data not shown). The latter result was expected because B13 recognizes an epitope in the C2 domain that is poorly exposed on correctly folded gp120 (111). Control sera from nonvaccinated, HIV-1-infected individuals bound to correctly folded MN and SF-2 rgp120s with titers about 10-fold higher than those binding to the denatured forms of these proteins (data not shown), consistent with previous results (105, 109). Sera from the rgp120 vaccinees also showed preferential reactivity with correctly folded rgp120, although the correctly folded/denatured rgp120 titer ratio was a little lower than that seen with sera from HIV-1-infected individuals. It is clear, however, that both MN and SF-2 rgp120s do preferentially induce antibodies to correctly folded, rather than denatured, forms of monomeric gp120. Therefore, it is unlikely that the HIV-1 infections seen in this cohort arose because of an inherent inability of the immunogens to induce antibodies that are able to recognize the correctly folded gp120 monomer. However, it is possible that the antibodies raised to the monomeric gp120 immunogen recognize poorly (or perhaps not at all) relevant structures on the native, oligomeric forms of HIV-1 envelope glycoproteins, since they exist on infectious virions.
(ii) Neutralizing antibodies.
Titers of antibodies to monomeric gp120 are not predictive of primary-virus neutralization titers (48, 108, 110, 147, 148). We therefore tested whether serum taken from each vaccinee before HIV-1 infection could neutralize the HIV-1 strain isolated from the same individual soon after infection. The isolated viruses were cultured only in mitogen-stimulated PBMC (see below) to avoid selection of neutralization-sensitive strains. Neutralization of these primary viruses was assessed in a well-characterized assay that uses mitogen-stimulated PBMC as target cells and p24 antigen output as a measure of virus replication (17, 144). Vaccinee sera were tested at a 1:8 dilution, since higher concentrations may lead to nonspecific inhibition of cell growth or virus replication. At this dilution, no serum sample from any vaccinee at any time point prior to infection was able to reduce the infectivity of the autologous isolate by 90% (data not shown). A few sera from some individuals sporadically caused 50% neutralization, but the significance of a twofold reduction in HIV-1 infectivity is questionable.
Several explanations for the failure of the MN and SF-2 rgp120 vaccines to induce antibodies able to neutralize the infecting strains of HIV-1 seem feasible. One is that the antibody response to the immunogens may be directed mostly at nonneutralizing epitopes. Alternatively, the immunogens may induce primary-virus neutralizing antibodies at an inadequate titer. Another possibility is that the infecting strains of HIV-1 are unusually resistant to neutralization; primary viruses have a wide spectrum of sensitivity to neutralization by HIV-1-positive sera, MAbs, and soluble CD4-based reagents (25, 27, 93, 98, 146). Therefore, it is possible that the rgp120 vaccines allowed transmission of only relatively resistant viruses.
To address the latter hypothesis, we tested the sensitivity of HIV-1 strains isolated from infected vaccine recipients to neutralization by a panel of test reagents. The panel comprised three human MAbs (IgG1b12, 2G12, and 2F5) and a tetrameric form of the CD4-IgG molecule (CD4IgG2) shown in independent studies to have the broadest and most potent primary-virus neutralizing activity yet described (23, 35, 144). We also included a commercially available human MAb (447/52-D) against the third variable (V3) region of gp120 (55), although this neutralizes very few primary isolates (35, 108). Except for 447/52-D (which was in limited supply), this reagent panel was tested against eight HIV-1 strains isolated from individuals who received at least three immunizations with MN or SF-2 rgp120s (MN recipients C07, C10, C11, and C17; SF-2 recipients C13, C16, C20, and C21). As controls, we selected an isolate from a placebo recipient (C18), five strains isolated from nonvaccinated individuals with acute HIV-1 infection, and three strains from individuals who had had a partial vaccination course, receiving only one or two immunizations with MN or SF-2 rgp120s (C04, C06, and C09). In view of the limited immune response to the immunogen in these individuals before infection (Fig. 2), the viruses isolated from C04, C06, and C09 provide a control group for the viruses isolated from those participants who received a complete vaccination course. Also included in the experiment were viruses isolated from four of the putative transmitters of HIV-1 to four of the infected vaccinees (pC04, pC13, pC16, and pC18). Furthermore, historical data on the neutralization sensitivity of 12 other clade B primary strains isolated from non-acute-phase individuals were also available for comparison (144). The sequences of all the HIV-1 strains tested, including the historical controls, were randomly distributed in a phylogenetic tree analysis, indicating that there was no selection bias (data not shown).
Overall, compared to the control groups, there were few differences in the neutralization sensitivities of the group of viruses isolated from those infected vaccinees who received a complete course of vaccinations (Table 3). A similar conclusion was reached when the 90% infectious dose values for the groups of isolated viruses were compared (data not shown). One of the test MAbs (2G12) was noticeably less effective against the control group comprising the viruses isolated from the placebo subjects, the infected subjects who received a partial vaccination course, and the acutely infected subjects than it was against the other vaccinee isolates and the historic control isolates (Table 3). Whether this truly represents a relative insensitivity of viruses isolated from control subjects to neutralization by this MAb is uncertain. It is not likely that the rgp120 vaccines have selected for 2G12-sensitive viruses, although this possibility exists. The small number of isolates in this study limits the significance of any conclusions that can be drawn about putative vaccine-induced selection pressures on viral phenotype.
TABLE 3.
Neutralization of vaccinee and control isolates by MAbs and CD4IgG2
| Isolate | Descriptiona | 90% infectious dose neutralization titer (μg/ml) with:
|
||||
|---|---|---|---|---|---|---|
| CD4IgG2 | MAb:
|
|||||
| IgG1b12 | 2G12b | 2F5 | 447/52-D | |||
| AD6 | Acute | 12 | 6.6 | >50 | <2 | >50 |
| AD13 | Acute | >50 | >50 | >50 | 9.7 | >50 |
| AD60 | Acute | 39 | >50 | >50 | 9.1 | 48 |
| AD75 | Acute | 35 | 9.3 | >50 | 40 | NDc |
| AD74 | Acute | 49 | >50 | >50 | 40 | ND |
| C18 | Placebo | 48 | >50 | >50 | 46 | >50 |
| C06 | Early MN | 45 | 41 | 13 | 46 | >50 |
| C04 | Early SF-2d | 46 | >50 | 2.7 | 46 | >50 |
| C09 | Early SF-2d | 9.3 | 5.9 | <2 | 25 | >50 |
| C07 | Late MN | 36 | 30 | >50 | 31 | >50 |
| C10 | Late MN | >50 | >50 | 18 | 46 | >50 |
| C11 | Late MN | 7.3 | 4.2 | >50 | 8 | 31 |
| C17 | Late MN | <2 | 7.6 | <2 | 35 | 45 |
| C13 | Late SF-2 | 45 | >50 | <2 | 41 | >50 |
| C20 | Late SF-2 | 5.1 | >50 | >50 | 9.1 | ND |
| C21 | Late SF-2 | 41 | 9 | >50 | 26 | ND |
| pC04 | Partner for C04 | >50 | >50 | 10 | 47 | ND |
| pC13 | Partner for C13 | 44 | >50 | >50 | 8 | >50 |
Acute denotes isolates obtained from nonvaccinated individuals with presumed primary infection. The designations of early and late isolates are based on the number of immunizations each individual received prior to infection (see Fig. 1 and 2).
Apparently discrepant neutralization titers are highlighted.
ND, not determined.
Early-infection isolates; i.e., there was no antibody response to vaccine prior to virus isolation.
We conclude from this experiment that virus strains able to infect multiply immunized MN and SF-2 rgp120 recipients are not unusually resistant to neutralization per se. Indeed, the most neutralization-resistant strain of those we studied was from individual C18, a placebo recipient. We noted one difference in the neutralization sensitivity among pairs of viruses isolated from infected vaccinees and their putative partners: isolate C13 was sensitive to MAb 2G12, whereas isolate pC13 was almost completely resistant to this MAb (Table 3). However, viral sequence analysis later indicated that isolates pC13 and C13 were genetically unrelated and that pC13 was unlikely to have been the true donor of the virus to C13 (see below). Thus, the neutralization data for this pair of isolated viruses were not discrepant but were actually indicative of the presence of two distinct viral strains in these two patients, a fact later confirmed by phylogenetic analysis.
Mucosal antibodies.
The levels of antigen-specific and total antibodies in external secretions, including parotid saliva, pre-ejaculate, and seminal plasma, and in secretions collected by vaginal washing, uterine cervical wicking, nasal washing, and rectal wicking were determined. Results of ELISA analyses of secretions from nine infected vaccinees, two placebo recipients, and two putative transmitters are presented in Table 4. Antibodies of the IgG isotype specific for MN rgp120 were detected in the genital tract secretions of three infected vaccinees (C05, C09, and C10) and one putative transmitter (pC12). IgG antibodies were also detected in seminal fluid from three males and a vaginal wash and cervical wick from one female. One male vaccinee positive for IgG antibodies in seminal plasma (C10) had IgA anti-MN rgp120 antibodies in his parotid saliva as detected by ELISA. The isotype association of HIV-1-specific antibodies in these secretions reflected the predominant isotype present in the respective secretions. Thus, total immunoglobulins in both male and female genital tract secretions were predominantly of the IgG isotype. External secretions from the remaining infected vaccinees did not contain antigen-specific antibodies at levels above the cutoff (100 ng/ml). When sufficient volumes of external secretions were available, the samples were also analyzed by Western blotting to confirm the results obtained by ELISA and to determine the specificity of antibodies (Table 5). IgG antibodies to gp120 were detected in one sample of seminal plasma (C09), in one sample from a putative transmitter (pC12), and in cervicovaginal secretions from one female (C05). Parotid saliva from C10 contained IgG, but not IgA, antibodies to gp120, gp41, and p24. With the exception of C10 (for whom IgA antibodies were detected in parotid saliva by ELISA but not by Western blotting), concordant results were obtained. These data indicate that systemic rgp120 immunization of these volunteers was ineffective at inducing mucosal immune responses manifested by the presence of secretory IgA antibodies.
TABLE 4.
Antigen-specific antibodies and total immunoglobulins in mucosal secretions
| Volunteer | Immunogen | Visit no. | Specimen | Concn of antigen-specific immunoglobulin (ng/ml)
|
Total immunoglobulin concn (ng/ml)
|
||
|---|---|---|---|---|---|---|---|
| IgA | IgG | IgA | IgG | ||||
| C05 | SF-2 | 6 mo | Vaginal wash | 0 | 243 | 0 | 129,680 |
| 6 mo | Cervical wick | 0 | 436 | 0 | 688 | ||
| 8 | Cervical wick | 0 | 2,597 | NDa | ND | ||
| C09 | SF-2 | 2 | Parotid saliva | 33 | 0 | 79,644 | 1,486 |
| 2 | Semen | 0 | 109 | 37,498 | 64,780 | ||
| C10 | MN | 2 | Seminal plasma | 36 | 207 | 17,403 | 35,247 |
| 3 | Parotid saliva | 239 | 18 | 800,100 | 5,346 | ||
| 5 | Parotid saliva | 180 | 43 | 482,220 | 10,262 | ||
| 6 | Parotid saliva | 113 | 35 | 264,090 | 4,280 | ||
| 6 | Parotid saliva | 154 | 24 | 360,650 | 3,658 | ||
| C12 | Placebo | 2 | Parotid saliva | 27 | 0 | 117,760 | 856 |
| 4 | Parotid saliva | 23 | 0 | 114,700 | 838 | ||
| 5 | Parotid saliva | 19 | 0 | 85,011 | 1,442 | ||
| 6 | Parotid saliva | 18 | 0 | 60,247 | 2,779 | ||
| 8 | Parotid saliva | 19 | 0 | 215,407 | 856 | ||
| pC12 | 1 | Parotid saliva | 60 | 0 | 491,530 | 3,122 | |
| 1 | Semen | 0 | 223 | 41,632 | 58,012 | ||
| C16 | SF-2 | 2 | Parotid saliva | 22 | 0 | 133,450 | 1,983 |
| 2 | Pre-ejaculate | 0 | 0 | 1,244 | 735 | ||
| 2 | Rectal wick | 68 | 10 | 589,450 | 30,275 | ||
| 2 | Seminal plasma | 27 | 18 | 134,130 | 113,710 | ||
| 5 | Nasal wash | 28 | 10 | 86,715 | 9,021 | ||
| 5 | Parotid saliva | 22 | 0 | 238,120 | 1,871 | ||
| 6 | Parotid saliva | 15 | 0 | 197,370 | 6,202 | ||
| 6 | Pre-ejaculate | 0 | 0 | ND | ND | ||
| 6 | Rectal wick | 0 | 0 | ND | ND | ||
| 8 | Parotid saliva | 0 | 0 | ND | ND | ||
| pC16 | 1 | Parotid saliva | 10 | 11 | 54,465 | 979 | |
| 1 | Pre-ejaculate | 0 | 0 | 24 | 11 | ||
| 1 | Rectal wick | 0 | 21 | 7,975 | 1,652 | ||
| 8 | Parotid saliva | 21 | 0 | 166,321 | 227 | ||
| C17 | MN | 2 | Parotid saliva | 61 | 5 | 240,400 | 3,133 |
| 5 | Parotid saliva | 45 | 8 | 155,370 | 2,084 | ||
| 6 | Parotid saliva | 23 | 13 | 206,800 | 2,368 | ||
| 8 | Parotid saliva | 29 | 12 | 235,876 | 2,532 | ||
| C18 | Placebo | 2 | Parotid saliva | 0 | 0 | 161,160 | 4,089 |
| 2 | Seminal plasma | 0 | 13 | 35,288 | 100,690 | ||
| 5 | Parotid saliva | 33 | 0 | 189,890 | 1,301 | ||
| 6 | Parotid saliva | 29 | 0 | 393,177 | 778 | ||
| 8 | Parotid saliva | 26 | 2 | 365,619 | 808 | ||
| C20 | SF-2 | 5 | Parotid saliva | 44 | 3 | 588,480 | 12,621 |
| 6 | Parotid saliva | 28 | 0 | 399,988 | 901 | ||
| C21 | SF-2 | 1 | Parotid saliva | 27 | 3 | 115,010 | 1,190 |
| 5 | Parotid saliva | 26 | 4 | 486,308 | 1,997 | ||
| 6 | Parotid saliva | 27 | 10 | 882,237 | 3,589 | ||
| 8 | Parotid saliva | 0 | 41 | ND | ND | ||
| C24 | MN | 2 | Parotid saliva | 38 | 13 | 171,498 | 3,370 |
| 5 | Parotid saliva | 50 | 25 | 213,037 | 5,714 | ||
| C25 | MN | 2 | Parotid saliva | 39 | 17 | 1,370,000 | 18,332 |
| 5 | Parotid saliva | 20 | 0 | 252,208 | 2,081 | ||
ND, not determined.
TABLE 5.
Western blot analysis of samples of external secretions from selected individuals
| Volunteer | Visit no. | Specimen type | gp banda
|
Antibody used | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 160 | 120 | 66 | 55 | 51 | 41 | 31 | 24 | 17 | ||||
| PC12 | 1C | Semen | + | − | − | − | − | − | − | ++ | ± | IgG |
| C09 | 2 | Semen | + | − | − | − | − | − | − | − | − | IgG |
| C05 | 8 | Cervical wick | + | ± | ± | ± | ± | ± | − | ++ | + | Ig |
| C05 | 6 mo | Cervical wick | ++ | ± | ++ | ± | + | + | − | ++ | ± | IgG |
| C10 | 5 | Parotid saliva | + | − | − | − | − | ± | − | + | − | IgG |
| C10 | 2 | Semen | − | − | − | − | − | − | − | ± | − | Ig |
| C10 | 5 | Parotid saliva | − | − | − | − | − | − | − | − | − | IgA |
+, band seen; −, no band seen; ±, faint band seen.
Limiting-dilution analysis of CTL precursors.
CTL precursor frequency analysis was performed to determine whether vaccination with soluble rgp120 might have skewed the postinfection response against envelope antigens. Table 6 shows the results for nine of the individuals from whom B-LCL were established. Background precursor frequencies against the control vaccinia virus (NYCBH) were generally low for each individual (<50 of 106 cells). A result was considered significant if >50 of 106 cells (corresponding to a precursor frequency of 1 in 20,000) were scored positive with a background level of <50 per 106 cells. Alternatively, if the background was between 50 and 100 per 106 cells, a result of >150 per 106 cells was considered significant. Five of nine subjects (C06, C10, C11, C13, and C21) showed Gag-specific CTL responses for at least one of the time points studied. Two subjects (C06 and C20) generated reverse transcriptase-specific CTL precursors, and two (C06 and C21) had Nef-specific precursors. Env-specific CTL responses were infrequent and were not vigorous. The highest Env-specific response was detected in subject C06, but this was specific for the LAI envelope and not the MN strain with which this individual had been immunized. In fact, none of the rgp120-vaccinated individuals had detectable Env-specific CTL responses to the immunizing strain of HIV-1. One placebo recipient (C18) was, ironically, the only individual with a postinfection cellular immune response to the MN strain. There were no SF2-specific CTL responses.
TABLE 6.
CTL precursor frequencies of infected vaccineesa
| Volunteer | Study day | Immunogen | Frequency per 106 PBMC of CTL precursors vsb:
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NYCBH | Gag | RT | IIIb | MN | RF | SF2 | Nef | |||
| C06 | 537 | MN | 1 | 296 | ND | ND | 22 | 35 | ND | ND |
| 664 | MN | 2 | 46 | 1,528 | ND | 23 | 1 | 3 | 22 | |
| 664 | MN | 34 | 33 | 1,288 | 347 | 48 | 58 | 42 | 250 | |
| 664 | MN | 50 | 133 | 1,499 | 106 | 21 | 28 | 26 | 136 | |
| C09 | 642 | SF-2 | 1 | 26 | 81 | 15 | 8 | 11 | 20 | 4 |
| C10 | 679 | MN | 15 | 415 | ND | 20 | 22 | 729 | ND | ND |
| 909 | MN | 16 | 7 | 27 | 94 | 3 | 22 | 1 | 1 | |
| C11 | 834 | MN | 35 | 278 | ND | 37 | 6 | 36 | 30 | ND |
| C13 | 656 | SF-2 | 15 | 86 | ND | 40 | 26 | 40 | ND | ND |
| 763 | SF-2 | 70 | 196 | ND | 36 | 43 | 31 | 13 | ND | |
| C18 | 832 | Placebo | 1 | 4 | 3 | 5 | 62 | 2 | 2 | 7 |
| C20 | 730 | SF-2 | 3 | 16 | 57 | 13 | 39 | 46 | 13 | 14 |
| 730 | SF-2 | 2 | 13 | 14 | 23 | 18 | 17 | 30 | 10 | |
| C21 | 917 | SF2 | 5 | 147 | 41 | 9 | 2 | 9 | 5 | 134 |
| 917 | SF2 | 1 | 159 | 17 | 26 | 3 | 13 | 3 | 148 | |
| C24 | 1172 | MN | 29 | 13 | 46 | 36 | 29 | 20 | 3 | 33 |
Determined by the method of Koup et al. (84).
For a background value against the NYCBH control of <50/106 PBMC, a precursor frequency of 50/106 PBMC above background is considered significant (corresponding to 1/20,000 cells). For a background between 50 and 100/106 PBMC (e.g., subject C13), a precursor frequency of >150/106 is considered significant. Significant values are in boldface.
Phenotypes and growth characteristics of infecting HIV-1 strains.
HIV-1 strains were isolated in PBMC cocultures from the 18 infected vaccinees and from 6 of the putative transmitters. For only one subject (C16) was depletion of CD8+ T cells prior to culture required to obtain a virus isolate, indicating that virus replication in vitro was sufficient to overcome CD8+-T-cell-mediated suppressive effects in the majority of cases. All 24 viruses grew well in mitogen-stimulated PBMC; representative growth rates for several strains are shown in Fig. 3. The in vitro replication rates of viruses isolated from the infected vaccine recipients were not significantly different from those of isolates from the putative transmitters and from nonvaccinated, acutely infected individuals (Fig. 3 and data not shown). It is common for HIV-1 strains isolated soon after primary infection to replicate relatively slowly in culture (26, 45, 46). Our results indicate that it is unlikely that prior immunization with rgp120 selected for the transmission of only poorly replicating HIV-1 strains to the infected vaccinees.
FIG. 3.

Replication kinetics of viral isolates from infected vaccinees in activated PBMC. The replication kinetics of selected HIV-1 isolates from the infected vaccinees were evaluated in cultures of PHA-stimulated normal donor PBMC. Cells were infected with 100 TCID50 of each isolate, and the levels of HIV-1 p24 antigen were measured in culture supernatants on days 0, 3, 7, 10, and 14 after inoculation. Isolate pC13 is from the partner of C13; isolates AD6 and AD13 were obtained from nonvaccinated acute seroconvertors.
The phenotypes of the 18 viruses isolated from the vaccinees were determined by their abilities to induce the formation of syncytia in the MT-2 T-cell line. Sixteen of the viruses isolated had an NSI phenotype, whereas the viruses isolated from C04 and C20 were SI strains (data not shown). An SI virus isolate was also obtained from pC04, the putative transmitter to C04. The SI strains carried positively charged amino acids in the V3 loop; this is typically associated with the SI phenotype (Fig. 4) (47). Individuals C04 and C20 underwent relatively rapid declines in CD4 T-cell counts (57), consistent with their infection by a virulent, SI strain of HIV-1 (67). A minor proportion (5 to 20% in different studies) of cases of HIV-1 infection in nonvaccinated individuals involves transmission of SI strains (4, 46, 89, 130, 137, 158), consistent with the proportion observed here (2 of 18), so the isolation of SI strains from individuals C04 and C20 was not necessarily a vaccine-related phenomenon.
FIG. 4.
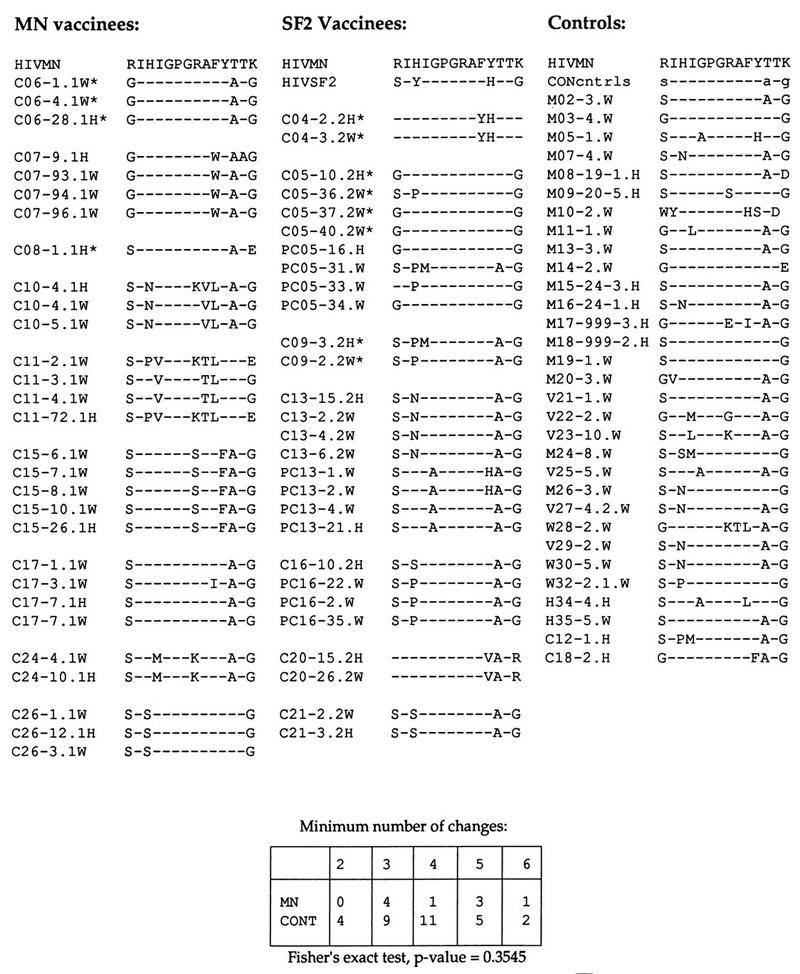
The antigenic domain in the V3 loop. All available sequences are included, grouped by individual, and aligned with the MN consensus sequence. An example of a matrix for a Fisher’s exact test, used to examine the possibility of greater diversity in the MN recipient strains relative to MN compared to the control (CONT) set, is shown at the bottom. The number of amino acid changes relative to the vaccine strain sequence for the closest sequence in each of the vaccinees and for each of the controls was tallied (138, 139). For example, C06 had three changes relative to MN in this region. The number of individuals with viruses carrying a given number of mutations was tallied for the MN vaccine recipient group and for the controls, and the two groups were not significantly different by this measure. The positions that tend to correlate with the SI phenotype are the first and last amino acids of this region, and the two SI isolates from among the vaccine recipients (C04 and C20) carry positively charged arginine and lysine residues in these positions, respectively. Dashes indicate gaps in the sequence alignments.
Coreceptor use by the infecting HIV-1 strains.
Macrophage-tropic, NSI isolates of HIV-1 preferentially use CCR5 as a coreceptor for infection (26, 33, 136) and are phenotypically characteristic of the majority of HIV-1 strains isolated during primary infection (158). We assessed the coreceptor requirements of three NSI isolates (C07, C08, and C13) and one SI isolate (C20) from the infected vaccinees, along with three NSI isolates from putative transmittors (pC05, pC18, and pC21) and three additional NSI isolates from nonvaccinated individuals, obtained during primary HIV-1 infection (AD60, AD74, and AD75). We found that NSI isolates from the infected vaccinees, donors, and nonvaccinated controls all used exclusively CCR5 for infection, while the SI isolate used a broader range of coreceptors, including CCR5, CCR2b, CCR3, and CXCR4 (data not shown). Although the number of isolates tested was too small to draw formal conclusions, these results suggest that prior immunization with rgp120 does not result in the selection of phenotypic variants with distinct coreceptor requirements. Rather, the isolates from the infected vaccinees displayed phenotypic profiles and coreceptor usage patterns consistent with most sexually transmitted strains of HIV-1 (29, 65).
Virus burdens in infected vaccine recipients.
Irrespective of any effect of the immunization regimen, the 18 individuals we examined became infected with HIV-1. The most direct evidence for this was the detection of HIV-1 virion-associated RNA in the plasma of each individual by a sensitive and quantitative PCR-based assay (72, 102). The levels of plasma HIV-1 virion-associated RNA for each of the participants, superimposed on their antibody responses to the immunogens, are shown in Fig. 2. Although there was considerable variation between individuals, in general we observed a sharp increase in the viral burden soon after infection followed by a decline over time. However, because of infrequent sampling intervals, we had no way of determining the true peak viral burdens, thus preventing analysis of this parameter. In all cases, the HIV-1 RNA levels in the plasma reached an approximate steady state, ranging from 750 to 48,000 copies/ml at 9 to 12 months after infection.
Recent studies have shown that the levels of HIV-1 virion-associated RNA sustained in the plasma following acute infection are more predictive of the rate of disease progression than is the peak of viremia (92, 102, 129, 154). We compared the levels of HIV-1 RNA found in the plasma 9 to 12 months after infection for the group of vaccinees who had received a complete course of immunization with those of the partial vaccination and placebo groups and found no significant difference in plasma viral burden (P = 0.08, Wilcoxon rank sum test). The lowest level of plasma HIV-1 virion-associated RNA was recorded for C12, a placebo recipient (Fig. 2). No significant difference in the viral burdens of those individuals immunized with MN rgp120 and those immunized with SF-2 rgp120 was noted (P = 1.0, Wilcoxon rank sum test) (Fig. 5). Individuals with a high viral burden in plasma following primary infection have been shown to progress more rapidly to AIDS (102, 122). Whether prior immunization with HIV-1 rgp120 will alter the rate of clinical progression in the vaccinees, however, is beyond the scope of the present study.
FIG. 5.
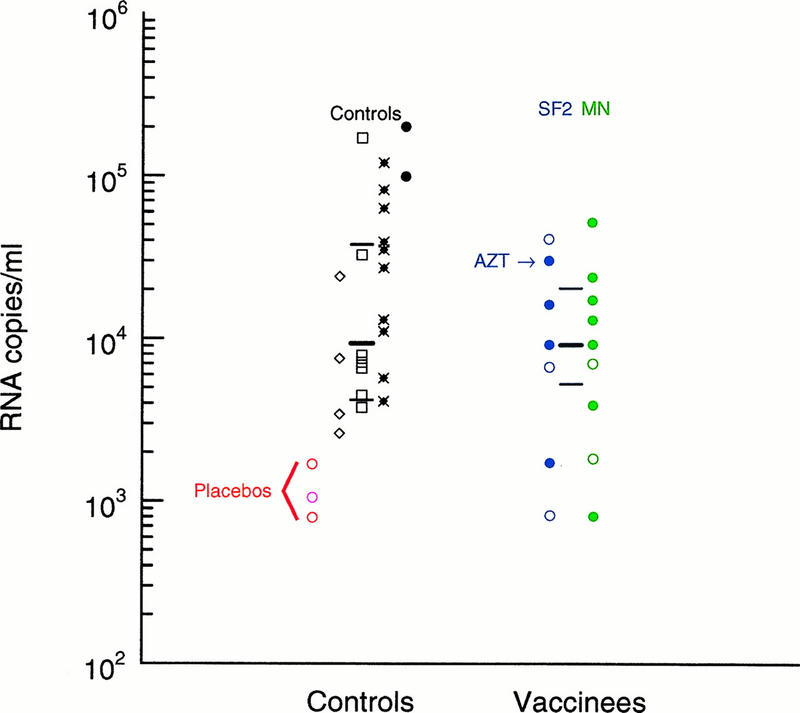
Comparison of the viral loads of infected vaccinated individuals and matched controls at 9 to 12 months after the estimated time of infection. Viral RNA copy numbers in sera or plasma from HIV-1 infected individuals who had been given an SF2 rgp120 vaccine are labeled in blue, and those who had received MN rgp120 are in green. Individuals who had received three or four vaccinations prior to infection are indicated by closed circles; those who had less than three vaccinations are indicated by open circles. The single individual who had documented antiretroviral agent (AZT) use prior to this sampling period is indicated with an arrow. In cases for which two samples from a single individual were available during this 3-month period, the average value was used. Twenty-three controls matched for risk factors for infection, gender, age (±10 years), and year of seroconversion were obtained from four different cohorts, excluding samples that were derived from patients with prior use of antiretroviral agents and patients for whom no samples were available in the appropriate time frame of 9 to 12 months postseroconversion. Three samples from individuals given placebo vaccinations prior to infection were included with the controls and are marked as open circles in the first column of controls. The red circles are for placebo recipients from the 401 study, and the pink circle is a placebo recipient from another U.S. vaccine trial. The other four columns display control RNA values from the four different cohorts: the TRUNK cohort (◊), the ALIVE cohort (□), the MACS cohort (•×), and HIVNET (•). The medians and interquartile ranges for the complete control set and the complete vaccine recipient set are indicated by the thick and thin horizontal black bars, respectively.
We also compared the plasma RNA levels of the infected vaccinees at 9 to 12 months postseroconversion with those of controls derived from several cohorts of nonvaccinated individuals monitored through the acute stage of HIV-1 infection (Fig. 5). Controls were selected from among individuals who had not been given antiretroviral therapy, who were sampled in the comparable time frame of 9 to 12 months postinfection, and who were matched for age, presumed risk of exposure, and the year of primary infection. Also included in the control set were three placebo vaccination cases, two from the 401 study and one from an earlier vaccine trial (57). The placebo recipients as a set had the lowest viral burdens among the controls. One of the infected vaccinees (C20) had received antiretroviral therapy (zidovudine [AZT]) prior to 9 months postinfection, as indicated in Fig. 5; this individual still had a relatively high viral RNA level (28,000 RNA copies/ml) and was retained in the analysis (Fig. 2). Nonparametric statistics were used for the comparison, because the distribution of RNA levels in the control group did not fit a normal or log-normal distribution by a Kolmogorov-Smirnov test for normality, probably due to differences in sample collection procedures among the different cohorts (Fig. 5).
The highest viral RNA values were found among the nonplacebo control cases (Fig. 5), but a Wilcoxon rank statistic did not indicate that the infected vaccine cases had significantly lower values than the controls (P = 0.12). The overall median viral burdens for the infected vaccine cases and the controls were comparable (8,500 and 9,325 RNA copies/ml, respectively). A similar conclusion was reached in an independent study of the infected vaccinees and controls (57). The median plasma RNA value for the infected vaccine recipient group and the controls was intermediary relative to the results from other cohorts. A median HIV-1 plasma RNA burden of 13,020 copies/ml was found for a group of 180 homosexual or bisexual men enrolled in the Pittsburgh MACS study between April 1984 and March 1985 (102); a median value of 5,871 copies/ml was found in a study of 165 hemophiliacs 12 to 36 months after the presumed date of infection (122); and Garcia et al. found a median viral burden of 9,331 copies/ml for asymptomatic individuals at a median time of 48 months after detection of infection (52). We did note that there were fewer high-viral-load burdens among the infected rgp120 recipients than among the highest quartile in the matched control group (Fig. 5). However, artifacts due to differences in sample collection protocols at different clinical sites could contribute to this observation, and its overall significance is unclear.
The viral burden analyses should be considered with several qualifications in mind, all of which apply in general to comparisons of viral loads between cohorts. First, several different cohorts were included among the controls, and two trials contributed to the rgp120 vaccinee cohort. Differing methods of sample storage and collection (e.g., serum versus plasma), or perhaps even differences in the underlying biology (e.g., intravenous drug use versus sexual transmission), may subtly influence comparisons, because we noted distinctive distributions of RNA values in the different control cohorts (Fig. 5). Second, the distribution of viral burdens for a cohort may be influenced by the age of the subjects (122) or by the frequency of acute HIV-1 infection in enrollees who undergo symptomatic primary infection. These individuals tend to have higher sustained RNA levels (21, 52, 62, 67, 92, 126) and may be more likely to be enrolled in certain natural history cohorts. Inspection of control subjects’ clinical records, however, indicated that there was no recorded bias for symptomatic acute infection or age among our control set. Third, the intrinsic variability of the plasma RNA assay and within-subject variation together add significant noise to the data. It has been proposed that sustained changes in viral RNA levels within a patient must be at least 3-fold (log 0.5) (131) or 2.6-fold (log 0.41) (125) to be considered indicative of a biologically relevant change. Differences between the median values of the vaccinee and control cohorts were less pronounced than this (Fig. 5).
Some individuals (C04, C05, C06, C07, C09, and C12) continued to receive booster immunizations after infection had occurred (Fig. 1 and 2). In principle, this could be a concern because immunization with other antigens has been reported to be associated with transient increases in the viral burden (123, 140). In our study, postinfection immunization was sometimes associated temporally with an increase in the viral burden (for C04, C05, C07, and C12), but any causal relationship between these events cannot be determined unequivocally because of the infrequency of sample collection.
Viral sequence analysis.
DNA sequences of the gp120 and gp160 regions of env from uncultured and cultured PBMCs, respectively, from each of the infected vaccinees and their matched controls (see above) were obtained. These are presented as deduced amino acid sequences in Fig. 6. Between one and five sequences were generated for each of the vaccine recipients, and a single representative sequence was generated for each of the matched controls. Whenever possible, vaccinee sequences were generated by using two different strategies in parallel in two different laboratories. A sequence from each vaccinee was generated from cultured PBMC. Cloning and sequencing directly from the study subjects’ PBMC were performed when there was a target copy number adequate for direct amplification by PCR (i.e., in 15 of 18 cases).
FIG. 6.


Amino acid alignment of gp120 protein sequences. Only a single sequence is shown per individual vaccine recipient, although multiple sequences were determined. Yellow boxes indicate antigenic regions in V2, V3, and C4 that were analyzed in detail. Gray shading indicates distinctive amino acid signature sites. Purple boxes indicate the most distinctive motifs identified with MotifScan. All sequences are aligned to the consensus sequence of the control set, labeled CON. The sequence designations include the study subject identification number, the clone number, and either an H (if sequenced at the University of Alabama at Birmingham) or a W (if sequenced at Northwestern University). Dashes indicate identity with the B subtype sequence at the top of the alignment; periods indicate insertions made to maintain the alignment; pound signs indicate frameshift mutations.
Similarity scores were generated for the 883-bp gap-stripped nucleotide sequence set, comprising the 95 env sequences obtained from the infected vaccinees and the matched case controls. No unexpected interpatient similarities or within-subject divergences were found (91). Uncorrected distances are presented here, because this measure was used as a rapid screen to test for anomalies among the viral sequences. The closest between-subject distance for nonrelated people was found to be 4.4% (median, 9.7%). The most-divergent within-subject sequences from among the vaccinees were found in C05, with a distance of 4.8%; C05 was first sampled for sequencing approximately 1 year postinfection, which was atypically late relative to the rest of the cohort. The median within-subject distance was 1.7%, with a median estimated time from seroconversion of 85 days at sampling. Four HIV-1-infected partners of infected vaccine recipients were sampled. Three of these epidemiologically linked donor-recipient pairs had highly similar sequences, as would be expected: between 2.8 and 5.8% for pC05 and C05, between 2.5 and 3.5% for pC16 and C16, and between 0.7 and 2.5% for pC12 and C12. These relationships were supported by phylogenetic analyses which demonstrated monophyletic clustering (see below). However, pC13 and C13, who comprised another epidemiologically linked pair, had quite distinct viral sequences that ranged in distance from 8.0 to 8.6%. The sequences were not associated in the phylogenetic analysis, suggesting that transmission had not, in fact, occurred between these two individuals (see Fig. 7).
FIG. 7.
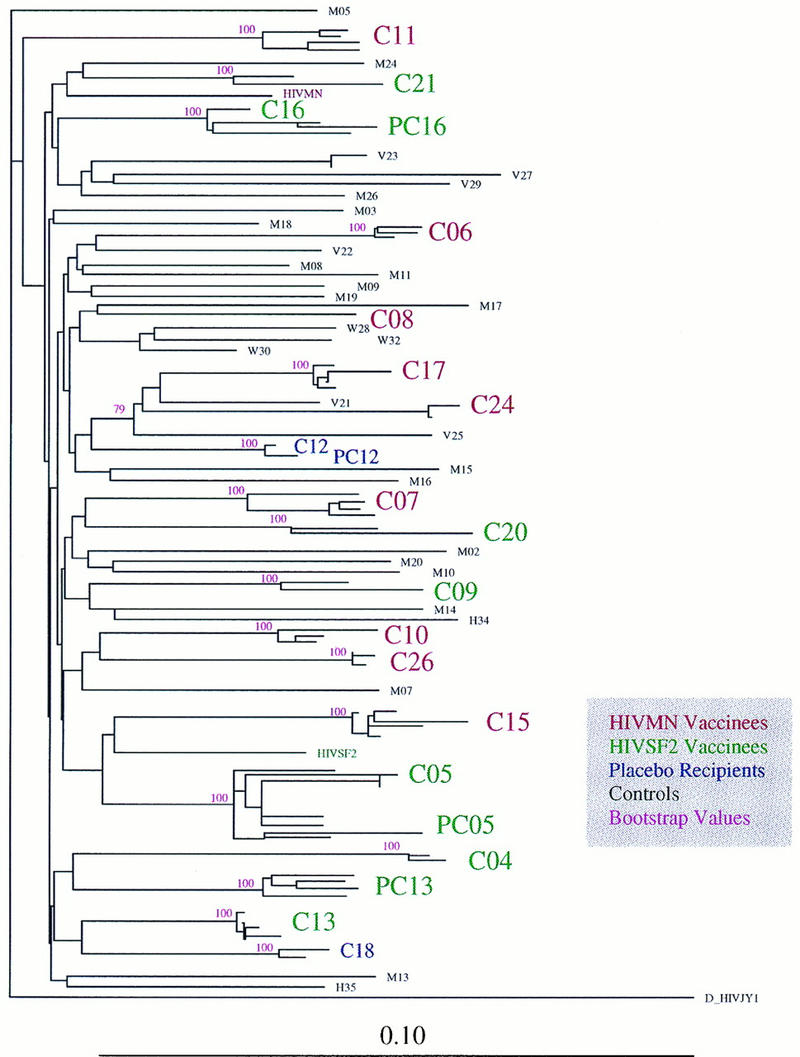
Vaccine breakthrough sequences and controls: neighbor-joining phylogenetic tree, with bootstrap values, showing the relationships between the viral sequences from vaccinated individuals and matched controls. Partners of vaccinated individuals are indicated with a P (e.g., C05 and PC05 are vaccinee C05 and C05’s partner, respectively). Bootstrap values of greater than 50 are shown. Vaccine strains MN and SF2 are included. A D subtype viral sequence was used as an outgroup. Multiple sequences for the vaccinees and their partners are included, and all sequences from the same patient cluster in 100 of 100 bootstrap resampling replicates.
Phylogenetic analyses.
Phylogenetic reconstructions of the relationships between viral sequences from the infected vaccinees and those from the matched controls confirmed the authenticity of the samples: in neighbor-joining trees, each of the within-subject sequence sets formed a distinctive clade preserved in 100 of 100 resamplings in bootstrap analysis (Fig. 7). Three of the four epidemiologically linked pairs, all except vaccine recipient C13 and his partner pC13, were closely associated in phylogenetic analysis, again preserved in 100 of 100 bootstrap tests. A comparison with matched case controls (Fig. 7) and a number of contemporary and archival viral sequences (data not shown) showed that all vaccinee and control sequences in this study were from the B clade and that they formed an unstructured topological pattern, with one notable exception. Sequences from four individuals (two infected vaccine recipient cases, C17 and C24, and two control cases, V21 and V25) were associated both in the neighbor-joining tree shown in Fig. 7, through bootstrap analysis (in 79 of 100 bootstrap tests), and in a maximum-likelihood phylogenetic reconstruction (data not shown). Retrospective examination of clinical records, undertaken by special request because of the observed clustering pattern, revealed that the four study subjects with phylogenetically linked viral sequences each had intravenous drug use as their primary risk factor for infection, and all four attended the same clinic in the same city.
There is no evidence suggesting that an unusual lineage, or a cluster of related strains, dominated the set of viral sequences obtained from the infected vaccine recipients. Furthermore, the genetic distances between the vaccine strains (SF2 or MN) and the vaccinees’ viral sequences were not unusually high. The corrected sequence distances for the vaccine strains were extracted from the Kimura two-parameter distance matrix used to generate the neighbor-joining tree shown in Fig. 7. The distances between the viral sequences in the vaccine recipients and their vaccine sequence were not significantly greater than the distances to the controls, as determined by a standard two-sample t test. The median distance between MN and the control viral sequences was 9.1%, and the median distance between MN and the sequences from the infected MN rgp120 vaccine recipient cases was 9.2%. Similarly, the median distance between SF2 and the control viral sequences was 9.3%, and the median distance between SF2 and the sequences from the infected SF2 rgp120 vaccine recipient cases was 8.4%. Thus, the viruses from the infected vaccine recipients are not genetically more distant from the vaccine strain than are the control sequences, indicating that there was no unusual genetic selection pressure on the viruses transmitted to the rgp120 vaccinees.
Signature sites.
To determine if there were any patterns in the protein sequences that were distinctive among the viral sequences from the infected vaccine recipients, two methods were used to scan for signature patterns in single amino acid positions. The program ENTROPY was used to screen for any single-amino-acid positions that were significantly more variable in the infected-vaccinee sequence set than in the control set (82). The program VESPA (77) was used to detect changes in the most common amino acid in a position, comparing viruses from the infected vaccinees to those of the controls. The control data set had 31 sequences, including the 29 matched controls and the 2 placebo recipients. The sequence set from the infected MN vaccine recipients included a single sequence from each of the nine MN rgp120 vaccinees, and the sequence set from the infected SF2 vaccine recipients had seven such sequences. The sequence sets from the infected vaccine recipients who received MN or SF2 were considered both separately and in combination.
Three sites meeting the minimum criteria to be considered as potential signature sites (see Materials and Methods) were identified and are indicated in Fig. 6. The first site, which was found to be distinctive for SF2 vaccinees but not for MN vaccinees, was located in the signal peptide, so it is unlikely to be biologically meaningful. The second site was found in the C1 domain of gp120. The viral sequences from the MN and SF2 rgp120 data sets both had changes at this position relative to the control set (Fig. 6). Both the MN and SF2 vaccine strains carry a threonine at this position. Comparison of the viral sequences from the MN and the SF2 rgp120 data sets, as well as the control data set, for the presence or absence of a threonine at this position indicated a potentially interesting difference: 0 of 9 MN rgp120 recipients, 1 of 7 SF2 rgp120 recipients, and 7 of 11 controls carried a threonine at this position (P = 0.0025 when comparing SF2 and MN rgp120 vaccinee strains combined to the controls; Fisher’s exact test). However, only 24 of 81 sequences of the B subtype in the Los Alamos database were found to have a threonine at this site, dramatically reducing the significance of the above finding (P = 0.19 when comparing SF2 and MN to the database sequences; Fisher’s exact test). Alanine, not threonine, was the most common amino acid at this position in the B subtype sequences in the database (found in 34 of the 81 sequences) and was also the most common amino acid at this position in the vaccinees’ viral sequences. The biological significance of these observations is unclear.
The third potentially interesting signature site was an N-linked glycosylation site (defined as an N-X-S/T sequon) at the base of the V2 loop. It was present in only 2 of 7 sequences from SF2 rgp120 recipients but was conserved among the matched-control viral sequences (26 of 31) and in the clade B sequences in the database (71 of 103 sequences) (117). It is, however, also conserved among MN rgp120 recipient sequences (eight of nine), so it is not obvious that the variation observed at this site in the SF2 rgp120 recipients is of any biological relevance.
The gp120 protein is among the most heavily glycosylated proteins known, and glycosylation patterns can influence the antigenicity of a protein (93, 119). Other than the one glycosylation site noted above, no particular glycosylation patterns were specific to the infected vaccine recipient cases. There was an average of 24 potential N-linked glycosylation sites in the gp120 proteins for each set of sequences among the controls and the MN and SF2 rgp120 vaccine groups, with an overall range of 19 to 29 such sites for the entire set of sequences.
Analysis of signature motifs.
Although most antibody binding sites have a conformational component (71, 73, 109, 111, 113), we considered whether distinct patterns could be found that were suggestive of escape mutations over short contiguous stretches of gp120. In principle, a single substitution at any one of three or four sites in the core of an antibody epitope might abrogate binding of antibodies to a particularly immunogenic domain, such as the V3 loop. To address this question systematically, and to develop a better understanding of how frequently one might find distinctive motifs by chance alone, we developed a program called MotifScan. By using a sliding window, all viral sequences from controls and vaccinees were compared to the vaccine strains to assess variation in motifs ranging in length from 4 to 8 amino acids. Here, we will refer specifically to the results of the hexamer analysis. Motifs for each sequence were scored as having either identity or nonidentity relative to the appropriate vaccine strain, and the scores were tallied. The controls were then compared to the vaccine recipients by a Fisher’s exact test. This analysis and the single-site signature analysis are not independent, and thus it is not surprising that two of the single-site signature positions are embedded in distinctive motifs (Fig. 6). By this method, two sites in SF2 rgp120 vaccine recipient sequences and three sites in the MN vaccinee sequences were significant (Fig. 6). The most distinctive site in the SF2 vaccinee viral sequence set consisted of the overlapping hexamers in the sequence stretch LPCRIKQI (located in C4), and the second most distinctive site overlaps the glycosylation signature site at the base of the V2 loop, TPLCVTLN; neither of these regions was distinctive in viruses from the MN rgp120 vaccinees. In viruses from the MN vaccinees, there were three distinctive regions: the motif VIRSENF in the C2 domain was the most distinctive, followed by ATEKLWV, overlapping the C1 signature site, and GPGRAFY in the V3 loop; these three regions were not distinctive in viruses from the SF2 vaccinees.
Only one of these motifs the GPGRAFY motif at the tip of the V3 loop, overlaps a known immunogenic domain (Fig. 4). The GPGRAF and PGRAFY motifs had P values of 0.034 and 0.05, respectively, in the Fisher’s exact test used to rank the motifs in the MN rgp120 vaccinees for variability relative to the MN rgp120 vaccine strain. This variation occurs in an antigenic domain and so merits note, but upon consideration of additional data, the potential significance of variation in this motif is diminished. First, when Monte Carlo randomizations of the sequences were done, we found that 80 of 100 randomly selected sequence data sets yielded motifs somewhere in gp120 with a P value of <0.034. This means that comparably distinctive motifs were found by chance alone in the majority of random data sets. Second, SF2 shares the GPGRAF sequence exactly with MN, yet viruses from the infected rgp120 SF2 vaccinees showed no particular variation in this motif. Third, the only way statistically significant variation in this motif can be observed is by excluding from analysis the two individuals who received only two MN rgp120 vaccinations prior to infection, whose viral sequences both carry the MN-like GPGRAF motif. Yet both these individuals had anti-V3 MN antibodies and a neutralizing antibody response to MN virus prior to infection (57). In comparison, three of the vaccinees who had been given three or more MN rgp120 vaccinations, who had variations from the GPGRAF motif, had no detectable anti-V3 MN antibodies and no MN neutralization response prior to infection (57). Thus, exclusion of the two cases with only two vaccinations prior to infection may not be a biologically appropriate way to consider the V3 sequence data.
Analysis of antigenic linear domains.
The three most potent and broadly reactive anti-gp120 neutralizing MAbs bind either to complex discontinuous epitopes that are inappropriate for sequence analysis (IgG1b12 and 2G12) or to a linear domain in gp41 (2F5) that is not present in rgp120 vaccines. To screen for potential protein differences in antigenic domains, we were confined to examining linear regions, which are more amenable to analysis. Therefore, we selected linear regions in V2, V3, and C4 (Fig. 6) which react with MAbs that are capable of neutralizing TCLA viral strains but that have limited breadth and potency against primary isolates (75). Examples of MAbs that have some ability to bind to these peptides and to neutralize virus are as follows: for V2, G3-4 (64) and BAT085 (50); for C4, G3-508 and G3-299 (141); and for V3, 447-D (55). Figure 6 shows the location of each of these epitopes on the gp120 sequence. Because the V3 region of gp120 has historically been considered the most important linear domain for HIV-1 neutralization (98, 109, 148) and is the most intensively studied of these linear binding domains (75), it was selected as the representative region for display as shown in Fig. 4.
Two methods were used to examine the variability of these regions in the vaccinee and control data sets relative to the sequences of the vaccine strains. First, contingency tables were generated based on a simple tally of the number of amino acid substitutions for each sequence within the short protein domain relative to the MN or SF2 sequences (Fig. 4). Comparison of the viral sequences from vaccinees given a complete set of three or four vaccinations to their corresponding vaccine strain showed no significant increase in the number of substitutions in these domains relative to the control set of viral sequences (Fisher’s exact test) (for an example, see Fig. 4). Second, because there is a gradation between amino acid substitutions that are conservative and relatively subtle and those that are very distinctive, a second test was performed based on generation of a protein similarity score by using the Henikoff protein structure-based amino acid substitution matrix (61, 82).
The distributions of protein similarity scores of the controls and of the postvaccine infection cases, relative to the vaccine strain, were compared by using a one-sided Wilcoxon test to see if the vaccinee viruses were more highly modified in the three regions under consideration. This approach revealed a trend toward more highly divergent sequences in the V2 domain among SF2 rgp120 recipients relative to SF2 (P = 0.015) that was statistically significant when either the 4 SF-2 rgp120 recipients receiving a complete immunization course were considered or all 11 SF-2 rgp120 vaccinees were included. However, this trend in V2 was not observed in MN rgp120 recipients relative to MN. In contrast, a trend toward greater diversity in MN recipients was found in the V3 domain (P = 0.025), but the significance was lost if all nine MN rgp120 vaccinees were included (P = 0.15), and this trend was not observed in the SF2 rgp120 recipients. In the C4 domain, which is in a relatively conserved part of gp120, no such tendency for greater divergence was found. Thus, this neutralizing-domain linear-epitope analysis revealed trends suggestive of greater variation in vaccinee sequences in V2 and V3. However, the distinctions between the viral sequences from MN and SF2 rgp120 recipients were not consistent, which renders their significance questionable.
DISCUSSION
HIV-1 subunit vaccines based on recombinant forms of the monomeric gp120 glycoprotein were first developed over 10 years ago. Their creation was a logical consequence of the assumption that neutralizing antibodies were likely to be a correlate of protection against HIV-1 and of experimental observations that most serum HIV-1-neutralizing antibody activity was directed at gp120 (58, 60, 98, 110). It is still a central tenet of HIV-1 vaccine design that it would be desirable for a vaccine to induce broadly neutralizing antibodies able to prevent HIV-1 transmission or to significantly limit systemic dissemination of the virus. It also remains true that gp120 contains most, but not all, of the neutralizing-antibody epitopes identified to date (71, 110). Most studies of the gp120 vaccines in chimpanzees have indicated that the apparent correlate of protection against infection is the anti-gp120 antibody response, more specifically the response to the V3 region (7, 8, 15, 38, 53, 54, 143). Therefore, much of our effort has focused on characterizing the antibody response to gp120 in the infected vaccinees.
Assuming that a preexisting anti-gp120 antibody response alone is sufficient for protection of humans from HIV-1 infection, which is far from certain, the most probable explanation for infection of these vaccine recipients is that the subunit rgp120 immunogens did not induce antibodies of sufficient quality, abundance, or specificity to afford protection. Most of the infected vaccinees who received the complete course of vaccination did develop a reasonable antibody response to the immunogen (C20 was an exception in this regard). However, the anti-gp120 response was relatively type restricted compared to infection-induced responses, in that there was better reactivity with the vaccine rgp120 than with a different rgp120. It is possible that many of the antibodies induced by the rgp120 proteins were directed at the more variable epitopes on gp120, a feature which is not necessarily advantageous for neutralization of heterologous primary viruses under field conditions. However, some cross-reactive rgp120 monomer binding antibodies could clearly be detected in most vaccinees prior to infection. Although the peak antibody titers in many vaccinees approached infection-induced anti-rgp120 titers (Table 2), the immune responses to the immunogens were generally transient. This meant that exposure to the infecting HIV-1 strain rarely occurred near the time when the anti-rgp120 response to booster immunizations was maximal, although C16 and C17 were notable exceptions. This could be significant, since successful protection in the HIV-1 LAI chimpanzee model was found when the animals were challenged at the time of their maximal antibody response to the immunogen (7, 8, 15, 38, 53, 54, 143). However, even under these favorable conditions, using a neutralization-sensitive, vaccine sequence-matched HIV-1 strain, protection was found to occur in only about half the challenged chimpanzees. Conditions in the human trials are less favorable to successful protection in several respects, such as the timing of challenge with respect to immunization and the heterologous nature of the challenge. Furthermore, mucosal antibodies in genital tract secretions induced by systemic immunization were either absent or present at low levels. Assuming that HIV-1 infection in these vaccinees was acquired by the mucosal route, the potential protective effect of local antibodies was not manifest.
What is relevant to protection is not the ability of a vaccine such as rgp120 to induce antibodies reactive with itself but the ability of the immunogens to induce antibodies capable of binding to the infecting strain and neutralizing it. A correlate of virus neutralization is antibody reactivity with the native, oligomeric form of the envelope glycoproteins (48, 132). We did not test the ability of the rgp120 vaccines to induce oligomer-reactive antibodies; however, using assays based on TCLA viruses, MN and SF-2 rgp120s have been shown to induce in humans significant titers of antibodies able to neutralize the HIV-1 strain from which the immunogen was derived (6, 9, 59, 68, 96, 156). Information on the neutralizing-antibody titers elicited against the vaccine strain in the individuals we have studied is reported elsewhere (57, 99). It is now clear, however, that passage of HIV-1 in T-cell lines selects for variants with an abnormal sensitivity to neutralization that is not typical of primary viruses (96, 98, 108, 110, 148). This may reflect, in part, adaptation of HIV-1 to use the CXCR4 coreceptor after passage through cell lines instead of the CCR5 coreceptor that is used by most primary, NSI strains to enter PBMC (26, 85, 136). Consequently, it is not now clear whether the induction by the rgp120 vaccines of antibodies able to neutralize HIV-1 MN or HIV-1 SF-2 has any real significance for the protection of humans against primary viruses under in vivo conditions.
We found that the subunit rgp120 vaccines failed to induce, in any individual, antibodies capable of neutralizing (by 90%) the primary HIV-1 strain isolated from that individual during the acute phase of infection or soon thereafter. This finding is in accord with those of others, who observed that a spectrum of primary viruses were strongly resistant to neutralization by serum antibodies from rgp120 vaccinees (96, 98, 156). We showed, however, that most HIV-1 strains isolated from the infected vaccinees could be neutralized by human MAbs and by a CD4-based reagent with sensitivities that were comparable to those of two sets of control primary HIV-1 strains. This suggests that the preexisting anti-gp120 antibody response has not exerted a selection pressure which permits infection of the vaccine recipients only by especially neutralization-resistant HIV-1 strains.
The absence of HIV-1-specific IgA antibodies in external secretions of systemically immunized and infected individuals is not surprising; from numerous studies using other microbial antigens, it is well known that systemic immunization is usually not effective in the induction of secretory IgA antibodies (for a review, see reference 103). Furthermore, it has been the experience of the AVEG that systemically administered HIV-1 vaccines have not been effective inducers of mucosal IgA anti-HIV-1 antibodies (unpublished data). In HIV-1-infected individuals, mucosal antibodies are mainly of the IgG isotype (3, 20).
Although the soluble rgp120 immunogens were not designed to induce CTL activity, there is evidence that they can do so to a limited extent (3, 32, 59). We were unable to assess the ability of the rgp120 vaccines to induce CTL activity prior to infection because of the nature of the biological samples provided to us. However, the envelope CTL responses after infection were infrequent and, when present, were not vigorous among the infected vaccinees. This is not surprising, since the soluble rgp120 proteins would not be expected to enter the class I pathway that allows for presentation to CD8+ CTL. The frequency of Gag responders (55%) was similar to that found for a cohort of HIV-1-infected men from the San Francisco Men’s Health Study (150). Although the degree of Env responsiveness was low in this group of individuals, this is consistent with previously published reports (129) and our own unpublished data (150). The lack of a suitable control group limits speculation on the role that a prior humoral immune response may have had in the generation of Env-specific CD8+ CTL, as has been described for an animal model (157).
By definition, the MN and SF-2 rgp120 vaccines did not prevent infection in any of the individuals we studied, but it is also important to evaluate whether vaccination might ameliorate disease in the infected recipients. The individuals in this cohort have not been studied long enough for any clinically beneficial or adverse effects of vaccination to be identified, and there are too few participants for a definitive conclusion to be reached in any case. However, one parameter we could usefully measure was the virus burden. Prechallenge vaccination of chimpanzees with LAI rgp120 was found to reduce postinfection plasma viremia in some HIV-1LAI-infected animals (143), and passive administration of the anti-gp41 virus-neutralizing MAb 2F5 to chimpanzees prior to challenge with a primary HIV-1 isolate also reduced the viral burden (24). Furthermore, there is now compelling evidence linking the level of persistent plasma virion-associated RNA to disease progression (92, 102, 122, 129, 154). There was a slight trend toward fewer infected vaccinees with very high viral burdens, but there was no significant difference in the overall distribution of viral burdens among the infected vaccinees compared to a control group of symptomatic acutely infected individuals and matched case controls at 9 to 12 months postinfection. The median plasma viral burdens for the infected vaccine recipients and the controls in our study of 8,500 and 9,325 RNA copies/ml, respectively, are comparable to those found by others (52, 102, 122). The conclusion that rgp120 vaccination did not significantly affect the postinfection viral burden has also been reached by others (57).
We determined the phenotypes and growth characteristics of the strains isolated from the infected vaccinees. In all but two cases (C04 and C20), the strains possessed the NSI phenotype, in that they were unable to replicate and form syncytia in the MT-2 T-cell line. The coreceptor usage patterns for several selected isolates were consistent with the phenotype determinations. SI strains were isolated from individuals C04 and C20; it is shown elsewhere that these individuals underwent particularly rapid declines in their CD4 cell counts (57), a phenomenon associated with the presence of SI virus (70, 89, 130). The presence of SI strains in individuals C04 and C20 is not necessarily an adverse effect of rgp120 vaccination, since a minority of transmitted strains in unvaccinated individuals have the SI phenotype (46, 89, 130, 158). Determination of the growth rates of isolates from infected vaccinees in PBMC in vitro revealed that there was nothing unusual about their replication competence; indeed, some of the isolated viruses grew quite vigorously and showed distinct cytopathic effects when cultured in vitro. The phenotype and virus culture data do not, therefore, support the idea that vaccination permitted the transmission only of weakly replicating HIV-1 strains or, conversely, of strains that were especially virulent.
Genetic sequencing and phylogenetic analysis of the HIV-1 strains from infected vaccine recipients indicated that they were not infected with either an unusual lineage or atypically divergent viruses, relative to contemporary clade B viral strains from the United States, the presumed country of infection of all the trial participants. We also systematically examined protein sequence variation, both by scanning gp120 amino acid sequences for signatures and by specifically analyzing linear domains previously identified to be potentially immunogenic. Our goal was to identify distinctive patterns in protein variability that could be considered indicative of vaccine-induced selective pressure among the infected vaccine recipients. However, it should be emphasized that with small sample sizes, moderate effects will not generally provide very low P values. Furthermore, these statistical tests were done in the context of an open-ended exploratory data analysis, and multiple tests confound the interpretation of borderline significance estimates. Thus, the statistical measures used here provide the basis for a ranking system rather than a reliable indication of selection.
Given these limitations, we identified the most-distinctive amino acid positions and motifs in gp120 from sequences obtained from infected vaccine recipients relative to the controls. Once a region or site was identified as being among the most distinctive, we used other biological and statistical considerations to evaluate the relevance of the site, some of which are summarized below. For example, the most variable motifs were no more unusual than could be found by analysis of randomly selected sequences. Second, sites that appeared to be distinctive among vaccine recipients compared to the control group were not necessarily distinctive when compared to the B subtype sequences from the recent Los Alamos Human Retroviruses and AIDS Database. Third, the potentially interesting sites or motifs identified were specific for viruses obtained from either the set of MN rgp120 vaccinees or the SF2 rgp120 vaccinees; there was no concordance between regions that were distinctive for the two sets. Finally, the sequence analysis results need to be considered in the context of the neutralizing-antibody assays: vaccinee sera collected prior to infection did not neutralize primary isolates, and strains infecting vaccinees were not particularly resistant to the effects of potent neutralizing MAbs. Taken together, these factors argue against specific vaccine-induced selection pressures.
The V3 amino acid sequences of the strains isolated from the MN rgp120 recipients did not precisely match that of MN rgp120, and MN has a motif at the tip of the V3 loop (GPGRAFY) that is similar to the United States’ B subtype consensus sequence (90, 118). Additionally, when using an amino acid substitution matrix that estimates protein sequence similarities based on the alignments of the antigenic regions in V3, a trend toward greater distance from the vaccine strain among the viral sequences from MN vaccine recipients was observed. These observations should be qualified. Only the viruses obtained from MN rgp120 vaccinees showed greater variability in this region, and in particular the association was found only in patients given three or more vaccinations prior to infection. If one considered the SF2 cases, which share the GPGRAF motif at the tip of the V3 loop with MN, or the full set of MN vaccinees, greater-than-expected variability in the V3 motifs was not evident. The MN rgp120 vaccinees whom had only two vaccinations prior to infection had detectable anti-V3 antibody prior to infection, while three of those who received three or more vaccinations prior to infection did not (57). Thus, the divergence from the MN sequence in the V3 regions of some vaccinees is quite possibly attributable purely to chance. It should also be noted that MN and SF-2 are TCLA strains isolated in 1983 to 1984, so the sequence divergence from them in a variable region is an inevitable consequence of more than a decade of evolution of HIV-1 in vivo combined with the consequence of selection in cell lines in vitro.
In the context of analyzing potential vaccine-induced selection pressures, it is also important to point out that many recent studies concerning the V3 loop have brought its relevance to vaccine protection into question. First, there is evidence that while antibodies to the V3 region can potently neutralize HIV-1 TCLA strains, they are not of paramount importance for neutralization of primary strains (96, 98, 110, 148). Second, HIV-1 V3 sequences are continually diversifying, so the proportion of contemporary strains in the United States that have a V3 sequence closely matching the MN V3 sequence is inexorably diminishing, and most individuals, after primary infection, carry a virus with detectable variation in this region (78, 118). A particularly graphic example of increasing divergence in the V3 loop crown was provided by a recent study of virus strains circulating in Memphis, Tenn. (128). Of viruses collected early in 1993, 78% of the sequenced strains contained the GPGRAF motif, while approximately 1 year later, this proportion had decreased to 22%, with a wide spectrum of motifs being found instead. This degree of sequence divergence, in the absence of any putative vaccine-induced selection pressure, should be recalled when considering the sequences of the vaccine and control isolates (Fig. 4). Third, the GPGRAFY motif is not common globally (13). Fourth, neutralization serotype studies (97, 114, 121, 152) all indicate that V3 antibodies do not contribute to cross-neutralization in any dominant sense. Thus, it seems unlikely that the rgp120 vaccines have exerted selection pressure on the mixtures of strains to which the vaccinees may have been exposed such that only strains with a V3 loop significantly divergent from that of the immunogen were transmitted.
The purpose of our study was not to determine the efficacy of the MN and SF-2 rgp120 vaccines, and the number of cases of infected vaccinees that we have studied to date is too small to draw conclusions about the efficacy of these immunogens. We note, however, that the distribution of infected individuals among the MN rgp120, SF-2 rgp120, and placebo groups was similar to that in the test population as a whole (Table 1). Furthermore, the total number of infections of study subjects enrolled in the 201 cohort represents an annual infection rate of about 2.0 cases per 100 person-years, which is not unexpected for a high-risk cohort in the United States. We were unable to obtain evidence that rgp120 vaccination has had any significant impact on the in vitro or in vivo characteristics of the infecting HIV-1 strains. More effort needs to be focused on identifying the forms of immunogens that best present neutralizing antibody and CTL epitopes to the human immune system, defining the kinetics and magnitude of responses needed for protection, and determining how immunogens can best be formulated to induce protective immune responses, rather than on further evaluation of the rgp120 and rgp160 vaccines.
ACKNOWLEDGMENTS
We thank the AVEG for conducting the MN and SF-2 rgp120 clinical trials and for the provision of samples. We thank W. Weibull (TORCH Study), J. Phair, A. Saah, R. Detels, C. Rinaldo, Jr., A. Muñoz, L. Jacobson (all of MACS), S. Buchbinder (San Francisco City Clinic Cohort), D. Vlahov (ALIVE study), J. Geodert (National Cancer Institute), and G. Woody (Philadelphia VA HIVNET study) for providing data and specimens for control subjects. We are grateful to Genentech, Inc., and Chiron, Inc., for supplying samples of the rgp120 immunogens. We thank Hermann Katinger, Dennis Burton, George Lewis, and Paul Maddon for the gifts of MAbs and CD4IgG2. We also thank the staff at each CHIP and AVEG site that contributed to this study: K. Sheridan, A. Pomales, J. Leu, S.-W. Poon, and J. Binley of The Aaron Diamond AIDS Research Center of The Rockefeller University; A. Ploss, B. Hertzfeld, P. Otto, S. Wu, and T. Myers of Northwestern University Medical School; D. Wolpert, C. Macken, J. Bingham, and A. Halpern of Los Alamos National Laboratory; N. Jones and A. Shea of Harvard University School of Medicine; G. Shaw, P. Smith, M. Mulligan, S. J. Prince, R. Kulhavy, R. Casteel, S. Ghosh, Z. Moldoveanu, S. Duncan, M. Smith, J. Decker, B. Lambert, S. Campbell-Hill, S. Morrison, B. Jian, L. Li, M. Mosteller-Barnum, K. Agnew, L. Castell, and H. Nguyen of The University of Alabama at Birmingham School of Medicine; P. Wright, P. Spearman, D. Karzon, M. A. Harbison, L. Wagner, K. Rybzyck, K. Crumbo, M. Braeuner, D. Owens, G. Rees, F. Robinson, L. Horton, I. Kuli-Zade, R. Smith, R. Cornell, and J. Keltner of Vanderbilt University School of Medicine; G. Gorse, R. Belshe, D. Kennedy, S. Frey, H. Isreal, C. Berry, B. Reed, T. Spitz, T. Pacatti, G. Patel, M. Mandava, T. Grant, and K. Feurer of St. Louis University School of Medicine; J. Lambert, D. Schwartz, C. Hilton, A. Funkhouser, and M. Johnson of Johns Hopkins University School of Medicine; M. Keefer, T. Evans, W. Bonnez, R. Reichman, L. Demeter, S. Erb, M. A. Pugliese, and J. Nichols of the University of Rochester School of Medicine and Dentistry; L. Corey, M. J. McElrath, D. Berger, H. Stacey, and L. Burke of University of Washington at Seattle School of Medicine; T. Matthews, D. Montefiori, K. Weinhold, C. McDanal, T. Greenwell, D. Davison, D. Woodford, T. Harding, A. Pilgrim, and A. Gaitin of Duke University Medical Center; P. Fast, A. Schultz, J. McNamara, B. Savarese, S. Wescott, M. C. Walker, B. Mathieson, W. Rida, D. Lawrence, and N. Ketter of the National Institute of Allergy and Infectious Diseases; and P. Barr, M. Wolff, D. Stablein, C. Smith, D. Brown, N. Lomax, and T. Voss of EMMES Corporation.
This work was supported by the National Institutes of Health under grant AI45218, the Correlates of HIV-1 Immune Protection Contract.
REFERENCES
- 1.Allaway G P, Davis-Bruno K L, Beaudry G A, Garcia E B, Wong E L, Ryder A M, Hasel K W, Gauduin M-C, Koup R A, McDougal J S, Maddon P J. Expression and characterization of CD4-IgG2, a novel heterotetramer which neutralizes primary HIV-1 isolates. AIDS Res Hum Retroviruses. 1995;11:533–540. doi: 10.1089/aid.1995.11.533. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S F, Gish W, Miller W, Myers E W. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Artenstein A W, VanCott T C, Sitz K V, Robb M L, Wagner K F, Veit S C D, Rogers A F, Garner R P, Byron J W, Burnett P R, Birx D L. Mucosal immune responses in four distinct compartments of women infected with human immunodeficiency virus type 1: a comparison by site and correlation with clinical information. J Infect Dis. 1997;175:265–271. doi: 10.1093/infdis/175.2.265. [DOI] [PubMed] [Google Scholar]
- 4.Åsjö B, Morfeldt-Månson L, Albert J, Biberfeld G, Karlsson A, Lidman K, Fenyö E M. Replicative capacity of human immunodeficiency virus from patients with varying severity of HIV infection. Lancet. 1986;i:660–662. [PubMed] [Google Scholar]
- 5.Baenziger J, Liu Y, Walker C. Proceedings of the Seventh Annual Meeting of the National Cooperative Vaccine Development Groups for AIDS, Reston, Va. 1994. Cytotoxic T lymphocyte (CTL) responses in baboons and humans immunized with a recombinant HIV-1 gp120 vaccine; p. 88. [Google Scholar]
- 6.Belshe R B, Graham B S, Keefer M C, Gorse G J, Wright P, Dolin R, Matthews T, Weinhold K, Bolognesi D P, Sposto R, Stablein D M, Twaddell T, Berman P W, Gregory T, Izu A E, Walker M C, Fast P for the NIAID AIDS Vaccine Clinical Trials Network. Neutralizing antibodies to HIV-1 in seronegative volunteers immunized with recombinant gp120 from the MN strain of HIV-1. JAMA. 1994;272:475–480. doi: 10.1001/jama.272.6.475. [DOI] [PubMed] [Google Scholar]
- 7.Berman P W, Gregory T J, Riddle L, Nakamura G R, Champe M A, Porter J P, Wurm F M, Hershberg R D, Cobb E K, Eichberg J W. Protection of chimpanzees from infection by HIV-1 after vaccination with recombinant glycoprotein gp120 but not gp160. Nature. 1990;345:622–625. doi: 10.1038/345622a0. [DOI] [PubMed] [Google Scholar]
- 8.Berman P W, Groopman J E, Gregory T, Clapham P R, Weiss R A, Ferriani R, Riddle L, Shimasaki C, Lucas C, Lasky L A, Eichberg J W. Human immunodeficiency virus type 1 challenge of chimpanzees immunized with recombinant envelope glycoprotein gp120. Proc Natl Acad Sci USA. 1988;85:5200–5204. doi: 10.1073/pnas.85.14.5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berman P W, Matthews T J, Riddle L, Champe M, Hobbs M R, Nakamura G R, Mercer J, Eastman D J, Lucas C, Langlois A J, Wurm F M, Gregory T J. Neutralization of multiple laboratory and clinical isolates of human immunodeficiency virus type 1 (HIV-1) by antisera raised against gp120 from the MN isolate of HIV-1. J Virol. 1992;66:4464–4469. doi: 10.1128/jvi.66.7.4464-4469.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berman P W, Murthy K K, Wrin T, Vennari J C, Cobb E K, Eastman D J, Champe M, Nakamura G R, Davison D, Powell M F, Bussiere J, Francis D P, Matthews T, Gregory T J, Obijeski J F. Protection of MN rgp120-immunized chimpanzees from heterologous infection with a primary isolate of human immunodeficiency virus type 1. J Infect Dis. 1996;173:52–59. doi: 10.1093/infdis/173.1.52. [DOI] [PubMed] [Google Scholar]
- 11.Berman P W, Riddle L, Nakamura G, Haffar O K, Nunes W M, Skehel P, Byrn R, Groopman J, Matthews T, Gregory T. Expression and immunogenicity of the extracellular domain of the human immunodeficiency virus type 1 envelope glycoprotein, gp160. J Virol. 1989;63:3489–3498. doi: 10.1128/jvi.63.8.3489-3498.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binley J M, Klasse P J, Cao Y, Jones I, Markowitz M, Ho D D, Moore J P. Differential regulation of the antibody responses to Gag and Env proteins of human immunodeficiency virus type 1. J Virol. 1997;71:2799–2809. doi: 10.1128/jvi.71.4.2799-2809.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blouin J C, Guzman E A, Foley B. Global variation in the HIV-1 V3 region. In: Myers G, Korber B, Foley B, Jeang K-T, Mellors J W, Wain-Hobson S, editors. Human retroviruses and AIDS compendium. III. Los Alamos, N.Mex: Los Alamos National Laboratory; 1996. pp. 29–41. [Google Scholar]
- 14.Bolognesi D P, Corey L, Vermund S H, Hoth D F. HIV vaccine development: a progress report. Ann Intern Med. 1994;8:603–611. doi: 10.7326/0003-4819-121-8-199410150-00008. [DOI] [PubMed] [Google Scholar]
- 15.Bruck C, Thiriart C, Fabry L, Francotte M, Pala P, Van Opstal O, Culp J, Rosenberg M, De Wilde M, Heidt P, Heeney J. HIV-1 envelope-elicited neutralizing antibody titres correlate with protection and virus load in chimpanzees. Vaccine. 1994;12:1141–1148. doi: 10.1016/0264-410x(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 16.Burton D R, Pyati J, Koduri R, Thornton G B, Sawyer L S W, Hendry R M, Dunlop N, Nara P L, Lamacchia M, Garratty E, Stiehm E R, Bryson Y J, Moore J P, Ho D D, Barbas C F., III Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science. 1994;266:1024–1027. doi: 10.1126/science.7973652. [DOI] [PubMed] [Google Scholar]
- 17.Cao Y, Qin L, Zhang L, Safrit J, Ho D D. Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N Engl J Med. 1995;332:201–208. doi: 10.1056/NEJM199501263320401. [DOI] [PubMed] [Google Scholar]
- 18.Capon D J, Chamow S M, Mordenti J, Marsters S A, Gregory T, Mitsuya H, Byrn R A, Lucas C, Wurm F M, Groopman J E, Smith D H. Designing CD4 immunoadhesins for AIDS therapy. Nature. 1989;337:525–531. doi: 10.1038/337525a0. [DOI] [PubMed] [Google Scholar]
- 19.Carmichael A, Jin X, Sissons P, Borysiewicz L. Quantitative analysis of the human immunodeficiency virus type 1 (HIV-1)-specific cytotoxic T lymphocyte (CTL) response at different stages of HIV-1 infection: differential CTL responses to HIV-1 and Epstein-Barr virus in late disease. J Exp Med. 1993;177:249–256. doi: 10.1084/jem.177.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casteel L R, Allen S, Kulhavy R, Zulu I, Omara J, Mestecky J. Proceedings of the 9th Annual Meeting of the National Cooperative Vaccine Development Groups for AIDS. 1997. HIV-specific antibodies in external secretions of seronegative and seropositive high-risk individuals, abstr. 73; p. 178. [Google Scholar]
- 21.Chakrabarti L, Cumont M-C, Montagnier L, Hurtrel B. Variable course of primary simian immunodeficiency virus infection in lymph nodes: relationship to disease progression. J Virol. 1994;68:6634–6642. doi: 10.1128/jvi.68.10.6634-6643.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chesney M A, Lurie P, Coates T J. Strategies for addressing the social and behavioral challenges of prophylactic HIV vaccine trials. J Acquired Immune Defic Syndr. 1995;9:30–35. [PubMed] [Google Scholar]
- 23.Conley A J, Kessler II J A, Boots L J, Tung J-S, Arnold B A, Keller P M, Shaw A R, Emini E A. Neutralization of divergent human immunodeficiency virus type 1 variants and primary isolates by IAM-41-2F5, an anti-gp41 human monoclonal antibody. Proc Natl Acad Sci USA. 1994;91:3348–3352. doi: 10.1073/pnas.91.8.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conley A J, Kessler II J A, Boots L J, McKenna P M, Schleif W A, Emini E A, Mark III G E, Katinger H, Cobb E K, Lunceford S M, Rouse S R, Murthy K K. The consequence of passive administration of an anti-human immunodeficiency virus type 1 neutralizing monoclonal antibody before challenge of chimpanzees with a primary virus isolate. J Virol. 1996;70:6751–6758. doi: 10.1128/jvi.70.10.6751-6758.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Connor R I, Ho D D. Human immunodeficiency virus type 1 variants with increased replicative capacity develop during the asymptomatic stage before disease progression. J Virol. 1994;68:4400–4408. doi: 10.1128/jvi.68.7.4400-4408.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connor R I, Sheridan K E, Ceradini D, Choe S, Landau N R. Change in coreceptor use correlates with disease progression in HIV-1 infected individuals. J Exp Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daar E S, Li X L, Moudgil T, Ho D D. High concentrations of recombinant soluble CD4 are required to neutralize primary human immunodeficiency virus type 1 isolates. Proc Natl Acad Sci USA. 1990;87:6574–6578. doi: 10.1073/pnas.87.17.6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daniel M D, Mazzara G P, Simon M A, Sehgal P K, Kodama T, Panicali D L, Desrosiers R C. High-titer immune responses elicited by recombinant vaccinia virus priming and particle boosting are ineffective in preventing virulent SIV infection. AIDS Res Hum Retroviruses. 1994;10:839–851. doi: 10.1089/aid.1994.10.839. [DOI] [PubMed] [Google Scholar]
- 29.Dean M, Carrington M, Winkler C, Huttley G A, Smith M W, Allikmets R, Goedert J J, Buchbinder S P, Vittinghoff E, Gomperts E, Donfield S, Vlahov D, Kaslow R, Saah A, Rinaldo C, Detels R Hemophilia Growth and Development Study; Multicenter AIDS Cohort Study; Multicenter Hemophilia Cohort Study; San Francisco City Cohort; ALIVE Study. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion of the CKR5 structural allele. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 30.Deng H K, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton R E, Hill C M, Peiper S C, Schall T J, Littman D R, Landau N R. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 31.de St. Groth F. The evaluation of limiting dilution assays. J Immunol Methods. 1982;49:R11–R23. doi: 10.1016/0022-1759(82)90269-1. [DOI] [PubMed] [Google Scholar]
- 32.Doe B, Liu Y, Steimer K S, Walker C M. Priming of cytotoxic T lymphocyte responses with recombinant HIV envelope proteins in murine and primate models. In: Girard M, Valette L, editors. Retroviruses of human AIDS and related animal diseases, 7th Colloque des Cent Gardes. Marnes-La-Coquette, France: La Fondiation Marcel Mèrieau; 1994. pp. 321–325. [Google Scholar]
- 33.Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 34.D’Souza M P, Harden V A. Chemokines and HIV-1 second receptors. Confluence of two fields generates optimism in AIDS research. Nat Med. 1996;2:1293–1300. doi: 10.1038/nm1296-1293. [DOI] [PubMed] [Google Scholar]
- 35.D’Souza, M. P., D. Livnat, J. A. Bradac, S. H. Bridges, the AIDS Clinical Trials Group Antibody Selection Working Group, and collaborating investigators. Evaluation of monoclonal antibodies to human immunodeficiency virus type 1 primary isolates by neutralization assays: performance criteria for selecting candidate antibodies for clinical trials. J. Infect. Dis., in press. [DOI] [PubMed]
- 36.Eddy S, Mitchison G, Durbin R. Maximum discrimination hidden Markov models of sequence consensus. J Comp Biol. 1995;2:9–23. doi: 10.1089/cmb.1995.2.9. [DOI] [PubMed] [Google Scholar]
- 37.El-Amed Z, Murthy K K, Higgins K, Cobb E K, Haigwood N L, Levy J A, Steimer K S. Resistance of chimpanzees immunized with recombinant gp120 SF-2 to challenge by HIV-1 SF-2. AIDS. 1995;9:1313–1322. doi: 10.1097/00002030-199512000-00003. [DOI] [PubMed] [Google Scholar]
- 38.Emini E A, Schlief W A, Nunberg J H, Conley A J, Eda Y, Tokiyoshi S, Putney S D, Matsushita S, Cobb K E, Jett C M, Eichberg J M, Murthy K K. Prevention of HIV-1 infection in chimpanzees by gp120 V3 domain-specific monoclonal antibody. Nature. 1991;355:728–730. doi: 10.1038/355728a0. [DOI] [PubMed] [Google Scholar]
- 39.Faulkner D V, Jurka A. Multiple aligned sequence editor (MASE) Trends Biochem Sci. 1988;13:321–326. doi: 10.1016/0968-0004(88)90129-6. [DOI] [PubMed] [Google Scholar]
- 40.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 41.Felsenstein J. Confidence on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 42.Felsenstein J. Phylogenies from molecular sequences: inference and reliability. Annu Rev Genet. 1988;22:521–565. doi: 10.1146/annurev.ge.22.120188.002513. [DOI] [PubMed] [Google Scholar]
- 43.Felsenstein J. PHYLIP—phylogenetic inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 44.Felsenstein J. PHYLIP (phylogenetic inference package) version 3.4. Seattle: Department of Genetics, University of Washington; 1993. . (Distributed by the author.) [Google Scholar]
- 45.Ferbas J, Daar E S, Grovit-Ferbas K, Lech W J, Detels R, Giorgi J V, Kaplan A H. Rapid evolution of human immunodeficiency virus strains with increased replicative capacity during the seronegative window of primary infection. J Virol. 1996;70:7285–7289. doi: 10.1128/jvi.70.10.7285-7289.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fiore J R, Björndal A, Peipke K A, di Stefano M, Angarano G, Pastore G, Gaines H, Fenyö E M, Albert J. The biological phenotype of HIV-1 is usually retained after sexual transmission. Virology. 1994;204:297–303. doi: 10.1006/viro.1994.1534. [DOI] [PubMed] [Google Scholar]
- 47.Fouchier R A M, Groenink M, Kootstra N A, Tersmette M, Huisman H G, Miedema F, Schuitemaker H. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J Virol. 1992;66:3183–3187. doi: 10.1128/jvi.66.5.3183-3187.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fouts T R, Binley J M, Trkola A, Robinson J E, Moore J P. Neutralization of the human immunodeficiency virus type 1 primary isolate JR-FL by human monoclonal antibodies correlates with antibody binding to the oligomeric form of the envelope glycoprotein complex. J Virol. 1997;71:2779–2785. doi: 10.1128/jvi.71.4.2779-2785.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fultz P N, Nara P, Barre-Sinoussi F, Chaput A, Greenberg M L, Muchmore E, Kieny M-P, Girard M. Vaccine protection of chimpanzees against challenge with HIV-1-infected peripheral blood mononuclear cells. Science. 1992;256:1687–1690. doi: 10.1126/science.256.5064.1687. [DOI] [PubMed] [Google Scholar]
- 50.Fung M S C, Sun C R Y, Gordon W L, Liou R-S, Chang T W, Sun W N C, Daar E S, Ho D D. Identification and characterization of a neutralization site within the second variable region of human immunodeficiency virus type 1 gp120. J Virol. 1992;66:848–856. doi: 10.1128/jvi.66.2.848-856.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao F, Morrison S G, Robertson D L, Thornton C L, Craig S, Karlsson G, Sodroski J, Morgado M, Galvao-Castro B, von Briesen H, Beddows S, Weber J, Sharp P M, Shaw G M, Hahn B H the WHO and NIAID Networks for HIV Isolation and Characterization. Molecular cloning and analysis of functional envelope genes from human immunodeficiency virus type 1 sequence subtypes A through G. J Virol. 1996;70:1651–1667. doi: 10.1128/jvi.70.3.1651-1667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia F, Vidal C, Gatell J M, Miro J M, Soriano A, Pumarola T. Viral load in asymptomatic patients with CD4+ lymphocytes above 500 × 106/l. AIDS. 1997;11:53–57. doi: 10.1097/00002030-199701000-00008. [DOI] [PubMed] [Google Scholar]
- 53.Girard M, Kieny M-P, Pinter A, Barré-Sinoussi F, Nara P, Kolbe H, Kusumi K, Chaput A, Reinhart T, Muchmore E, Roneo J, Kaczorek M, Gomard E, Gluckman J-C, Fultz P N. Immunization of chimpanzees confers protection against challenge with human immunodeficiency virus. Proc Natl Acad Sci USA. 1991;88:542–546. doi: 10.1073/pnas.88.2.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Girard M, Meignier B, Barré-Sinoussi F, Kieny M-P, Matthews T, Muchmore E, Nara P L, Wei Q, Rimsky L, Weinhold K, Fultz P N. Vaccine-induced protection of chimpanzees against infection by a heterologous human immunodeficiency virus type 1. J Virol. 1995;69:6239–6248. doi: 10.1128/jvi.69.10.6239-6248.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gorny M K, Conley A J, Karwowska S, Buchbinder A, Xu J-Y, Emini E A, Koenig S, Zolla-Pazner S. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J Virol. 1992;66:7538–7542. doi: 10.1128/jvi.66.12.7538-7542.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gorse G J, Frey S E, Patel G, Newman F K, Belshe R B the NIAID AIDS Vaccine Clinical Trials Network. Vaccine-induced antibodies to native and recombinant human immunodeficiency virus type 1 envelope glycoproteins. Vaccine. 1994;12:912–918. doi: 10.1016/0264-410x(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 57.Graham, B. S., M. J. McElrath, R. I. Connor, D. H. Schwartz, G. J. Gorse, M. C. Keefer, M. J. Mulligan, T. J. Matthews, S. M. Wolinsky, D. C. Montefiori, S. H. Vermund, J. S. Lambert, L. Corey, R. B. Belshe, R. Dolin, P. F. Wright, B. T. Korber, M. C. Wolff, P. E. Fast, AIDS Vaccine Evaluation Group, and Correlates of HIV Immune Protection Group. Analysis of intercurrent human immunodeficiency virus type 1 infections in phase I and II trials of candidate AIDS vaccines. J. Infect. Dis., in press. [DOI] [PubMed]
- 58.Graham B S, Wright P F. Candidate AIDS vaccines. N Engl J Med. 1995;333:1331–1339. doi: 10.1056/NEJM199511163332007. [DOI] [PubMed] [Google Scholar]
- 59.Graham B S, Keefer M C, McElrath M J, Gorse G J, Schwartz D H, Weinhold K, Matthews T J, Esterlitz J R, Sinangil F, Fast P E the NIAID AIDS Vaccine Evaluation Group. Safety and immunogenicity of a candidate HIV-1 vaccine in health adults: recombinant glycoprotein (rgp120) Ann Intern Med. 1996;125:270–279. doi: 10.7326/0003-4819-125-4-199608150-00003. [DOI] [PubMed] [Google Scholar]
- 60.Haynes B F. Scientific and social issues of human immunodeficiency virus vaccine development. Science. 1993;260:1279–1286. doi: 10.1126/science.8493572. [DOI] [PubMed] [Google Scholar]
- 61.Henikoff S, Henikoff J G. Performance evaluation of amino acid substitution matrices. Proteins Struct Funct Genet. 1993;17:49–61. doi: 10.1002/prot.340170108. [DOI] [PubMed] [Google Scholar]
- 62.Henrard D R, Daar E, Farzadegan H, Clark S J, Phillips J, Shaw G M, Busch M P. Virologic and immunologic characterization of symptomatic and asymptomatic primary HIV-1 infection. J Acquired Immune Defic Syndr. 1995;9:305–310. [PubMed] [Google Scholar]
- 63.Hillis D M, Bull J J. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol. 1993;42:182–189. [Google Scholar]
- 64.Ho D D, Fung M S C, Cao Y, Li X L, Sun C, Chang T W, Sun N-C. Another discontinuous epitope on glycoprotein gp120 that is important in human immunodeficiency virus type 1 neutralization is identified by a monoclonal antibody. Proc Natl Acad Sci USA. 1991;88:8949–8952. doi: 10.1073/pnas.88.20.8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang Y, Paxton W A, Wolinsky S M, Neumann A U, Zhang L, He T, Kang S, Ceradini D, Jin Z, Yazdanbaksh K, Kuntsman K, Erickson D, Landau N R, Phair J, Ho D D, Koup R A. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2:1240–1243. doi: 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- 66.Issel C J, Horohov D W, Lea D F, Adams W V, Jr, Hagius S D, McManus J M, Allison A C, Montelaro R C. Efficacy of inactivated whole-virus and subunit vaccines in preventing infection and disease caused by equine infectious anemia virus. J Virol. 1992;66:3398–3408. doi: 10.1128/jvi.66.6.3398-3408.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jurrians S, van Gemen B, Weverling G J, van Strup D, Nara P, Coutinho R, Koot M, Schuitemaker H, Goudsmit J. The natural history of HIV-1 infection: virus load and virus phenotype in determinants of clinical course. Virology. 1994;204:223–233. doi: 10.1006/viro.1994.1526. [DOI] [PubMed] [Google Scholar]
- 68.Kahn J O, Sinangil F, Baenziger J, Mumar N, Wynne D, Coleman R L, Steimer K S, Dekker C L, Chernoff D. Clinical and immunologic responses to human immunodeficiency virus (HIV) type 1 SF2 gp120 subunit vaccine combined with MF59 adjuvant with or without muramyl tripeptide dipalmitoyl phosphatidylethanolamine in non-HIV-infected human volunteers. J Infect Dis. 1994;170:1288–1291. doi: 10.1093/infdis/170.5.1288. [DOI] [PubMed] [Google Scholar]
- 69.Kahn J O, Steimer K S, Baenziger J, Duliege A-M, Feinberg M, Elbeik T, Chesney M, Mumar N, Chernoff D, Sinangil F. Clinical, immunologic, and virologic observations related to human immunodeficiency virus (HIV) type 1 infection in a volunteer in an HIV-1 vaccine clinical trial. J Infect Dis. 1995;171:1343–1347. doi: 10.1093/infdis/171.5.1343. [DOI] [PubMed] [Google Scholar]
- 70.Keet I P M, Krijnen P, Koot M, Lange J M A, Miedema F, Goudsmit J, Coutinho R A. Predictors of rapid progression to AIDS in HIV-1 seroconvertors. AIDS. 1993;7:51–57. doi: 10.1097/00002030-199301000-00008. [DOI] [PubMed] [Google Scholar]
- 71.Kent, K. A., and J. Robinson. 1996. Antigenic determinants on HIV-1 envelope glycoproteins: a Dickens of a time with an Oligomer Twist. AIDS 10(Suppl. A):S107–S114. [PubMed]
- 72.Khadir A, Coutlee F, Saint-Antoine P, Olivier C, Voyer H, Kessous-Elbaz A. Clinical evaluation of Amplicor HIV-1 test for detection of human immunodeficiency virus type 1 proviral DNA in peripheral blood mononuclear cells. J Acquired Immune Defic Syndr. 1995;9:257–263. [PubMed] [Google Scholar]
- 73.Klasse P J. Physico-chemical analyses of the humoral immune response to HIV-1: quantification of antibodies, their binding to viral antigens and neutralization of viral infectivity. In: Korber B, Brander C, Moore J, D’Souza P, Walker B, Koup R, Haynes B, Myers G, editors. HIV molecular immunology databases. IV. Los Alamos, N.Mex: Los Alamos National Laboratory; 1996. pp. 22–49. [Google Scholar]
- 74.Koenig S, Earl P, Powell D, Pantaleo G, Merli S, Moss B, Fauci A S. Group-specific, major histocompatibility complex class I-restricted cytotoxic responses to human immunodeficiency virus 1 (HIV-1) envelope proteins by cloned peripheral blood T cells from an HIV-1-infected individual. Proc Natl Acad Sci USA. 1988;85:8638–8642. doi: 10.1073/pnas.85.22.8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Korber B, Brander C, Walker B, Koup R, Moore J P, Haynes B, Myers G. HIV molecular immunology database 1995. Los Alamos, N.Mex: Theoretical Biology and Biophysics, Los Alamos National Laboratory; 1995. [Google Scholar]
- 76.Korber, B., B. H. Hahn, and S. M. Wolinsky. Unpublished data.
- 77.Korber B, Myers G. Signature patterns analysis: a method for assessing viral sequence relatedness. AIDS Res Hum Retroviruses. 1992;8:1549–1558. doi: 10.1089/aid.1992.8.1549. [DOI] [PubMed] [Google Scholar]
- 78.Korber B, Wolinsky S, Haynes B, Kuntsman K, Levy R, Furtado M, Otto P, Myers G. HIV-1 intrapatient sequence diversity in the immunogenic V3 region. AIDS Res Hum Retroviruses. 1992;8:1461–1465. doi: 10.1089/aid.1992.8.1461. [DOI] [PubMed] [Google Scholar]
- 79.Korber B T M, Farber R M, Wolpert D H, Lapedes A S. Covariation of mutations in the V3 loops of human immunodeficiency virus type 1 envelope protein: an information theoretical analysis. Proc Natl Acad Sci USA. 1993;90:2176–2180. doi: 10.1073/pnas.90.15.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Korber B T M, Kunstman K J, Patterson B K, Furtado M, McEvilly M M, Levy R, Wolinsky S M. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3 region of the envelope protein of brain-derived sequences. J Virol. 1994;68:7467–7481. doi: 10.1128/jvi.68.11.7467-7481.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korber B T M, Learn G, Mullins J I, Hahn B H, Wolinsky S M. Protecting HIV databases. Nature. 1995;378:242–244. doi: 10.1038/378242a0. [DOI] [PubMed] [Google Scholar]
- 82.Korber B T M, MacInnes K, Smith R F, Myers G. Mutational trends in V3 loop protein sequences observed in different genetic lineages of human immunodeficiency virus type 1. J Virol. 1994;68:6730–6744. doi: 10.1128/jvi.68.10.6730-6744.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koup R A, Safrit J T, Cao Y, Andrews C A, McLeod G, Borkowsky W, Farthing C, Ho D D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koup R A, Pikora C A, Luzuriaga K, Brettler D B, Day E S, Mazzara G P, Sullivan J L. Limiting dilution analysis of cytotoxic T lymphocytes to human immunodeficiency virus gag antigens in infected persons: in vitro quantitation of effector cell populations with p17 and p24 specificities. J Exp Med. 1991;174:1593–1600. doi: 10.1084/jem.174.6.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kozak S L, Platt E J, Madani N, Ferro F E, Jr, Peden K, Kabat D. CD4, CXCR-4, and CCR-5 dependencies for infection by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J Virol. 1997;71:873–882. doi: 10.1128/jvi.71.2.873-882.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kozlowski P A, Jackson S. Serum IgG subclasses and molecular forms in HIV infection: selective increase in monomer and apparent restriction of the antibody response to IgA1 antibodies mainly directed at env glycoproteins. AIDS Res Hum Retroviruses. 1992;8:1773–1780. doi: 10.1089/aid.1992.8.1773. [DOI] [PubMed] [Google Scholar]
- 87.Kutteh W H, Prince S J, Hammond K R, Kutteh C C, Mestecky J. Variations in immunoglobulins and IgA subclasses of human uterine cervical secretions around the time of ovulation. Clin Exp Immunol. 1996;104:538–542. doi: 10.1046/j.1365-2249.1996.36742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kwok S, Higuchi R. Avoiding false positives with PCR. Nature. 1989;339:237–238. doi: 10.1038/339237a0. [DOI] [PubMed] [Google Scholar]
- 89.Lafeuillade A, Tamalet C, de Micco P, Thiebaut C. Virological analysis of primary HIV-1 infection with rapid CD4 depletion. AIDS. 1995;9:1380–1381. doi: 10.1097/00002030-199512000-00015. [DOI] [PubMed] [Google Scholar]
- 90.LaRosa G J, Davide J P, Weinhold K, Waterbury J A, Profy A T, Lewis J A, Langlois A J, Dreesman G R, Boswell R N, Shadduck P, Holley C H, Karplus M, Bolognesi D P, Matthews T J, Emini E A, Putney S D. Conserved sequence and structural elements in the HIV-1 principal neutralizing determinant. Science. 1990;249:932–935. doi: 10.1126/science.2392685. [DOI] [PubMed] [Google Scholar]
- 91.Learn G H, Jr, Korber B T M, Foley B, Hahn B H, Wolinsky S M, Mullins J I. Maintaining the integrity of human immunodeficiency virus sequence databases. J Virol. 1996;70:5720–5730. doi: 10.1128/jvi.70.8.5720-5730.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee T-H, Sheppard H W, Reis M, Dondero D, Osmond D, Busch M P. Circulating HIV-1 infected cell burden from seroconversion to AIDS: importance of post seroconversion viral load on disease course. J Acquired Immune Defic Syndr. 1994;7:381–388. [PubMed] [Google Scholar]
- 93.Leonard C K, Spellman M W, Riddle L, Harris R J, Thomas J N, Gregory T J. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J Biol Chem. 1990;265:10373–10382. [PubMed] [Google Scholar]
- 94.Lurie P, Bishaw M, Chesney M A, Cooke M, Lemos Fernandes M E, Hearst N, Katongole-Mbidde E, Koetsawang S, Lindan C P, Mandel J, Mkloyi M, Coates T J. Ethical, behavioral, and social aspects of HIV vaccine trials in developing countries. JAMA. 1994;271:295–301. [PubMed] [Google Scholar]
- 95.Mascola J R, McNeil J G, Burke D S. AIDS vaccines. Are we ready for human efficacy trials? JAMA. 1994;272:488–489. doi: 10.1001/jama.272.6.488. [DOI] [PubMed] [Google Scholar]
- 96.Mascola J R, Snyder S W, Weislow O S, Belay S M, Belshe R B, Schwartz D H, Clements M L, Dolin R, Graham B S, Gorse G J, Keefer M C, McElrath M J, Walker M C, Wagner K F, McNeil J G, McCutchan F E, Burke D S The National Institute of Allergy and Infectious Diseases AIDS Vaccine Evaluation Group. Immunization with envelope subunit vaccine products elicits neutralizing antibodies against laboratory-adapted but not primary isolates of human immunodeficiency virus type 1. J Infect Dis. 1996;173:340–348. doi: 10.1093/infdis/173.2.340. [DOI] [PubMed] [Google Scholar]
- 97.Mascola J R, Louder M K, Surman S R, Vancott T C, Yu X F, Bradac J, Porter K R, Nelson K E, Girard M, McNeil J G, McCutchan F E, Birx D L, Burke D S. Human immunodeficiency virus type 1 neutralizing antibody serotyping using serum pools and an infectivity reduction assay. AIDS Res Hum Retroviruses. 1996;12:1319–1328. doi: 10.1089/aid.1996.12.1319. [DOI] [PubMed] [Google Scholar]
- 98.Matthews T J. Dilemma of neutralization resistance of HIV-1 field isolates and vaccine development. AIDS Res Hum Retroviruses. 1994;10:631–632. doi: 10.1089/aid.1994.10.631. [DOI] [PubMed] [Google Scholar]
- 99.McElrath M J, Corey L, Clements M L, Belshe R, Dolin R, Graham B, Fast P, Matthews T, Francis D, Duliege A M the NIAID AIDS Vaccine Trials Network. Abstracts of the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy. Washington, D.C: American Society for Microbiology; 1994. A phase II HIV prophylactic vaccine trial in seronegative volunteers: expanded safety and immunogenicity evaluation of two recombinant gp120 vaccines, abstr. I241; p. 238. [Google Scholar]
- 100.McElrath M J, Corey L, Greenberg P D, Matthews T J, Montefiori D C, Rowen L, Hood L, Mullins J. Human immunodeficiency virus type 1 infection despite prior immunization with a recombinant envelope vaccine regimen. Proc Natl Acad Sci USA. 1996;93:3972–3977. doi: 10.1073/pnas.93.9.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McKeating J A, McKnight A, Moore J P. Differential loss of envelope glycoprotein gp120 from virions of human immunodeficiency virus type 1 isolates: effect on infectivity and neutralization. J Virol. 1991;65:852–860. doi: 10.1128/jvi.65.2.852-860.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mellors J W, Rinald C R, Jr, Gupta P, White R M, Todd J A, Kingsley L A. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 103.Mestecky J. The common mucosal immune system and current strategies for induction of immune responses in external secretions. J Clin Immunol. 1987;7:265–276. doi: 10.1007/BF00915547. [DOI] [PubMed] [Google Scholar]
- 104.Mestecky J, Kilian M. Immunoglobulin A (IgA) Methods Enzymol. 1985;116:37–75. doi: 10.1016/s0076-6879(85)16005-2. [DOI] [PubMed] [Google Scholar]
- 105.Moore J P. The reactivities of HIV-1 positive human sera with solid-phase V3 loop peptides can be poor predictors of their reactivities with V3 loops on native gp120 molecules. AIDS Res Hum Retroviruses. 1993;9:209–219. doi: 10.1089/aid.1993.9.209. [DOI] [PubMed] [Google Scholar]
- 106.Moore J P, Anderson R. The WHO and why of HIV vaccine trials. Nature. 1994;372:313–314. doi: 10.1038/372313a0. [DOI] [PubMed] [Google Scholar]
- 107.Moore J P, Cao Y, Ho D D, Koup R A. Development of the anti-gp120 antibody response during seroconversion to human immunodeficiency virus type 1. J Virol. 1994;68:5142–5155. doi: 10.1128/jvi.68.8.5142-5155.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Moore J P, Cao Y, Qing L, Sattentau Q J, Pyati J, Koduri R, Robinson J, Barbas III C F, Burton D R, Ho D D. Primary isolates of human immunodeficiency virus type 1 are relatively resistant to neutralization by monoclonal antibodies to gp120, and their neutralization is not predicted by studies with monomeric gp120. J Virol. 1995;69:101–109. doi: 10.1128/jvi.69.1.101-109.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moore J P, Ho D D. Antibodies to discontinuous or conformationally sensitive epitopes on the gp120 glycoprotein of human immunodeficiency virus type 1 are highly prevalent in sera of infected humans. J Virol. 1993;67:863–875. doi: 10.1128/jvi.67.2.863-875.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moore, J. P., and D. D. Ho. 1995. HIV-1 neutralization: the consequences of viral adaptation to growth on transformed T cells. AIDS 9(Suppl. A):S117–S136. [PubMed]
- 111.Moore J P, Sattentau Q J, Wyatt R, Sodroski J. Probing the structure of the human immunodeficiency virus surface glycoprotein gp120 with a panel of monoclonal antibodies. J Virol. 1994;68:469–484. doi: 10.1128/jvi.68.1.469-484.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moore J P, Wallace L A, Follett E A C, McKeating J A. An ELISA for antibodies to the envelope glycoprotein of divergent strains of HIV-1. AIDS. 1989;3:155–163. doi: 10.1097/00002030-198903000-00006. [DOI] [PubMed] [Google Scholar]
- 113.Moore J P, Sodroski J. Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. J Virol. 1996;70:1863–1872. doi: 10.1128/jvi.70.3.1863-1872.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Moore J P, Cao Y, Leu J, Qin L, Korber B, Ho D D. Inter- and intraclade neutralization of human immunodeficiency virus type 1: genetic clades do not correspond to neutralization serotypes but partially correspond to gp120 antigenic serotypes. J Virol. 1996;70:427–444. doi: 10.1128/jvi.70.1.427-444.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Murthy K K, Cobb E K, El-Amad Z, Ortega H, Hsueh F C, Satterfield W, Lee D R, Kalish M L, Haigwood N L, Kennedy R C, Steimer K S, Schultz A, Levy J A. Titration of a vaccine stock preparation of human immunodeficiency virus type 1 SF-2 in cultured lymphocytes and in chimpanzees. AIDS Res Hum Retroviruses. 1996;12:1341–1348. doi: 10.1089/aid.1996.12.1341. [DOI] [PubMed] [Google Scholar]
- 116.Muster T, Guinea R, Trkola A, Purtscher M, Klima A, Steindl F, Palese P, Katinger H. Cross-neutralizing antibodies against divergent human immunodeficiency virus type 1 isolates induced by the gp41 sequence ELDKWAS. J Virol. 1994;68:4031–4034. doi: 10.1128/jvi.68.6.4031-4034.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Myers G, Farmer A. Hidden Markov models and HIV analysis. J Comp Biol. 1996;2:24–29. [Google Scholar]
- 118.Myers G, Korber B T M, Berzofsky J A, Smith R F, Pavlakis G N. Human retroviruses and AIDS database 1992. Los Alamos, N.Mex: Theoretical Biology, Los Alamos National Laboratory; 1992. [Google Scholar]
- 119.Myers G, Lenroot R. HIV glycosylation and what does it portend? AIDS Res Hum Retroviruses. 1992;8:1459–1460. doi: 10.1089/aid.1992.8.1459. [DOI] [PubMed] [Google Scholar]
- 120.National Institute of Allergy and Infectious Diseases. Collection and processing of mucosal specimens. Bethesda, Md: Division of AIDS, National Institute of Allergy and Infectious Diseases; 1996. [Google Scholar]
- 121.Nyambi P N, Nkengasong J, Lewi P, Andries K, Janssens W, Fransen K, Heyndrickx L, Piot P, van der Groen G. Multivariate analysis of human immunodeficiency virus type 1 neutralization data. J Virol. 1996;70:6235–6243. doi: 10.1128/jvi.70.9.6235-6243.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O’Brien T R, Blattner W A, Waters D, Eyster E, Hilgartner H W, Cohen A R, Luban N, Hatzakis A, Aledort L M, Rosenberg P S, Miley W J, Kroner B L, Goedert J J. Serum HIV-1 RNA levels and time to development of AIDS in the Multicenter Hemophilia Cohort Study. JAMA. 1996;276:105–110. [PubMed] [Google Scholar]
- 123.O’Brien W A, Grovit-Ferbas K, Namazi A, Ovcak-Derzu S, Wang H-J, Park J, Yeramian C, Mao S-H, Zack J A. Human immunodeficiency virus type 1 replication can be increased in peripheral blood of seropositive patients after influenza vaccination. Blood. 1995;86:1082–1088. [PubMed] [Google Scholar]
- 124.Olsen G J, Matsuda H, Hagstrom R, Overbeek R. fastDNAml: a tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput Appl Biosci. 1994;10:41–48. doi: 10.1093/bioinformatics/10.1.41. [DOI] [PubMed] [Google Scholar]
- 125.Paxton W B, Coombs R W, McElrath M J, Keefer M C, Hughes J, Sinangil F, Chernoff D, Demeter L, Williams B, Corey L The National Institute of Allergy and Infectious Diseases AIDS Evaluation Group. Longitudinal analysis of quantitative virologic measures in human immunodeficiency virus-infected subjects with >400 CD4 lymphocytes: implications for applying measurements to individual patients. J Infect Dis. 1997;175:247–254. doi: 10.1093/infdis/175.2.247. [DOI] [PubMed] [Google Scholar]
- 126.Pedersen C, Lindhardt B O, Jensen B L. Clinical course of primary HIV-1 infection: consequences for subsequent course of infection. Br Med J. 1989;299:154–157. doi: 10.1136/bmj.299.6692.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Public Health Service. HIV infection in vaccine trial volunteers. Bethesda, Md: National Institute of Allergy and Infectious Diseases; 1994. [Google Scholar]
- 128.Rencher S D, Lockey T D, Slobod K S, Hurwitz J L. Drift from the GPGRAF HIV-1 envelope V3 crown sequence in a North American inner city. AIDS Res Hum Retroviruses. 1997;13:527–528. doi: 10.1089/aid.1997.13.527. [DOI] [PubMed] [Google Scholar]
- 129.Rinaldo C, Huang X-L, Fan Z, Ding M, Beltz L, Logar A, Panicali D, Mazzara G, Liebmann J, Cottrill M, Gupta P. High levels of anti-human immunodeficiency virus type 1 (HIV-1) memory cytotoxic T-lymphocyte activity and low viral load are associated with lack of disease in HIV-1-infected long-term nonprogressors. J Virol. 1995;69:5838–5842. doi: 10.1128/jvi.69.9.5838-5842.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roos M T L, Lange J M A, de Goede R E Y, Coutinho R A, Schellekens P T A, Miedema F, Tersmette M. Viral phenotype and immune response in primary human immunodeficiency virus type 1 infection. J Infect Dis. 1992;165:427–432. doi: 10.1093/infdis/165.3.427. [DOI] [PubMed] [Google Scholar]
- 131.Saag M S, Holodniy M, Kuritzkes D R, O’Brien W A, Coombs R, Poscher M E, Jacobsen D M, Shaw G M, Richman D D, Volberding P A. HIV viral load markers in clinical practice. Nat Med. 1996;2:625–629. doi: 10.1038/nm0696-625. [DOI] [PubMed] [Google Scholar]
- 132.Sattentau Q J, Moore J P. Human immunodeficiency virus type 1 neutralization is determined by epitope exposure on the glycoprotein gp120 oligomer. J Exp Med. 1995;182:185–196. doi: 10.1084/jem.182.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scandella C J, Kilpatrick J, Lidster W, Parker C, Moore J P, Moore G K, Mann K A, Brown P, Coates S, Chapman B, Masiarz R R, Steimer K S, Haigwood N L. Non-affinity purification of recombinant gp120 for use in AIDS vaccine development. AIDS Res Hum Retroviruses. 1993;9:1233–1244. doi: 10.1089/aid.1993.9.1233. [DOI] [PubMed] [Google Scholar]
- 134.Schafer M E, Rhodes M, Prince S, Michalek S M, McGhee J R. A plastic intra-oral device for the collection of human parotid saliva. J Dent Res. 1977;56:728–733. doi: 10.1177/00220345770560070401. [DOI] [PubMed] [Google Scholar]
- 135.Siebelink K H J, Tijhaar E, Huisman R C, Huisman W, de Ronde A, Darby I H, Francis M J, Rimmelzwaan G F, Osterhaus A D M E. Enhancement of feline immunodeficiency virus infection after immunization with envelope glycoprotein subunit vaccines. J Virol. 1995;69:3704–3711. doi: 10.1128/jvi.69.6.3704-3711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Simmons G, Wilkinson D, Reeves J D, Dittmar M T, Beddows S, Weber J, Carnegie G, Desselberger U, Gray P W, Weiss R A, Clapham P R. Primary, syncytium-inducing human immunodeficiency virus type 1 isolates are dual-tropic and most can use either Lestr or CCR5 as coreceptors for virus entry. J Virol. 1996;70:8355–8360. doi: 10.1128/jvi.70.12.8355-8360.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sinnico A, Fora R, Sciandra M, Lucchini A, Carameelo P, Gioannini P. Risk of developing AIDS after primary acute HIV-1 infection. J Acquired Immune Defic Syndr. 1993;6:575–581. [PubMed] [Google Scholar]
- 138.Smith R F, Smith T F. Automatic generation of primary sequence patterns from sets of related protein sequences. Proc Natl Acad Sci USA. 1990;87:118–122. doi: 10.1073/pnas.87.1.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Smith R F, Smith T F. Pattern-induced multisequence alignment (PIMA) algorithm employing secondary structure-dependent gap penalties for use in comparative protein modeling. Protein Eng. 1992;5:35–41. doi: 10.1093/protein/5.1.35. [DOI] [PubMed] [Google Scholar]
- 140.Strapans S I, Hamilton B L, Follansbee S E, Elbeik T, Barbosa P, Grant R M, Feinberg M B. Activation of virus replication after vaccination of HIV-1-infected individuals. J Exp Med. 1995;182:1727–1737. doi: 10.1084/jem.182.6.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sun N-C, Ho D D, Sun C R Y, Liou R-S, Gordon W, Fung M S C, Li X-L, Ting R C, Lee T-H, Chang N T, Chang T-W. Generation and characterization of monoclonal antibodies to the putative CD4-binding domain of human immunodeficiency virus type 1 gp120. J Virol. 1989;63:3579–3585. doi: 10.1128/jvi.63.9.3579-3585.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Takahashi H, Merli S, Putney S D, Houghten R, Moss B, Germain R N, Berzofsky J A. A single amino acid interchange yields reciprocal CTL specificities for HIV-1 gp160. Science. 1989;246:118–121. doi: 10.1126/science.2789433. [DOI] [PubMed] [Google Scholar]
- 143.Ten Haaft P, Cornellissen M, Goudsmit J, Koornstra W, Dubbes R, Niphius N, Peeters M, Thiriart C, Bruck C, Heeney J L. Virus load in chimpanzees infected with human immunodeficiency virus type 1: effect of preexposure vaccination. J Gen Virol. 1995;76:1015–1020. doi: 10.1099/0022-1317-76-4-1015. [DOI] [PubMed] [Google Scholar]
- 144.Trkola A, Pomales A B, Yuan H, Korber B, Maddon P J, Allaway G P, Katinger H, Barbas III C F, Burton D R, Ho D D, Moore J P. Cross-clade neutralization of primary isolates of human immunodeficiency virus type 1 by human monoclonal antibodies and tetrameric CD4-IgG. J Virol. 1995;69:6609–6617. doi: 10.1128/jvi.69.11.6609-6617.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasan K, Sodroski J, Moore J P, Katinger H. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol. 1996;70:1100–1108. doi: 10.1128/jvi.70.2.1100-1108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Trkola, A., and J. P. Moore. Unpublished data.
- 147.Van Cott T C, Bethke F R, Burke D S, Redfield R R, Birx D L. Lack of induction of antibodies specific for conserved, discontinuous epitope of HIV-1 envelope glycoproteins by candidate AIDS vaccines. J Immunol. 1995;155:4100–4110. [PubMed] [Google Scholar]
- 148.Van Cott T C, Polonis V R, Loomis L D, Michael N L, Nara P L, Birx D L. Differential role of V3-specific antibodies in neutralization assays involving primary and laboratory-adapted isolates of HIV-1. AIDS Res Hum Retroviruses. 1995;11:1379–1392. doi: 10.1089/aid.1995.11.1379. [DOI] [PubMed] [Google Scholar]
- 149.Walker B D, Flexner C, Paradis T J, Fuller T C, Hirsch M S, Schooley R T, Moss B. HIV-1 reverse transcriptase is a target for cytotoxic T lymphocytes in infected individuals. Science. 1988;240:64–66. doi: 10.1126/science.2451288. [DOI] [PubMed] [Google Scholar]
- 150.Walker, B. D., et al. Unpublished data.
- 151.Wang S Z-S, Rushlow K E, Issel C J, Cook R F, Cook S J, Raabe M L, Chong Y-H, Costa L, Montelaro R C. Enhancement of EIAV replication and disease by immunization with a baculovirus-expressed recombinant envelope surface glycoprotein. Virology. 1994;199:247–251. doi: 10.1006/viro.1994.1120. [DOI] [PubMed] [Google Scholar]
- 152.Weber J, Fenyö E-M, Beddows S, Kaleebu P, Björndal Å the WHO Network for HIV Isolation and Characterization. Neutralization serotypes of human immunodeficiency virus type 1 field isolates are not predicted by genetic subtype. J Virol. 1996;70:7827–7832. doi: 10.1128/jvi.70.11.7827-7832.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wolinsky S M, Wike C, Korber B T M, Hutto C, Parks W P, Rosenblum L L, Kunstman K J, Furtado M R, Munoz J L. Selective transmission of human immunodeficiency virus type-1 variants from mothers to infants. Science. 1992;255:1134–1137. doi: 10.1126/science.1546316. [DOI] [PubMed] [Google Scholar]
- 154.Wolinsky S M, Korber B T M, Neumann A U, Daniels M, Kunstman K J, Whetsell A J, Furtado M R, Cao Y, Ho D D, Safrit J T, Koup R A. Adaptive evolution of human immunodeficiency virus type 1 during the natural course of infection. Science. 1996;272:537–542. doi: 10.1126/science.272.5261.537. [DOI] [PubMed] [Google Scholar]
- 155.World Health Organization. Scientific and public health rationale for HIV vaccine efficacy trials. AIDS. 1995;9:WHO1–WHO4. [PubMed] [Google Scholar]
- 156.Wrin T, Nunberg J H. HIV-1 MN recombinant gp120 vaccine serum, which fails to neutralize primary isolates of HIV-1, does not antagonize neutralization by antibodies from infected individuals. AIDS. 1994;8:1622–1623. doi: 10.1097/00002030-199411000-00017. [DOI] [PubMed] [Google Scholar]
- 157.Yap K L, Ada G L. The effect of specific antibody on the generation of cytotoxic T lymphocytes and the recovery of mice from influenza virus infection. Scand J Immunol. 1979;10:325–332. doi: 10.1111/j.1365-3083.1979.tb01358.x. [DOI] [PubMed] [Google Scholar]
- 158.Zhu T, Mo H, Wang N, Nam D, Cao Y, Koup R A, Ho D D. Genotypic and phenotypic characterization of HIV-1 in patients with primary infection. Science. 1993;261:1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]