

Abstract
To determine the molecular mechanism(s) of respiratory syncytial virus (RSV)-induced intercellular adhesion molecule-1 (ICAM-1) upregulation in respiratory epithelial cells (REC; A549 cell cultures), we investigated the roles of the transcription factors NF-κB and C/EBP. Increases in ICAM-1 message required de novo mRNA synthesis. ICAM-1 promoter constructs (luciferase reporter gene) transfected into A549 monolayers demonstrated promoter activation following RSV infection. Activation was abolished by site-specific mutation of the NF-κB (−228) or C/EBP (−239) sites. These data support the critical role of the activation of NF-κB and C/EBP in RSV-induced ICAM-1 expression by REC.
Respiratory syncytial virus (RSV) is the most important viral pathogen in infancy and early childhood (11, 21). Animal models of RSV bronchiolitis demonstrate acute airway obstruction associated with intense interstitial and peribronchial infiltrates (10, 27), features that are similar to observations in human autopsy specimens (1) and lavage samples from infants with RSV (5, 20). Studies in infants demonstrate that RSV infection causes production of a number of cytokines (4, 19, 20). Moreover, infection of airway epithelial cells by RSV upregulates the expression of intercellular adhesion molecule-1 (ICAM-1) (25, 28). The role of ICAM-1 in lung inflammation has been inferred from in vitro and in vivo studies with monoclonal antibodies (23, 31) and substantiated by studies of bronchial biopsies from asthmatics (18, 29). In the context of respiratory virus infection, the production of chemoattractive cytokines and ICAM-1 could provide a means of recruiting and retaining inflammatory cells within the airway, contributing to RSV-mediated lung injury.
Several groups have investigated the transcriptional regulation of the ICAM-1 gene and demonstrated differences in tumor necrosis factor alpha (TNF-α)- and gamma interferon (IFN-γ)-induced upregulation of ICAM-1 (13, 15, 16, 26, 30). RSV infection activates a number of transcription factors in respiratory epithelial cell cultures (6, 8, 17). The ICAM-1 promoter contains consensus binding sequences for several of these factors, implying but not proving that they contribute to virus-induced ICAM-1 activation. The studies described here were designed to identify the molecular mechanism of ICAM-1 gene activation following RSV infection in respiratory epithelial cell cultures.
Effects of RSV infection on ICAM-1 mRNA.
Initial studies were performed to examine ICAM-1 transcription following RSV infection. RNA was prepared from RSV-infected and control A549 cells following RSV exposure (RNeasy kit; Qiagen, Chatsworth, Calif.) and analyzed by Northern blot analysis (22) as described previously, probing with 32P-labeled cDNAs for ICAM-1 or β-actin (6, 7). These studies demonstrated a biphasic increase in mRNA levels, which peaked at 2 h following RSV exposure, decreased by 6 h, and increased again 24 h into infection (Fig. 1), similar to the biphasic activation of interleukin 8 (IL-8) following RSV infection (7). The cause of this biphasic activation is unclear, but the late phase requires viral replication (7). In order to determine whether the increase in mRNA resulted from new RNA or protein synthesis, cells were treated with actinomycin D (10 μg/ml) or cycloheximide (10 μg/ml). ICAM-1 mRNA was undetectable in RSV-infected cells following treatment with actinomycin D for 5 h, whereas cycloheximide treatment had no effect on ICAM-1 mRNA (Fig. 2).
FIG. 1.

Time course of RSV-induced ICAM-1 expression by Northern blot analysis. RNA was isolated from control and RSV-infected A549 cells at the indicated times, purified, and separated on glyoxal-agarose gels. RNA species were identified by Northern blot analysis with 32P-labeled ICAM-1 probe. ICAM-1 mRNA is detectable by 2 h, decreases by 6 h, and increases again at all subsequent time points following RSV infection in A549 cells. Results are representative of four experiments. C, control uninfected cells; rsv, hours postinfection with RSV.
FIG. 2.

RSV-induced ICAM-1 expression requires mRNA transcription. The increased mRNA expression seen following RSV infection requires increased gene transcription. ICAM-1 mRNA was increased in RSV-infected cells but not in control cells. Following actinomycin D treatment (10 μg/ml for 5 h) there was no detectable ICAM-1 mRNA. Cycloheximide treatment alone (10 μg/ml for 5 h) had no significant effect on ICAM-1 mRNA concentrations. Results are representative of three separate experiments.
ICAM-1 promoter activity after RSV infection.
To confirm that RSV-induced ICAM-1 upregulation occurred by activating gene transcription, the ICAM-1 5′-flanking region was analyzed with promoter/reporter gene constructs. The plasmid, pBH5000, which contains 5,000 bp of the 5′-proximal DNA fragment upstream to the ICAM-1 transcription start site, directing expression of the firefly luciferase reporter gene, was obtained (Christian Stratowa and Dwight Look [26, 30]) and transfected into A549 monolayers (6, 7). Lysates from these cells demonstrated greatly increased luciferase activity after RSV infection at all time points (the greatest luciferase activity was 6 h following virus exposure). Lysates from uninfected control cells demonstrated low levels of luciferase activity comparable to those of cells transfected with empty (promoterless) vector at all time points (Fig. 3). The time of peak activity differed between the Northern analysis and the reporter studies, most likely because of the difference between the time required for transcription, translation, and posttranslational modification of the reporter (luciferase) and the time required for transcription alone with Northern analysis. This accounts for the decreased levels of ICAM-1 mRNA when the activity of the reporter is increased. Also, the activities of both control (β-galactosidase [β-Gal]) and ICAM-1-luciferase constructs were markedly decreased by 24 h postinfection (48 h posttransfection) with this transient transfection model, making it difficult to perform analyses at later time points due to loss of plasmid or plasmid activity over time.
FIG. 3.

The 5′-flanking region of the ICAM-1 gene confers RSV responsiveness in A549 cells. A549 cells were transfected with pBH5000 and infected with RSV (multiplicity of infection, 1) for the indicated times, and then cell lysates were harvested for measurement of luciferase and β-Gal activities. ICAM-1 gene activation (measured as luciferase reporter gene activity) is significantly increased in A549 cells transfected with pBH5000 at all time points after RSV infection. However, luciferase activity is greatest at the 6-h time point, compared to the 2-h and 24-h time points. Results are the average of six replicate samples. Luciferase activity is normalized to the β-Gal activity of that sample in order to correct for transfection efficiency. Results are representative of two separate experiments. Error bars represent standard deviations. ∗∗, P is <0.01 compared to uninfected controls; †, P is <0.01 compared to 6-h RSV-infected cells.
Functional significance of the ICAM-1 NF-κB site after RSV infection.
Initial studies were focused on the NF-κB consensus sequence in the ICAM-1 promoter (position −228). Monolayers of A549 cells were transfected with reporter constructs containing the normal or mutated NF-κB sites (prepared by PCR with primers with normal or mutated sequences) (6, 7) (Fig. 4A). Cells transfected with the construct containing the normal NF-κB site had greatly enhanced luciferase activity after RSV infection (Fig. 4B). In contrast, the activation of the luciferase gene following RSV infection was significantly decreased in transfectants receiving mutated NF-κB promoter constructs (Fig. 4B), comparable to that in uninfected control cells and cells transfected with an empty vector (not shown).
FIG. 4.

Mutational analysis of the ICAM-1 promoter region: the roles of C/EBP and NF-κB in RSV-induced ICAM-1 activation. (A) Structure of NF-κB and C/EBP reporter plasmids used in promoter studies. This figure represents the cloning of the NF-κB and C/EBP reporter constructs used in the transfection studies. All constructs were cloned into the KpnI and SalI sites of the reporter plasmid pGL2-Basic. Capital letters in the bars indicate the sequence of the normal transcription factor consensus sequence; lowercase letters represent specific mutations introduced. The scheme is not drawn to scale. All cell lysates for experiments that follow were obtained 6 h following RSV infection. (B) Mutation of the NF-κB site at −228 eliminates RSV-induced ICAM-1 promoter activity. A549 cells were cotransfected with pGL2-Basic containing the appropriate NF-κB consensus sequence and a second plasmid containing the β-Gal reporter gene. Twenty-four hours following transfection, RSV was added to the cell monolayers and cell lysates were harvested for measurement of luciferase and β-Gal activities. RSV increased luciferase activity of A549 cells transfected with the plasmid reporter construct containing a normal NF-κB site. This increase in luciferase activity was eliminated in A549 cells transfected with the plasmid reporter construct containing a mutated NF-κB site. Results are the average of six individual samples, with each sample corrected for transfection efficiency relative to β-Gal activity, and are representative of three separate experiments. Error bars represent standard deviations. Samples were obtained 6 h following RSV exposure. ∗, P is <0.005 relative to control cells; †, P is <0.005 relative to normal NF-κB site. (C) Mutation of C/EBP site at −239 decreases RSV-induced ICAM-1 promoter activity. A549 cells were transfected with reporter plasmid constructs containing the normal or mutated C/EBP consensus sequences and infected with RSV as described above. ICAM-1 activation (measured as luciferase activity) was significantly increased in A549 cells transfected with the plasmid reporter construct containing a normal C/EBP site. This effect was lost when cells were transfected with reporter constructs containing a mutated C/EBP site. Results are the average of 4 to 6 samples, with each sample corrected for transfection efficiency relative to β-Gal activity, and are representative of three separate experiments. Error bars represent standard deviations. Samples were obtained 6 h following RSV exposure. ∗∗, P is <0.01 relative to controls (either uninfected cells or cells transfected with promotorless vector); ‡, P is <0.01 relative to RSV-infected cells transfected with the normal C/EBP consensus sequence.
Functional significance of the ICAM-1 C/EBP site after RSV infection.
The C/EBP site adjacent to the NF-κB site (−239 bp) was also examined. A549 cell monolayers transfected with pGL2-Basic luciferase reporter constructs containing normal and mutated C/EBP sites (prepared by PCR with primers containing normal or mutated C/EBP sequences) were infected with RSV 24 h later. Mutation of the C/EBP binding sequences significantly decreased promoter activation following RSV infection (Fig. 4A and C).
Comparison of RSV-induced and cytokine-induced ICAM-1 activation.
TNF-α- and IFN-γ-induced activation of ICAM-1 have been reported to require different transcription factors. To compare the mechanisms of transcriptional activation following RSV infection with TNF-α and IFN-γ treatments, cells transfected with the promoter constructs described above were incubated in the presence of cytokine or infected with RSV, and cell lysates were prepared and assayed for luciferase activity. Mutation of either the NF-κB (Fig. 5A) or C/EBP sites (Fig. 5B) significantly decreased ICAM-1 promoter activation following either TNF-α or RSV treatment. In contrast, mutation of either of these sites had no effect on IFN-γ-induced ICAM-1 activation. Ledebur et al. demonstrated that TNF-α-induced activation of the ICAM-1 promoter required the NF-κB site targeted in the current studies (13). In contrast, Look and coworkers (15, 16) demonstrated that the IFN-γ response element located at nucleotides −130 to −194 of the ICAM-1 5′-flanking region conferred IFN-γ responsiveness. The data presented here are consistent with the results of these previous studies indicating that the NF-κB and C/EBP binding sequences in the ICAM-1 promoter are required for both TNF-α-induced and RSV-induced ICAM-1 upregulation, whereas the IFN-γ response element was unaffected by these mutations (Fig. 5).
FIG. 5.
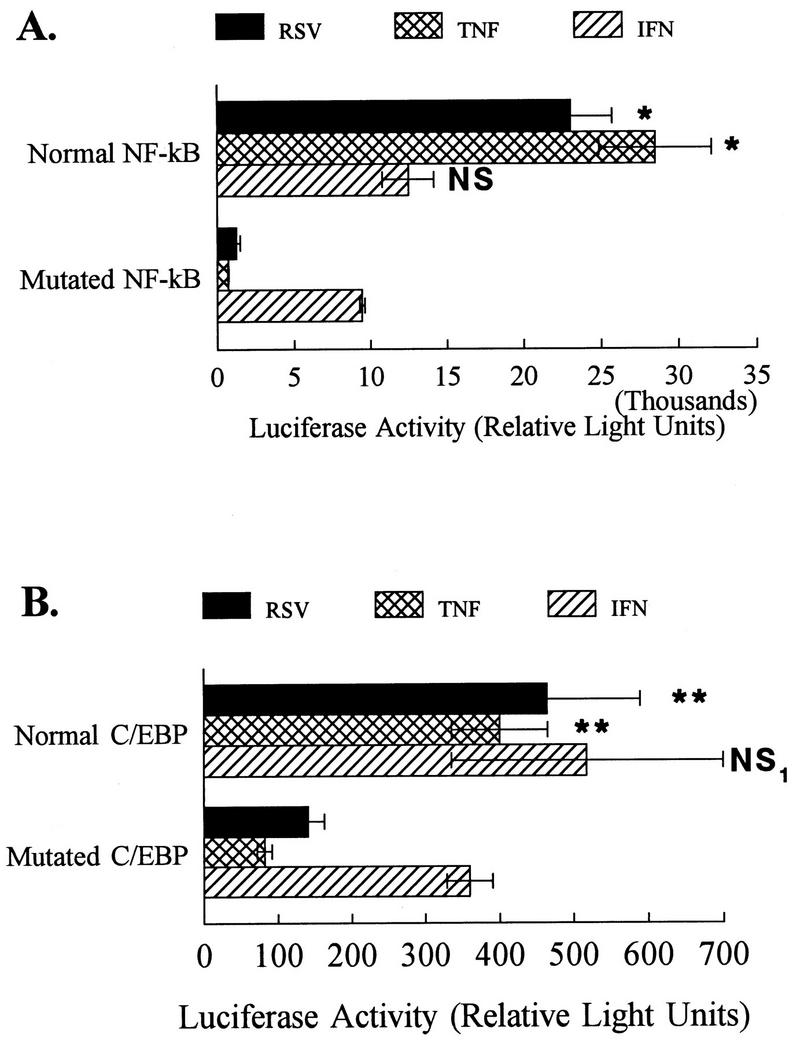
Mutational analysis of the ICAM-1 promoter region: roles of C/EBP and NF-κB in cytokine-induced ICAM-1 activation. (A) A549 cells were cotransfected with pGL2-Basic containing the appropriate NF-κB consensus sequence and a second plasmid containing the β-Gal reporter gene. Twenty-four hours following transfection, cell monolayers were infected with RSV (multiplicity of infection, 1) or treated with TNF-α (5 ng/ml) or IFN-γ (10 ng/ml) for 6 h prior to harvest of cell lysates. ICAM-1 gene activation was measured as luciferase activity of the reporter plasmids. RSV and TNF (known to increase ICAM-1 gene expression via the NF-κB site at −228) treatments increased luciferase activity of A549 cells transfected with the plasmid reporter construct containing a normal NF-κB site. This increase in luciferase activity was eliminated in A549 cells transfected with the plasmid reporter construct containing a mutated NF-κB site. In contrast, IFN-γ activation of the ICAM-1 promoter constructs was unaffected by mutation of the NF-κB binding site. Cell lysates were obtained for luciferase measurements of 6 h following virus exposure or cytokine treatment. Results are the average of three samples, with each sample corrected for transfection efficiency relative to β-Gal activity, and are representative of three separate experiments. Error bars represent standard deviations. ∗, P is <0.01 relative to normal NF-κB consensus sequence. NS, no significant differences between IFN-γ treatment of cells transfected with normal and mutated NF-κB consensus sequences. (B) Plasmid reporter constructs containing a normal and mutated C/EBP site remained responsive to IFN, but mutation of the C/EBP site reduced responsiveness to TNF-α or RSV treatment. A549 cells were transfected with reporter plasmid constructs containing the normal or mutated C/EBP consensus sequences and were then treated as described for panel A. ICAM-1 activation (measured as luciferase activity) was significantly increased in A549 cells transfected with the plasmid reporter construct containing a normal C/EBP site following infection with RSV or TNF-α treatment. This effect was lost when cells were transfected with reporter constructs containing a mutated C/EBP site. In contrast, responses to IFN-γ were unaltered by mutation of the C/EBP consensus sequences. Cell lysates were obtained for luciferase measurements 6 h following virus exposure or cytokine treatment. Results are the average of three samples, with each sample corrected for transfection efficiency relative to β-Gal activity, and are representative of three separate experiments. Error bars represent standard deviations. ∗∗, P is <0.01 relative to mutated C/EBP consensus sequences. NS1, no significant differences between IFN-γ treatment of cells transfected with normal and mutated C/EBP consensus sequences.
Researchers in several laboratories have demonstrated that activation of NF-κB and NF-IL-6 occurs following RSV infection of A549 monolayers (6, 8, 9, 12, 17). Electrophoretic mobility shift and antibody supershift assays demonstrated that NF-κB and NF-IL-6 activated following RSV infection (6, 9, 17). In addition, mutational and deletional analysis of the IL-8 promoter demonstrated that NF-IL-6 and NF-κB binding sites located at −94 and −50 bp were essential for RSV-induced IL-8 production. This arrangement of transcription factor binding sites is similar to the arrangement of C/EBP and NF-κB sites in the ICAM-1 promoter (26). RSV infection of the airway epithelium, therefore, utilizes similar mechanisms to regulate the orchestrated production of ICAM-1 and IL-8 (3).
Infections with viruses such as RSV trigger acute exacerbations of asthma (2, 24). RSV-induced NF-κB activation and the subsequent upregulation of ICAM-1 and chemokines could contribute to inflammatory cell recruitment (14, 31) and airway hyperreactivity following viral infection. Because of its pivotal role in inflammation, NF-κB will be an obvious target for new types of anti-inflammatory treatments for bronchiolitis or virus-induced asthma. Blocking the activation of NF-κB may prevent the early events in the inflammatory cascade, decreasing RSV-induced inflammation and subsequent lung injury.
In summary, we have shown that RSV regulates ICAM-1 gene expression at the transcriptional level, acting through the 5′-flanking region of the ICAM-1 gene. The transcription factors NF-κB (−228) and C/EBP (−239) are necessary for RSV-induced ICAM-1 upregulation, indicating one molecular mechanism of virus-induced airway inflammation.
Acknowledgments
These studies were supported in part by K08 HL02505 (J.M.S.), K08 HL03451 (M.A.F.), and grants from the American Lung Association and the Cystic Fibrosis Foundation.
REFERENCES
- 1.Aherne W, Bird T, Court S D, Gardner P S, McQuillin J. Pathological changes in virus infections of the lower respiratory tract in children. J Clin Pathol. 1970;23:7–18. doi: 10.1136/jcp.23.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bardin P G, Fraenkel D, Sanderson G, Dorward M, Johnston S, Holgate S. Increased sensitivity to the consequences of rhinoviral infection in atopic subjects. Chest. 1995;107:S157. doi: 10.1378/chest.107.3_supplement.157s. [DOI] [PubMed] [Google Scholar]
- 3.Barnes P J, Karin M. Nuclear Factor -κB—a pivotal transcription factor in chronic inflammatory diseases. New Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 4.Becker S, Reed W, Henderson F W, Noah T L. RSV infection of human airway epithelial cells causes production of the β-chemokine RANTES. Am J Physiol. 1997;16:L512–L520. doi: 10.1152/ajplung.1997.272.3.L512. [DOI] [PubMed] [Google Scholar]
- 5.Everard M L, Swarbrick A, Wrightham M, McIntyre J, Dunkley C, James P D, Sewell H F, Milner A D. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch Dis Child. 1994;71:428–432. doi: 10.1136/adc.71.5.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fiedler M A, Wernke-Dollries K, Stark J M. Mechanism of RSV-induced IL-8 gene expression in A549 cells prior to viral replication. Am J Physiol. 1996;271:L963–L971. doi: 10.1152/ajplung.1996.271.6.L963. [DOI] [PubMed] [Google Scholar]
- 7.Fiedler M A, Wernke-Dollries K, Stark J M. Respiratory syncytial virus increases IL-8 gene expression and protein release in A549 cells. Am J Physiol. 1995;13:L865–L872. doi: 10.1152/ajplung.1995.269.6.L865. [DOI] [PubMed] [Google Scholar]
- 8.Fiedler M A, Wernke-Dollries K, Stark J M. Inhibition of viral replication reverses respiratory syncytial virus-induced NF-κB activation and interleukin-8 gene expression in A549 cells. J Virol. 1996;70:9079–9082. doi: 10.1128/jvi.70.12.9079-9082.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garofalo R, Sabry M, Jamaluddin M, Yu R K, Casola A, Ogra P L, Brasier A R. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J Virol. 1996;70:8773–8781. doi: 10.1128/jvi.70.12.8773-8781.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegele R G, Robinson P J, Gonzalez S, Hogg J C. Production of acute bronchiolitis in guinea-pigs by human respiratory syncytial virus. Eur Respir J. 1993;6:1324–1331. [PubMed] [Google Scholar]
- 11.Henderson F W, Clyde W A, Jr, Collier A M, Denny F W, Senior R J, Sheaffer C I, Conley W G, III, Christian R M. The etiologic and epidemiologic spectrum of bronchiolitis in pediatric practice. J Pediatr. 1979;95:183–190. doi: 10.1016/s0022-3476(79)80647-2. [DOI] [PubMed] [Google Scholar]
- 12.Jamaluddin M, Garofalo R, Ogra P L, Brasier A R. Inducible translational regulation of the NF-IL6 transcription factor by respiratory syncytial virus infection in pulmonary epithelial cells. J Virol. 1996;70:1554–1563. doi: 10.1128/jvi.70.3.1554-1563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ledebur H C, Parks T P. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. J Biol Chem. 1995;270:933–943. doi: 10.1074/jbc.270.2.933. [DOI] [PubMed] [Google Scholar]
- 14.Leff A R, Hamann K J, Wegner C D. Inflammation and cell-cell interactions in airway hyperresponsiveness. Am J Physiol. 1991;260:L189–L206. doi: 10.1152/ajplung.1991.260.4.L189. [DOI] [PubMed] [Google Scholar]
- 15.Look D C, Pelletier M R, Holtzman M J. Selective interaction of a subset of interferon-gamma response element-binding proteins with the intercellular adhesion molecule-1 (ICAM-1) gene promoter controls the pattern of expression on epithelial cells. J Biol Chem. 1994;269:8952–8958. [PubMed] [Google Scholar]
- 16.Look D C, Rapp S R, Keller B T, Holtzman M J. Selective induction of intercellular adhesion molecule-1 by interferon-gamma in human airway epithelial cells. Am J Physiol. 1992;263:L79–L87. doi: 10.1152/ajplung.1992.263.1.L79. [DOI] [PubMed] [Google Scholar]
- 17.Mastronarde J G, He B, Monick M M, Mukaida N, Matsushima K, Hunninghake G W. Induction of interleukin (IL)-8 gene expression by respiratory syncytial virus involves activation of nuclear factor (NF)-κB and NF-IL-6. J Infect Dis. 1996;174:262–267. doi: 10.1093/infdis/174.2.262. [DOI] [PubMed] [Google Scholar]
- 18.Montefort S, Roche W R, Howarth P H, Djukanovic R, Gratziou C, Carroll M, Smith L, Britten K M, Haskard D, Lee T H, Holgate S T. Intercellular adhesion molecule-1 (ICAM-1) and endothelial leucocyte adhesion molecule-1 (ELAM-1) expression in the bronchial mucosa of normal and asthmatic subjects. Eur Respir J. 1992;5:815–823. [PubMed] [Google Scholar]
- 19.Noah T L, Becker S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am J Physiol. 1994;265:L472–L478. doi: 10.1152/ajplung.1993.265.5.L472. [DOI] [PubMed] [Google Scholar]
- 20.Noah T L, Henderson F W, Wortman I A, Devlin R B, Handy J, Koren H S, Becker S. Nasal cytokine production in viral acute upper respiratory infection of childhood. J Infect Dis. 1995;171:584–592. doi: 10.1093/infdis/171.3.584. [DOI] [PubMed] [Google Scholar]
- 21.Parrott R H, Kim H W, Arrobio J O, Hodes D S, Murphy B R, Brandt C D, Camargo E, Chanock R M. Epidemiology of respiratory syncytial virus infection in Washington, D.C. II. Infection and disease with respect to age, immunologic status, race and sex. Am J Epidemiol. 1973;98:289–300. doi: 10.1093/oxfordjournals.aje.a121558. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 23.Smith C W, Marlin S D, Rothlein R, Toman C, Anderson D C. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 1989;83:2008–2017. doi: 10.1172/JCI114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stark J M, Busse W W. Respiratory virus infection and airway hyperreactivity in children. Pediatr Allergy Immunol. 1991;2:95–110. [Google Scholar]
- 25.Stark J M, Godding V, Sedgwick J B, Busse W W. Respiratory syncytial virus infection enhances neutrophil and eosinophil adhesion to cultured respiratory epithelial cells. J Immunol. 1996;156:4774–4782. [PubMed] [Google Scholar]
- 26.Stratowa C, Audette M. Transcriptional regulation of the human intercellular adhesion molecule-1 gene: a short overview. Immunobiology. 1995;193:293–304. doi: 10.1016/S0171-2985(11)80558-9. [DOI] [PubMed] [Google Scholar]
- 27.Taylor G, Stott E J, Hughes M, Collins P. Respiratory syncytial virus infection in mice. Infect Immun. 1984;43:649–655. doi: 10.1128/iai.43.2.649-655.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tosi M F, Stark J M, Hamedani A, Smith C W, Gruenert D C, Huang Y T. Intercellular adhesion molecule-1 (ICAM-1)-dependent and ICAM-1-independent adhesive interactions between polymorphonuclear leukocytes and human airway epithelial cells infected with parainfluenza virus type 2. J Immunol. 1992;149:3345–3349. [PubMed] [Google Scholar]
- 29.Vignola A M, Campbell A M, Chanez P, Bousquet J, Paullacoste P, Michel F B, Godart P. HLA-DR and ICAM-1 expression on bronchial epithelial cells in asthma and chronic bronchitis. Am Rev Respir Dis. 1994;148:689–694. doi: 10.1164/ajrccm/148.3.689. [DOI] [PubMed] [Google Scholar]
- 30.Voraberger G, Schafer R, Stratowa C. Cloning of the gene for intercellular adhesion molecule-1 and the analysis of its 5′-regulatory region. J Immunol. 1991;148:2777–2786. [PubMed] [Google Scholar]
- 31.Wegner C D, Gundel R H, Reilly P, Haynes N, Letts L G, Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) in the pathogenesis of asthma. Science. 1990;247:456–459. doi: 10.1126/science.1967851. [DOI] [PubMed] [Google Scholar]