

Abstract
Fusion peptides are hydrophobic sequences located at the N terminus of the transmembrane (TM) envelope proteins of the orthomyxoviruses and paramyxoviruses and several retroviruses. The Moloney murine leukemia virus TM envelope protein, p15E, contains a hydrophobic stretch of amino acids at its N terminus followed by a region rich in glycine and threonine residues. A series of single amino acid substitutions were introduced into this region, and the resulting proteins were examined for their abilities to be properly processed and transported to the cell surface and to induce syncytia in cells expressing the ecotropic receptor. One substitution in the hydrophobic core and several substitutions in the glycine/threonine-rich region that prevented both cell-cell fusion and the transduction of NIH 3T3 cells when incorporated into retroviral vector particles were identified. In addition, one mutation that enhanced the fusogenicity of the resulting envelope protein was identified. The fusion-defective mutants trans dominantly interfered with the ability of the wild-type envelope protein to cause syncytium formation in a cell-cell fusion assay, although no trans-dominant inhibition of transduction was observed. Certain substitutions in the hydrophobic core that prevented envelope protein processing were also found. These data indicate that the N-terminal region of p15E is important both for viral fusion and for the correct processing and cell surface expression of the viral envelope protein.
The envelope protein of the ecotropic Moloney murine leukemia virus (MoMuLV) comprises two polypeptides, the surface (SU) glycoprotein gp70 and the transmembrane (TM) glycoprotein p15E (41, 42). These proteins are processed from a common precursor, Pr85, by a host cell protease during transport to the cell surface and remain associated following cleavage. Infection of susceptible cells by MoMuLV is initiated by a specific interaction between gp70 (19, 24, 37) and the ecotropic receptor ATRC-1 (1). The interaction of retroviral SU proteins with their cognate receptors is thought to induce a conformational change that is transmitted to the TM subunit, thereby triggering events that eventually lead to fusion of the viral and host cell membranes (26). Several studies have implicated the amino terminus of retroviral TM proteins as being important in catalyzing the fusion of viral and cellular membranes (2–4, 7, 9, 11, 17, 21, 32, 36, 38). By analogy to the fusion proteins of the orthomyxoviruses and paramyxoviruses, this region is referred to as the fusion peptide.
Fusion peptides are relatively hydrophobic regions, typically rich in alanine and glycine residues (38). It has been suggested that they may form sided helices, with bulkier, more hydrophobic residues on one side associating with the membrane and smaller residues, such as glycine and alanine, on the other (5, 7, 18, 38, 39), although this model has been contested (15). A candidate fusion peptide at the amino terminus of the human immunodeficiency virus type 1 (HIV-1) TM protein, gp41, was identified by virtue of its sequence homology and analogous position to the fusion peptide of the paramyxovirus ReSV F1 glycoprotein (13). It contains a stretch of hydrophobic amino acids with the sequence LFLGFLG, and the sequence F-x-G is also present in the fusion peptides of paramyxoviral F1 proteins. In addition, this whole region shows a periodicity of glycine residues (4, 7).
The N terminus of the MoMuLV TM protein, p15E, is also hydrophobic and rich in alanine, glycine, and leucine residues. A stretch of hydrophobic residues with the sequence LALLLGGL occurs 7 to 14 residues from the SU-TM cleavage site and could provide a fusion core. Previous mutational analyses of HIV-1 (2, 9, 11, 23), simian immunodeficiency virus (4), and AKV murine leukemia virus (MuLV) (21) have demonstrated the importance of the hydrophobic residues in this region, as well as the conserved glycine residues (3, 7).
Computer modeling predicts that the fusion core of gp41 is followed by an internally looping structure of 8 to 17 amino acids, enriched in serine and threonine residues (ST domain) (14). In MuLV, there exists a similarly located region that is rich in glycine and threonine residues. In HIV-1, this region has been implicated in the noncovalent association between gp41 and the SU protein gp120, as both mutations (7, 11) and insertions (23) in this region resulted in the loss of cell-associated gp120.
Retroviral vectors based on MuLV are currently the most commonly used system for human gene therapy. Considerable effort has been directed towards engineering the envelope protein to enable retargeting of virions to specific cell surface ligands (reviewed in reference 6). However, although binding of virions to specific cell surface antigens has been achieved, this does not result in efficient transduction. We are therefore interested in understanding in more detail the postbinding changes that occur in the envelope protein and the role of the putative fusion peptide in this process.
The organization of the envelope protein of MoMuLV and the amino acid sequence of the N terminus of the TM protein are shown schematically in Fig. 1. This region contains a stretch of hydrophobic residues that may constitute a hydrophobic core of a fusion peptide followed by a region rich in glycine and threonine residues (the GT region). In order to study the importance of this region for envelope function, we introduced both conservative and nonconservative substitutions into the hydrophobic core and the GT region and additionally mutated residues further downstream. Mutants were constructed in the envelope protein expression vector CEE+ (24), which expresses the MoMuLV envelope protein from a cytomegalovirus promoter, by oligonucleotide-directed in vitro mutagenesis (Amersham International plc).
FIG. 1.

The N terminus of MoMuLV TM (p15E) protein. The 38 N-terminal amino acids of p15E analyzed in this study (hatched box) and the gp70-p15E cleavage site (arrow) are indicated. Numbering is from the start of the processed envelope protein after removal of the 33-amino-acid signal sequence (33), with the start of p15E at residue E437. The specific mutations that were introduced are shown below the wild-type MoMuLV sequence.
Cell-cell fusion mediated by envelope protein mutants.
Retroviral envelope proteins can mediate both virus-cell fusion (fusion from without) and cell-cell fusion (fusion from within) (40). The cocultivation of NIH 3T3 cells expressing the MoMuLV envelope protein with the rat XC cell line causes cell-cell fusion and leads to the formation of syncytia (29). The ability of wild-type and mutant envelope proteins to direct cell-cell fusion was tested by transfecting 1 × 105 NIH 3T3 cells with 15 μg of envelope protein expression plasmids in gridded (2- by 2-mm) 60-mm-diameter petri dishes and overlaying the cells with 5 × 105 XC cells at 18 to 20 h posttransfection. Following incubation for a further 36 h, the cells were fixed and stained with 1% methylene blue in methanol and the syncytia were counted, as described previously (21, 29).
In addition, XC cells were cocultivated with GPL cells transiently transfected with envelope protein expression plasmids. GPL cells (27) are NIH 3T3 cells expressing MoMuLV Gag-Pol and containing the Neor retroviral vector LNL6. Transfection of an envelope protein expression plasmid into these cells results in the production of retroviral vector particles, so this assay is sensitive to both fusion from within and fusion from without.
The ability of the mutant envelope proteins to induce syncytia was compared to that of the wild-type envelope protein (Table 1). All mutations in the hydrophobic domain, except the L441F substitution, drastically reduced or abolished syncytium formation in both GPL and NIH 3T3 transfections. In the GT-rich domain, the A457F, G460E, and T461P substitutions prevented syncytium formation, and mutant G460V exhibited decreased fusion ability. In contrast, several mutants with substitutions of the threonines at positions 463 and 464 retained nearly wild-type levels of fusion activity. In the more C-terminal region, the conversion of hydrophobic residues to charged residues for both M467E and L475R prevented syncytium formation. However, the A468R substitution had no effect, and mutant Q474E was only partially defective. These data suggest that mutations throughout the N-terminal region of p15E can affect some aspect of the fusion process.
TABLE 1.
Properties of p15E mutants
| Envelope protein | Relative no. of syncytiaa
|
Surface expressionb | Relative titerc | |
|---|---|---|---|---|
| NIH 3T3 cells | GPL cells | |||
| None | 0.1 ± 0.1 | 0.2 ± 0.1 | 0 | 0 |
| CEE+ (wild type) | 100 ± 1 | 100 ± 8 | 100 ± 23 | 100 ± 57 |
| L441F | 40 ± 3 | 104 ± 14 | ND | 70 ± 30 |
| L443R | 0 | 0 | 43 ± 17 | 0 |
| L443A | 0 | 0.1 ± 0.1 | ND | 0 |
| L443I | 0 | 0.6 ± 0.3 | ND | 3.3 ± 2 |
| L445E | 0 | 0 | 129 ± 14 | 0 |
| G449L | 0 | 0 | 24 ± 8 | 0.03 ± 0.03 |
| G449W | 0 | 0.1 ± 0.1 | ND | 0.01 ± 0.01 |
| G449A | 0 | 0.5 ± 0.3 | ND | 0.05 ± 0.03 |
| G449S | 0 | 0.3 ± 0.3 | ND | 0.14 ± 0.07 |
| A457F | 0 | 0 | 80 ± 3 | 0.07 ± 0.03 |
| G460V | 24 ± 9 | 31 ± 6 | ND | 3.7 ± 3 |
| G460E | 0 | 0 | 92 | 0.02 ± 0.02 |
| T461P | 0 | 0 | 88 | 0 |
| T463A | 80 ± 11 | 47 ± 4 | ND | 9.2 ± 3 |
| T464A | 182 ± 11 | 337 ± 85 | 83 ± 3 | 107 ± 7 |
| T464I | 98 ± 16 | 108 ± 9 | ND | 23 ± 3 |
| T464K | 75 ± 1 | 77 ± 6 | ND | 25 ± 15 |
| M467E | 0 | 0.1 ± 0.1 | ND | 4.4 ± 3 |
| A468R | 147 ± 3 | 103 ± 5 | ND | 124 ± 59 |
| Q474E | 15 ± 1 | 56 ± 13 | ND | 9.3 ± 5 |
| L475R | 0 | 0 | 53 ± 12 | 0 |
NIH 3T3 or GPL cells transiently expressing envelope proteins were overlaid with XC cells, and syncytia (four or more nuclei) were counted in 20 grids (2 by 2 mm). The average numbers of syncytia ± standard errors were calculated from at least three independent experiments. Wild-type CEE+ envelope protein gave 240 (NIH 3T3 cells) and 294 (GPL cells) syncytia in 20 grids. Results for each mutant are presented normalized to these values.
Calculated as mean channel fluorescence of the sample − mean background channel fluorescence normalized to the value for CEE+. ND, not determined.
Wild-type CEE+ transfected into GPL cells gave an average titer on NIH 3T3 cells of (5 ± 2.6) × 104 CFU/ml (three experiments). Results for all mutants were averaged from at least three independent transfections and are normalized to this value, ± standard error.
Interestingly, mutant T464A exhibited an increased ability to induce syncytia in XC cells cocultivated with both NIH 3T3 cells and GPL cells (182 and 337% of wild-type levels, respectively). To further examine this mutant, we carried out a time course analysis of syncytium formation following the addition of XC cells to transfected GPL cells (Fig. 2a). Mutant T464A caused the appearance of syncytia after only 5 h of cocultivation, while the wild-type envelope protein and the fusion-defective mutant Q474E started to form syncytia only after 9 h in culture. At 12 to 15 h, T464A expression resulted in the appearance of large polynuclear syncytia with over 20 nuclei, while the wild-type protein formed smaller syncytia with fewer than 10 nuclei. After 24 h, the number of syncytia formed by mutant T464A was 2-fold higher than that formed by the wild type, while mutant Q474E formed 3.5-fold fewer syncytia than the wild type (Fig. 2b).
FIG. 2.

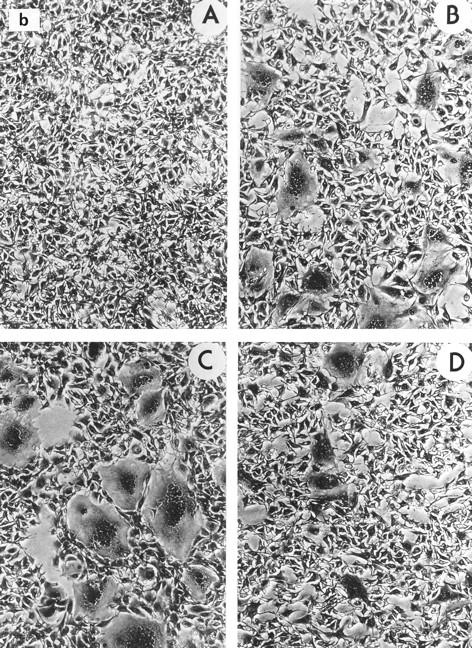
Syncytium formation by wild-type p15E and p15E mutants. (a) GPL cells were transfected with wild-type and mutant envelope protein-expressing vectors and cocultured with XC cells, and the syncytia were counted at various times. •, wild type; ▴, T464A; ▪, Q474E. The results are the averages from two independent experiments. (b) XC cells at the 24-h time point from one experiment were photographed at ca. ×50 magnification. A, negative control; B, wild type; C, T464; D, Q474E.
Transduction properties of mutants.
We further analyzed the ability of the mutant envelope proteins to cause virus-cell fusion by measuring the transduction efficiencies of retroviral vectors obtained from the transfected GPL cells. Culture supernatants were tested for their ability to confer G418 resistance to NIH 3T3 cells as previously described (27) by selection with 0.8 mg of G418 (Gibco/BRL) per ml for 8 to 10 days.
The transduction efficiency of each mutant relative to that of the wild type is shown in Table 1. In general, the results agreed with the fusion data. In the hydrophobic core, all of the mutants except L441F significantly decreased or abolished the viral titers, and in the GT-rich region, the nonfusogenic mutants A457F, G460E, and T461P were unable to transduce NIH 3T3 cells. The hyperfusogenic mutant, T464A, which produced two- to threefold more syncytia than the wild type in the cell-cell fusion assays, showed wild-type levels of transduction. Interestingly, two mutants that failed to induce syncytia in cocultivated XC cells (M467E and L443I) were able to transduce NIH 3T3 cells at levels that were 3 to 4% of the wild-type level, suggesting that the process of cell-cell fusion is more sensitive to envelope protein mutations than virus-cell fusion.
R-peptide cleavage does not enhance fusogenicity of defective mutants.
In viral particles, the cytoplasmic tail of MoMuLV envelope protein is cleaved by the viral protease to remove the C-terminal 16 amino acids (the R peptide). It has previously been shown that this truncation enhances the fusogenicity of the protein and results in syncytium formation when the truncated envelope protein is expressed in NIH 3T3 cells (30, 31). We therefore examined the effect of R-peptide truncation on wild-type envelope protein and fusion-defective and hyperfusogenic envelope protein mutants. The results, shown in Table 2, confirmed that expression of an R-less version of the wild-type protein resulted in massive syncytia in NIH 3T3 cells, as previously reported (30, 31). Furthermore, in agreement with the minimal effect seen in the XC cocultivation assays, the R-less versions of mutants L441F and A468R also produced maximum syncytium formation in NIH 3T3 cells. Removal of the R peptide of mutant Q474E, which displayed reduced syncytia in the XC cocultivation assay, resulted in an envelope protein that was still less fusogenic than the wild-type protein. Furthermore, an R-less version of the completely nonfusogenic mutant T461P did not result in any syncytia, suggesting that the defects in fusogenicity resulting from these mutations could not be compensated for by R-peptide truncation. Finally, the R-less version of the hyperfusogenic mutant, T464A, was no more fusogenic than the wild-type protein in this system.
TABLE 2.
Effect of R-peptide truncation on p15E mutants
| Envelope protein in LETRSNa |
Relative syncytium formationb |
|---|---|
| Wild type | 100 ± 13 |
| L441F | 80 ± 30 |
| T461P | 0 |
| T464A | 98 ± 6 |
| A468R | 96 ± 42 |
| Q474E | 59 ± 31 |
LETRSN contains an R-less version of MoMuLV envelope protein in which the MuLV long terminal repeat promoter drives the expression of MoMuLV envelope truncated at the R-peptide cleavage site at amino acid 616.
The relative number of syncytia ± standard error induced by NIH 3T3-XC cell cocultivation was calculated as described in Table 1, footnote a. The average number of syncytia induced by the wild-type R-less envelope protein was 1,073 ± 136 for 20 grids examined.
SU-TM protein processing and surface expression of mutant envelopes.
The envelope proteins of retroviruses are transported through the endoplasmic reticulum and the Golgi complex to the cell surface, where they are incorporated into budding virions. Cleavage of the precursor protein into SU and TM subunits is required for this transport process (12, 25). The position of the fusion peptide close to the SU-TM cleavage site made it a possibility that some of the nonfusogenic mutants were actually defective in envelope protein processing or cell surface expression. Accordingly, we examined the nature of the envelope protein present in lysates of transfected 293T cells (Fig. 3). Wild-type envelope protein was present as both the uncleaved precursor protein Pr85 and the cleaved gp70 SU subunit. However, for certain mutants (L443A/R, G449L/W/A/S, and L475R), only Pr85 could be detected, suggesting a block in the transport pathway and SU-TM cleavage. Not surprisingly, all of these mutants had previously been shown to be defective at promoting cell-cell fusion and titers (Table 1). In contrast, a second group of fusion-defective mutants (L445E, A457F, G460E, and T461P) that were processed normally, exhibiting both Pr85 and gp70 in their cell lysates, was identified (Fig. 3).
FIG. 3.

Western analysis of transfected-cell lysates. 293T cells were transfected with envelope protein expression plasmids, lysed 48 h later in 500 μl of lysis buffer (20 mM Tris-HCl [pH 7.5], 1% Triton X-100, 0.05% sodium dodecyl sulfate, 5 mg of sodium deoxycholate per ml, 150 mM NaCl, and 1 mM phenylmethylsulfonyl fluoride) for 10 min at 4°C, and centrifuged at 10,000 × g for 10 min to pellet nuclei. The cell lysates were resolved on a precast 8 to 16% gel, and the envelope protein precursor Pr85 and the processed SU subunit gp70 were detected by using a specific goat antiserum, as described previously (20). Lane 1, CEE+ wild-type protein; lane 2, L443A; lane 3, L443R; lane 4, L441F; lane 5, L445E; lane 6, G449L; lane 7, G449W; lane 8, G449A; lane 9, G449S; lane 10, T461P; lane 11, T463A; lane 12, A457F; lane 13, G460V; lane 14, G460E; lane 15, L475R; lane 16, Q474E; lane 17, A468R; lane 18, M467E; lane 19, T464K; lane 20, T464I; lane 21, T464A; lane 22, control (mock transfected).
We also measured the levels of envelope protein on the cell surface for some of the mutants that did not induce syncytia (Table 1). Cell surface envelope proteins were detected by indirect immunofluorescence and fluorescence-activated cell sorting (FACS) of transiently transfected 293T cells, as described elsewhere (20). For the processing-defective mutants L443R, G449L, and L475R, levels of envelope protein lower than for the wild type were detected, and this probably corresponded to uncleaved Pr85. In contrast, mutant envelope proteins L445E, A457F, and T461P, which were processed normally, were readily detectable on the cell surface. In addition, we saw that the hyperfusogenic mutant, T464A, was present on the cell surface at levels comparable to that of the wild-type protein, suggesting that its increased fusogenicity did not result simply from an increased level of cell surface expression.
Envelope protein incorporation into virions.
Envelope protein present at the cell surface is incorporated into viral particles during budding. To test whether the mutations we had made at the N terminus of p15E had any influence on this process, we analyzed the protein composition of viral particles obtained from 293T cells transfected with MoMuLV Gag-Pol and envelope protein expression vectors and purified from culture supernatants by centrifugation through 20% sucrose (20).
Most of the mutant viruses contained approximately wild-type levels of gp70 and p15E (Fig. 4). However, no gp70 or p15E protein could be detected for mutants L443R, G449L/A/S/W, or L475R, although some uncleaved precursor Pr85 appeared to be incorporated at low levels for mutants L443R and G449S/A/L. In addition, the A457F and L443A mutations reproducibly resulted in lower levels of envelope proteins being incorporated into virions. These results are in agreement with the analyses described above, as those mutants that exhibited defects in processing and cell surface expression were also not detected in virions.
FIG. 4.

Western analysis of virions. Retrovirus particles were generated by cotransfection of envelope protein expression plasmids and the Gag-Pol expression plasmid pHIT60 into 293T cells, essentially as described previously (20, 35). Virions were partially purified from culture supernatants by pelleting through a 20% sucrose cushion at 25,000 rpm and 4°C in an SW41 rotor for 4 h, and samples were resolved on sodium dodecyl sulfate–8 to 16% polyacrylamide gradient gels and transferred to membranes. The blots were probed with anti-gp70 and anti-p30 Gag antisera, as described previously (20). For the TM protein, both p15E and the processed (R-peptide-cleaved) p12E proteins were detected with either an anti-p15E antiserum (20) (lanes 1 to 15) or monoclonal antibody 42-114 (28) (lanes 16 to 26). For the upper panel, p15E and p12E run very closely together; the higher-migrating band (ns) is nonspecific. The two forms of TM protein are more clearly resolved in the lower panel. Lane 1, mock transfection; lane 2, CEE+ wild-type envelope protein; lanes 3 to 15, mutants L441F, L443R, L445E, G449L, A457F, G460E, T461P, T463A, T464A, M467E, A468R, Q474E, and L475R, respectively; lanes 16 and 17, mutants L443A and L443R, respectively; lanes 18 to 21, G449S, G449A, G449L, and G449W, respectively; lanes 22 and 23, G460E and G460V, respectively; lanes 24 to 26, T464K, T464I, and T464A, respectively. Some Pr85 can be seen for mutants L443R and G449S/A/L.
For the mutant envelope proteins that were efficiently incorporated into virions, no differences were seen in the relative levels of gp70 and p15E or in their mobilities on gels, suggesting that none of the mutations had gross effects on the site of SU-TM cleavage or SU-TM protein interactions. In addition, there were no apparent differences in the relative levels of p15E and the R-peptide-cleaved form, p12E. The hyperfusogenic mutant, T464A, also displayed wild-type levels of virion gp70, p15E, and p12E (Fig. 4, lane 11), so the hyperfusogenic phenotype of this mutant was not due to greater levels of envelope protein in virions or enhanced R-peptide cleavage.
Ecotropic receptor binding.
The analyses described above identified mutants L445E, A457F, G460E, and T461P as being primarily fusion defective. None of these mutants induced syncytia or resulted in transduction, despite being normally processed, expressed on the surface of cells, and incorporated into virions at wild-type levels. To confirm that the block to fusion was not occurring because of a defect in their ability to bind to the ecotropic receptor, we measured binding to NIH 3T3 cells by a FACS-based assay, as previously described (22, 43). As shown in Table 3, all of these mutants could bind to cells expressing the ecotropic receptor, indicating that these mutants are defective primarily at a postbinding step of the viral entry process.
TABLE 3.
Binding of fusion-defective mutants to the ecotropic receptor
| Envelope protein |
Relative bindinga |
|---|---|
| Wild type | 100 ± 10 |
| L445E | 91 ± 2 |
| A457F | 77 ± 12 |
| G460E | 111 ± 8 |
| T461P | 85 ± 4 |
Supernatants from Env-transfected GPL cells were incubated with 3 × 105 NIH 3T3 cells at 4°C for 2 h and then incubated with the anti-gp70 monoclonal antibody 83A25 (8) for 1 h at 4°C. The cells were washed, incubated with fluorescein-conjugated goat anti-rat immunoglobulin G for 30 min at 4°C, washed again, and fixed with 4% paraformaldehyde. Samples were analyzed on a FACStar Plus flow cytometer (Becton Dickinson, San Jose, Calif.). A mean channel number (MN) was obtained for each sample and converted to fluorescence intensity (f) according to the formula log(f) = aMN + b, in which a and b are constants derived from a linear regression of a standard curve generated by using fluorescein-labeled RCP-70-5 microbeads (Spherotech Inc., Libertyville, Ill.) with known fluorescence intensities. Background fluorescence was obtained from mock-transfected GPL cells and was subtracted from the experimental values after the fluorescence conversion. Viral binding was averaged from at least two independent experiments and was normalized relative to the value for the wild type, ± standard error.
Fusion-defective mutants trans dominantly interfere with cell-cell fusion.
Previous analyses of the fusion peptide region of HIV-1 identified one mutant that was also trans dominant when coexpressed with the wild-type protein (10). In addition, several deletion mutations of the N terminus of gp41 have also been shown to dominantly interfere with syncytium formation (32). We therefore examined whether the group of fusion-defective envelope proteins could function in trans to inhibit fusion. Accordingly, wild-type and mutant envelope proteins were coexpressed in GPL cells at ratios of 1:1 and 10:1, and syncytium formation in overlaid XC cells was examined (Table 4). At a ratio of 1:1, very few syncytia were observed (2.1 to 5.4% of the levels produced by the wild-type protein alone), and even at a 10:1 excess of the wild-type protein, only 21.5 to 38.5% of the wild-type level of syncytium formation was seen. These results are in close agreement with the percent inhibition observed at these ratios for the trans-dominant HIV-1 fusion peptide mutant (10). We also examined the effect of coexpression of the fusion mutants and the wild-type protein on the transduction ability of retroviral particles. In contrast to the situation in the cell-cell fusion assay, there was no indication of a trans-dominant effect on viral titer at either a 1:1 or a 10:1 ratio (data not shown).
TABLE 4.
Fusion peptide mutants dominantly interfere with wild-type envelope protein function
| Envelope protein(s)a | Relative syncytium formationb
|
|
|---|---|---|
| 1:1 ratio | 10:1 ratio | |
| CEE+ (wild type) alone | 100 ± 0.4 | 100 ± 1.8 |
| CEE+ with: | ||
| L445E | 4.5 ± 2.8 | 30.2 ± 1 |
| A457F | 5.5 ± 1.7 | 34.5 ± 1.7 |
| G460E | 2.7 ± 1.8 | 38.5 ± 5.7 |
| T461P | 2.1 ± 1 | 21.7 ± 2.7 |
For the 1:1 ratio, 7.5 μg each of CEE+ and the mutant envelope protein plasmids were cotransfected into NIH 3T3 cells; for the 10:1 ratio, 15 μg of CEE+ and 1.5 μg of mutant plasmid were cotransfected.
The relative number of syncytia induced by NIH 3T3-XC cocultivation was calculated as described in Table 1, footnote a. Averages of three independent transfections ± standard errors are shown.
Taken together, our data provide evidence that the amino terminus of MoMuLV TM protein, p15E, contains a fusion peptide. This result confirms and extends the previous mutational analysis of AKV MuLV by Jones and Risser (21). Substitution of amino acid L445 within a hydrophobic stretch at the N terminus of this region and several substitutions (A457F, G460E/V, and T461P) in an adjacent GT-rich region disrupted envelope protein-mediated cell-cell fusion and resulted in noninfectious retroviral particles when expressed in a packaging system. Furthermore, all of these proteins were incorporated into viral particles and could bind to the ecotropic receptor. This group of mutants is therefore defective primarily in the fusion step of the viral entry process (Table 5). Our analyses also identified a second group of mutants that were unable to process Pr85 to the SU and TM subunits and were not found in retroviral particles. This group included several mutants with substitutions at residues L443 and G449 in the hydrophobic core and the more C-terminal L475R substitution (Table 5). In a previous study of MoMuLV TM protein, it was also noted that the G449R substitution resulted in envelope protein that was not incorporated into virions (3).
TABLE 5.
Summary of findings for p15E mutants
| Envelope protein | SU/TM processinga | Incorporationb | Cell-cell fusionc | Titerc | Phenotype |
|---|---|---|---|---|---|
| L441F | + | + | +++ | +++ | WTd |
| L443R | − | − | − | − | Processing/incorporation defective |
| L443A | − | Low | − | − | Processing/incorporation defective |
| L443I | − | − | − | + | Processing/incorporation defective |
| L445E | + | + | − | − | Fusion defective |
| G449L | − | − | − | − | Processing/incorporation defective |
| G449W | − | − | − | − | Processing/incorporation defective |
| G449A | − | − | − | − | Processing/incorporation defective |
| G449S | − | − | − | − | Processing/incorporation defective |
| A457F | + | Low | − | − | Fusion defective |
| G460V | + | + | ++ | + | Nearly WT |
| G460E | + | + | − | − | Fusion defective |
| T461P | + | + | − | − | Fusion defective |
| T463A | + | + | +++ | + | Nearly WT |
| T464A | + | + | ++++ | +++ | Hyperfusogenic, but WT titer |
| T464I | + | + | +++ | ++ | Nearly WT |
| T464K | + | + | +++ | ++ | Nearly WT |
| M467E | + | + | − | + | No syncytia, but some titer |
| A468R | + | + | +++ | +++ | WT |
| Q474E | + | + | ++ | + | Nearly WT |
| L475R | − | − | − | − | Processing/incorporation defective |
+, SU and TM proteins are processed; −, only Pr85 is present.
+, substantial incorporation; −, no incorporation; Low, low level of incorporation.
++++, value greater than the wild-type value; +++, 50 to 100% of wild-type value; ++, 10 to 50% of wild-type value; +, 1 to 10% of the wild-type value.
WT, wild type.
Fusion peptides are hypothesized to insert into lipid bilayers, causing local membrane destabilization and initiating the fusion of host and viral membranes (5, 39). The hydrophobic nature of these peptides is important, and mutations that increase the overall hydrophobicity of these regions have been reported to increase fusogenicity, while polar or charged residues abolish fusion (4, 9, 11). Our data also confirm the importance of hydrophobicity in the fusion peptide, since the L445E substitution produced a fusion-defective mutant and the downstream M467E mutation prevented cell-cell fusion and reduced the ability of the protein to mediate transduction. Other substitutions at residue L445 could not be assessed for their effects on fusogenicity since these changes prevented the proper processing of the envelope protein.
Fusion peptides also contain glycine residues, which have been suggested to be important for fusion peptide function, possibly because of their influence on the secondary structure of the region (4, 7, 39). The effects of the several substitutions we made at position G449 on fusogenicity could not be analyzed since they prevented envelope protein processing. Further downstream, the mutation of G460 to a charged residue (glutamic acid) prevented fusion, while the substitution of valine had less effect. A similar substitution of arginine for MoMuLV G460 (3) also produced a protein that was determined to be defective in either binding or fusion.
We also investigated the role of the multiple threonine residues at the N terminus of MoMuLV p15E, located immediately downstream of the fusion peptide core in the GT-rich region. The substitution of alanine for T463 had little effect on cell-cell fusion, while the substitution T464A actually increased fusogenicity. Only the T461P mutation abolished fusion, and it is possible that a proline residue at this position had a detrimental effect on the overall secondary structure of this region. Similarly, the replacement of amino acid A457 with a bulky phenylalanine residue could have distorted local structure, causing the lack of fusogenicity in this mutant.
None of the mutant proteins that were incorporated into particles appear to have altered SU-TM protein interactions, as similar levels of SU and TM proteins were present in the partially purified virions. This included several mutations made in the GT region, which is in the position analogous to the HIV-1 ST region that has been implicated in SU-TM protein interactions (7, 11, 23). However, the T464A substitution in the GT region did result in an envelope protein with a hyperfusogenic phenotype. While this increased fusogenicity may have occurred by reducing the overall polar nature of this region, it is also possible that this substitution destabilized the interaction between the SU and TM subunits, making the proposed postbinding conformational change easier. In addition, mutant A468R also resulted in increased syncytium formation when expressed in NIH 3T3 cells. Jones and Risser (21) previously transfected an analogous AKV MuLV mutant into XC cells and found that it also produced a twofold-higher level of syncytium formation than the wild-type protein.
Greater effects on cell-cell fusion, as measured by cocultivation of XC cells with NIH 3T3 cells, than on virus-cell fusion, as measured by transduction, were seen for the mutant proteins L443I and M467E. Differential effects on cell-cell fusion and virus-cell fusion have previously been reported for MuLV (20), HIV-1 (34), and influenza virus hemagglutinin (16, 38). This result may reflect inherent differences between these two related but distinct processes, in particular in the relative number of fusion peptides that may be required to achieve fusion over the larger surface area of cell-cell fusion. In addition, while the fusion-defective mutants caused a trans-dominant inhibition of cell-cell fusion when coexpressed with the wild-type protein, we saw very little effect on virus-cell fusion, as measured by titer.
In summary, we have obtained evidence to suggest that the N terminus of MoMuLV TM protein p15E is important for envelope protein-mediated fusion and probably constitutes a fusion peptide. Furthermore, this region of p15E also influences the proper processing, cell surface expression, and subsequent incorporation into particles of the mature envelope protein.
Acknowledgments
We thank J. Ragheb for providing the LETRSN vector.
This work was supported by Genetic Therapy, Inc. (GTI)/Novartis and by NIH grant CA59318-04.
REFERENCES
- 1.Albritton L M, Tseng L, Scadden D, Cunningham J M. A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell. 1989;57:659–666. doi: 10.1016/0092-8674(89)90134-7. [DOI] [PubMed] [Google Scholar]
- 2.Bergeron L, Sullivan N, Sodroski J. Target cell-specific determinants of membrane fusion within the human immunodeficiency virus type 1 gp120 third variable region and gp41 amino terminus. J Virol. 1992;66:2389–2397. doi: 10.1128/jvi.66.4.2389-2397.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berkowitz R D, Goff S P. Point mutations in Moloney murine leukemia virus envelope protein: effects on infectivity, virion association, and superinfection resistance. Virology. 1993;196:748–757. doi: 10.1006/viro.1993.1532. [DOI] [PubMed] [Google Scholar]
- 4.Bosch M L, Earl P L, Fargnoli K, Picciafuoco S, Giombini S, Wong-Staal F, Franchini G. Identification of the fusion peptide of primate immunodeficiency viruses. Science. 1989;244:694–697. doi: 10.1126/science.2541505. [DOI] [PubMed] [Google Scholar]
- 5.Brasseur R, Cornet B, Burny A, Vandenbranden M, Ruysschaert J M. Mode of insertion into a lipid membrane of the N-terminal HIV gp41 peptide segment. AIDS Res Hum Retroviruses. 1988;4:83–90. doi: 10.1089/aid.1988.4.83. [DOI] [PubMed] [Google Scholar]
- 6.Cosset F-L, Russell S J. Targeting retrovirus entry. Gene Ther. 1996;3:946–956. [PubMed] [Google Scholar]
- 7.Delahunty M D, Rhee I, Freed E O, Bonifacino J S. Mutational analysis of the fusion peptide of human immunodeficiency virus type 1: identification of critical glycine residues. Virology. 1996;218:94–102. doi: 10.1006/viro.1996.0169. [DOI] [PubMed] [Google Scholar]
- 8.Evans L H, Morrison R P, Malik F G, Protis J, Britt W J. A neutralizable epitope common to the envelope glycoproteins of ecotropic, polytropic, xenotropic, and amphotropic murine leukemia viruses. J Virol. 1990;64:6176–6183. doi: 10.1128/jvi.64.12.6176-6183.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Felser J M, Klimkait T, Silver J. A syncytia assay for human immunodeficiency virus type 1 (HIV-1) envelope protein and its use in studying HIV-1 mutations. Virology. 1989;170:566–570. doi: 10.1016/0042-6822(89)90448-0. [DOI] [PubMed] [Google Scholar]
- 10.Freed E O, Delwart E L, Buchschacher G I, Jr, Panganiban A T. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc Natl Acad Sci USA. 1992;89:70–74. doi: 10.1073/pnas.89.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freed E O, Myers D J, Risser R. Characterization of the fusion domain of human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc Natl Acad Sci USA. 1990;87:4650–4654. doi: 10.1073/pnas.87.12.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freed E O, Risser R. The role of envelope glycoprotein processing in murine leukemia virus infection. J Virol. 1987;61:2852–2856. doi: 10.1128/jvi.61.9.2852-2856.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallaher W R. Detection of a fusion peptide sequence in the transmembrane protein of human immunodeficiency virus. Cell. 1987;50:327–328. doi: 10.1016/0092-8674(87)90485-5. [DOI] [PubMed] [Google Scholar]
- 14.Gallaher W R, Ball J M, Garry R F, Griffin M C, Montelaro R C. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res Hum Retroviruses. 1989;5:431–440. doi: 10.1089/aid.1989.5.431. [DOI] [PubMed] [Google Scholar]
- 15.Gallaher W R, Segrest J P, Hunter E. Are fusion peptides really “sided” insertional helices? Cell. 1992;70:531–532. doi: 10.1016/0092-8674(92)90423-a. [DOI] [PubMed] [Google Scholar]
- 16.Gething M J, Doms R W, York D, White J. Studies on the mechanism of membrane fusion: site-specific mutagenesis of the hemagglutinin of influenza virus. J Cell Biol. 1986;102:11–23. doi: 10.1083/jcb.102.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gray K D, Roth M J. Mutational analysis of the envelope gene of Moloney murine leukemia virus. J Virol. 1993;67:3489–3496. doi: 10.1128/jvi.67.6.3489-3496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harter C, James P, Bachi T, Semenza G, Brunner J. Hydrophobic binding of the ectodomain of influenza hemagglutinin to membranes occurs through the “fusion peptide.”. J Biol Chem. 1989;264:6459–6464. [PubMed] [Google Scholar]
- 19.Heard J M, Danos O. An amino-terminal fragment of the Friend murine leukemia virus envelope glycoprotein binds the ecotropic receptor. J Virol. 1991;65:4026–4032. doi: 10.1128/jvi.65.8.4026-4032.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Januszeski M M, Cannon P M, Chen D, Rozenberg Y, Anderson W F. Functional analysis of the cytoplasmic tail of Moloney murine leukemia virus envelope protein. J Virol. 1997;71:3613–3619. doi: 10.1128/jvi.71.5.3613-3619.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones J S, Risser R. Cell fusion induced by the murine leukemia virus envelope glycoprotein. J Virol. 1993;67:67–74. doi: 10.1128/jvi.67.1.67-74.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadan M J, Sturm S, Anderson W F, Eglitis M A. Detection of receptor-specific murine leukemia virus binding to cells by immunofluorescence analysis. J Virol. 1992;66:2281–2287. doi: 10.1128/jvi.66.4.2281-2287.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kowalski M, Potz J, Basiripour L, Dorfman T, Goh W C, Terwilliger E, Dayton A, Rosen C, Haseltine W, Sodroski J. Functional regions of the envelope glycoprotein of human immunodeficiency virus type 1. Science. 1987;237:1351–1355. doi: 10.1126/science.3629244. [DOI] [PubMed] [Google Scholar]
- 24.MacKrell A J, Soong N W, Curtis C M, Anderson W F. Identification of a subdomain in the Moloney murine leukemia virus envelope protein involved in receptor binding. J Virol. 1996;70:1768–1774. doi: 10.1128/jvi.70.3.1768-1774.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCune J M, Rabin L B, Feinberg M B, Lieberman M, Kosek J C, Weissman I L. Endoproteolytic cleavage of gp160 is required for the activation of human immunodeficiency virus. Cell. 1988;53:55–67. doi: 10.1016/0092-8674(88)90487-4. [DOI] [PubMed] [Google Scholar]
- 26.Moore J P, Jameson B A, Weiss R A, Sattentau Q J. The HIV-cell fusion reaction. In: Bentz J, editor. Viral fusion mechanisms. Boca Raton, Fla: CRC Press; 1993. pp. 233–279. [Google Scholar]
- 27.Morgan R A, Nussbaum O, Muenchau D D, Shu L, Couture L, Anderson W F. Analysis of the functional and host range-determining regions of the murine ecotropic and amphotropic retrovirus envelope proteins. J Virol. 1993;67:4712–4721. doi: 10.1128/jvi.67.8.4712-4721.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinter A, Honnen W J, Tung J, O’Donnell P V, Hammerling U. Structural domains of endogenous murine leukemia virus gp70s containing specific antigenic determinants defined by monoclonal antibodies. Virology. 1982;116:499–516. doi: 10.1016/0042-6822(82)90143-x. [DOI] [PubMed] [Google Scholar]
- 29.Ragheb J A, Anderson W F. Uncoupled expression of Moloney murine leukemia virus envelope polypeptides SU and TM: a functional analysis of the role of TM domains in viral entry. J Virol. 1994;68:3207–3219. doi: 10.1128/jvi.68.5.3207-3219.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ragheb J A, Anderson W F. pH-independent murine leukemia virus ecotropic envelope-mediated cell fusion: implications for the role of the R peptide and p12E TM in viral entry. J Virol. 1994;68:3220–3231. doi: 10.1128/jvi.68.5.3220-3231.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rein A, Mirro J, Haynes J G, Ernst S M, Nagashima K. Function of the cytoplasmic domain of a retroviral transmembrane protein: p15E-p2E cleavage activates the membrane fusion capability of the murine leukemia virus Env protein. J Virol. 1994;68:1773–1781. doi: 10.1128/jvi.68.3.1773-1781.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaal H, Klein M, Gehrmann P, Adams O, Scheid A. Requirement of N-terminal amino acid residues of gp41 for human immunodeficiency virus type 1-mediated cell fusion. J Virol. 1995;69:3308–3314. doi: 10.1128/jvi.69.6.3308-3314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shinnick T M, Lerner R A, Sutcliffe J G. Nucleotide sequence of Moloney murine leukemia virus. Nature (London) 1981;293:543–548. doi: 10.1038/293543a0. [DOI] [PubMed] [Google Scholar]
- 34.Simmons G, McKnight A, Takeuchi Y, Hoshino H, Clapham P R. Cell-to-cell fusion, but not virus entry, in macrophages by T-cell line tropic HIV-1 strains: a V3 loop-determined restriction. Virology. 1995;209:696–700. doi: 10.1006/viro.1995.1307. [DOI] [PubMed] [Google Scholar]
- 35.Soneoka Y, Cannon P M, Ramsdale E E, Griffiths J C, Romano G, Kingsman S M, Kingsman A J. A transient three plasmid expression system for the production of high titre retroviral vectors. Nucleic Acids Res. 1995;23:628–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spear P. Virus-induced cell fusion. New York, N.Y: Plenum Press; 1987. pp. 3–32. [Google Scholar]
- 37.Vogt M, Haggblom C, Swift S, Haas M. Envelope gene and long terminal repeat determine the different biological properties of Rauscher, Friend, and Moloney mink cell focus-inducing viruses. J Virol. 1985;55:184–192. doi: 10.1128/jvi.55.1.184-192.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.White J M. Viral and cellular membrane fusion proteins. Annu Rev Physiol. 1990;52:675–697. doi: 10.1146/annurev.ph.52.030190.003331. [DOI] [PubMed] [Google Scholar]
- 39.White J M. Membrane fusion. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- 40.White J M, Kielian M, Helenius A. Membrane fusion proteins of enveloped animal viruses. Q Rev Biophys. 1983;16:151–195. doi: 10.1017/s0033583500005072. [DOI] [PubMed] [Google Scholar]
- 41.Witte O N, Tsukamoto-Adey A, Weissman I L. Cellular maturation of oncornavirus glycoproteins: topological arrangement of precursor and product forms in cellular membranes. Virology. 1977;76:539–553. doi: 10.1016/0042-6822(77)90236-7. [DOI] [PubMed] [Google Scholar]
- 42.Witte O N, Wirth D F. Structure of the murine leukemia virus envelope glycoprotein precursor. J Virol. 1979;29:735–743. doi: 10.1128/jvi.29.2.735-743.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu H, Soong N W, Anderson W F. Binding kinetics of ecotropic (Moloney) murine leukemia retrovirus with NIH 3T3 cells. J Virol. 1995;69:6557–6562. doi: 10.1128/jvi.69.10.6557-6562.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]