

Abstract
Previous reports have demonstrated that the U1 cell line, a model for postintegration latency, is defective at the level of Tat function and can be rescued by exogenously provided Tat protein. Sequence analysis of tat cDNAs from the U1 cell line identified two distinct forms of Tat, in agreement with the fact that this cell line contains two integrated human immunodeficiency (HIV) proviruses. One Tat cDNA lacked an ATG initiation codon, while the other contained an H-to-L mutation at amino acid 13 (H13→L). Both tat cDNAs were defective in terms of transcriptional activation of long terminal repeat-luciferase reporter gene in transient-transfection experiments. Introduction of the H13→L mutation in a wild-type tat background caused a severe reduction in transcriptional activation. Introduction of the same mutation in an infectious HIV molecular clone caused a severely defective phenotype which could be rescued when the HIV proviral DNA was transfected in a Jurkat cell line stably expressing the Tat protein (Jurkat-Tat) or in Jurkat cells treated with tumor necrosis factor alpha. Infectious virus stocks generated in Jurkat-Tat cells were used to infect Jurkat cells and exhibited severely impaired growth which could also be rescued by infecting Jurkat-Tat cells. These observations define tat mutations as a mechanism for HIV postintegration latency.
It is now recognized that human immunodeficiency virus type 1 (HIV-1) replication is continuously active at all stages of the disease in infected individuals (8, 20). Recent experiments using combination antiviral therapy have shown that inhibition of new rounds of infection produces a rapid and dramatic decrease in virus levels in plasma and lymph node (14, 26). However, this rapid decrease is followed by a lower rate of decrease, which has been ascribed to the persistence of chronically or latently infected cells (4, 21). The lower rate of decrease is thought to reflect the turnover of these chronically or latently infected cells and is a critical target in our effort to cure individuals infected with HIV (4, 21). Recent experiments have also documented that a significant proportion of the latent integrated HIV-1 DNA in resting CD4+ T cells is defective (4). HIV-1 can exhibit two different forms of latency in infected CD4+ T cells, pre- and postintegration latency, depending on the state of the provirus DNA within the infected cell. Different culture systems have served as in vitro models for postintegration latency, and the study of these cells has provided important insight into the mechanism of HIV transcriptional regulation and pathogenesis. The U1 monocytic cell line is one the most-studied models of postintegration latency and was cloned from a population of chronically HIV-1-infected U937 cells (11, 12). This cell line contains two integrated HIV proviruses and, under basal conditions, exhibits a pattern of viral mRNA expression characterized by low levels of multispliced HIV-1 transcripts encoding the regulatory proteins (22). HIV expression can be induced at the transcriptional level in U1 cells following exposure to tumor necrosis factor alpha (TNF-α) or phorbol esters (11, 12). This activation occurs, at least in part, via the translocation of the transcription factor NF-κB to the nucleus (13).
Several mechanisms have been proposed to explain this latency phenotype at a molecular level. Evidence has been presented that the endogenous Tat proteins are not active in the context of the U1 cells and that at least one of the two U1 proviruses is transcriptionally competent, indicating that the cellular factors necessary for Tat activity are present and functional in U1 cells (1–3, 7, 9).
To confirm that our U1 cell line (obtained from the AIDS Research Reagent Program, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), and maintained at low-passage level from frozen stock) contained inducible latent HIV-1 proviruses, we examined the ability of exogenous Tat protein to induce viral expression in these cells. U1 cells treated with recombinant Tat protein (19) released p24 antigen in their supernatant in a dose-dependent manner (Fig. 1). Virus released after Tat treatment was noninfectious when used in a secondary infection assay in Jurkat cells, indicating that the virus released was most likely defective (data not shown). These observations were consistent with the possibility that the tat open reading frame was mutated in the two proviruses integrated in the U1 cells.
FIG. 1.

Induction of HIV-1 production in U1 cells by exogenous Tat protein. One million U1 cells were incubated for 24 h with different amounts of recombinant Tat protein (19) in the presence of protamine sulfate (100 μg/ml) (10). Viral production was estimated by measuring the level of p24 antigen in culture supernatant by a commercial enzyme-linked immunosorbent assay (Dupont/NEN).
To determine the sequences of the tat cDNAs of the U1 proviruses, we performed reverse transcriptase PCR (RT-PCR) on total mRNA extracted from U1 cells treated with 10 nM tetradecanoyl phorbol acetate for 16 h. Total RNA purified with the Trizol reagent (GIBCO/BRL) from tetradecanoyl phorbol acetate-treated U1 cells was oligo(dT) primed to generate cDNA and was amplified by PCR using the following HIV-specific primers flanking the tat open reading frame: sense, 5′-ACGTGGATCCTTATTCGACAGAGGAGAGCAAGGA-3′ (final nucleotide, position 5374; a new BamHI site, introduced for cloning purpose, is indicated in boldface type), and antisense, 5′-AGATCGACCCAGATGAGTGCTAAGGATCCATTCA-3′ (first nucleotide, position 8045). The amplified fragment was gel purified, BamHI digested, and cloned into the unique BamHI site of the pREP9 expression vector downstream of the Rous sarcoma virus promoter (InVitrogen). Twenty independent clones were sequenced by cycle sequencing (Applied Biosystems), and two different nucleotide sequences were identified, in agreement with the presence of two HIV-1 proviruses integrated in U1 cells. Alignment of the deduced Tat amino acid sequences (Fig. 2) showed them to be most closely related to the NY5 isolate (18) and identified a distinct single amino acid substitution for each of the proteins compared to the HIV NY5 Tat protein. One of the tat cDNAs (Tat1U1) is mutated at the start codon (ATG→ACG), changing the first methionine amino acid to a threonine (M1→T). The other tat cDNA (Tat2U1) harbored a mutation (CAT→CTT) changing a histidine residue at position 13 to a leucine residue (H13→L). None of these mutations affected the sequence of the Rev protein.
FIG. 2.

Sequences of the two tat cDNAs from the U1 proviruses. The deduced amino acid sequences (Tat1U1 and Tat2U1), presented in single-letter code, are aligned to the closely related sequence of the HIV-1 NY5 virus Tat protein (TatNY5).
To determine whether these two U1 Tat proteins were functionally active in vivo, we first tested their abilities to transactivate the HIV-1 promoter by cotransfection of a construct containing the complete HIV-1 long terminal repeat (LTR) (nucleotide [nt] 1 to 791) driving the luciferase reporter gene (pLTR-Luc) and vectors expressing the different Tat proteins. We compared the activity of a wild-type (wt) Tat (TatACH2) (9) to the activity of each of the Tat proteins from the U1 cell proviruses (pRep9/tat1U1 and pRep9/tat2U1). As expected, transfection of increasing amounts of the TatACH2 expression vector resulted in increased luciferase activity, with a maximum 68-fold transactivation (Fig. 3A). No significant transactivation of the HIV-1 promoter was observed after cotransfection with pRep9/tat1U1 (which contains the Tat1U1 gene with a mutated start codon), indicating a lack of Tat expression, since Tat contains no other initiation codon. The Tat2U1 protein (H13→L) was also defective and transactivated the HIV-1 LTR to a significantly lower degree than did wild-type Tat (three- to fourfold reduction) (Fig. 3A). Since there are several amino acid differences between the primary sequences of TatACH2 and Tat2U1 (H13→L) proteins, we wanted to confirm that the H13→L mutation was responsible for the decrease in transactivation activity of Tat2U1 (H13→L). The same mutation (H13→L) was therefore introduced into the HIVNL4-3 tat open reading frame by site-directed mutagenesis and tested in transient-transfection experiments. Introduction of this mutation caused a severe reduction in Tat transactivating activity in comparison to that of wt protein, as predicted, therefore confirming the importance of the His13 residue in Tat activity (Fig. 3B).
FIG. 3.

Tat proteins encoded by the U1 proviruses are functionally defective. (A) Jurkat cells (5 × 106) were cotransfected by the DEAE-dextran method (25) with 1 μg of an HIV-1 LTR-luciferase reporter vector (pLTR-Luc) and 0.05, 0.15, 0.5, or 1.5 μg of a vector expressing either TatACH2 (pRep9/TatACH2 [hatched bars]), Tat1U1 (pRep9/tat1U1 [white bars]), or Tat2U1 (pRep9/tat2U1 [black bars]). To maintain the same amount of transfected DNA and avoid squelching artifacts, the different amounts of Tat expression vectors cotransfected were complemented to 1.5 μg of total DNA by using the empty pRep9 vector. Cells were harvested 24 h after transfection and luciferase activity was measured according to the luciferase assay system (Promega). Values (arbitrary luminescence units) represent the means of triplicate samples ± standard deviations (error bars) and are normalized to protein concentrations. (B) The same protocol as that described for panel A was used for the Tat-expressing vectors which expressed either HIVNL4-3 Tat72 protein (hatched bars) or the HIVNL4-3 Tat72 protein containing the His13→L substitution (black bars).
To examine the effect of the tat H13→L mutation in the context of virus infection, this mutation was introduced into the HIV molecular clone, pILIC (16) by site-directed mutagenesis. A PvuII-PvuII fragment (nt 4501 to 7231; +1 = mRNA start site) containing the coding region for the first exon of the Tat protein was subcloned into pUC19. Site-directed mutagenesis was performed on this vector with the Transformer kit (Promega, Madison, Wis.) and the following oligonucleotides: 5′-GCCCTGGAAGCTTCCAGGAAGTC-3′ (the codon for the leucine 13 residue is shown in boldface type) and a selection oligonucleotide changing a unique AatII site in pUC19 vector to an EcoRV site, 5′-GTGCCACCTGATATCTAAGAAACC-3′. The PvuII-mutated fragment was fully resequenced, and a PflMI-StuI fragment corresponding to nt 5459 to 6380 was purified and reintroduced into the unique PflMI-StuI sites of pILIC. The two resulting proviral infectious clones (wt and mutated) were electroporated into three different cell lines: Jurkat or two Jurkat cell lines stably expressing either one-exon Tat (Tat72) or two-exon Tat (Tat101) (19). We observed that wt virus replicated in all three cell lines, albeit with accelerated kinetics in Jurkat-Tat72 and Jurkat-Tat101 (data not shown). In contrast, when the provirus HIV-TatH13→L was transfected, no virus production was detected as long as 4 weeks after transfection in Jurkat control cells. As expected, the HIV-TatH13→L virus replicated with kinetics identical to those of wt virus after transfection into Jurkat-Tat72 or Jurkat-Tat101 (data not shown). To determine whether this mutation was sufficient to mimic the latent phenotype observed with U1 cells, Jurkat cells were transfected by electroporation and stimulated after different times (day 3, 10, or 16 postinfection) with TNF-α, a cytokine previously reported to induce HIV expression in U1 cells (12, 15). Detectable virus production was noted between days 15 and 20 following transfection of wt HIV and was significantly accelerated and amplified when cells were treated with TNF-α at day 3, 10, or 16 postinfection (Fig. 4A). In contrast, no virus production was detected following transfection of the HIV-TatH13→L mutant up to day 38 (Fig. 4B). Treatment of cells transfected with this mutant clone with TNF-α at day 3, 10, or 16 induced virus production to a level similar to that observed following infection with wt virus. This observation therefore demonstrates that the HIV-TatH13→L mutation is sufficient to reproduce the latent phenotype characteristic of the HIV provirus integrated in U1 cells.
FIG. 4.
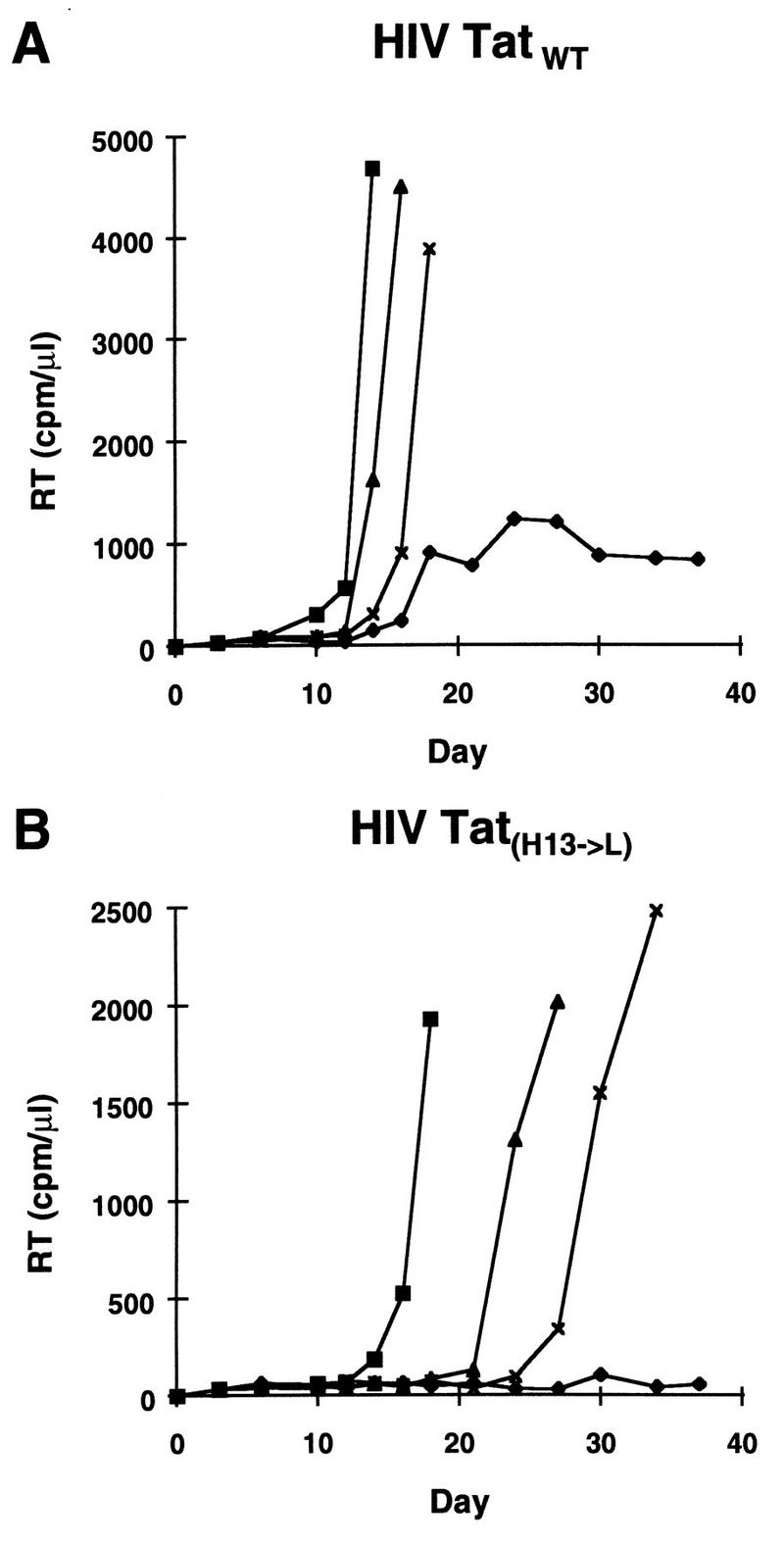
Replacement of histidine 13 with leucine confers a latent phenotype to an HIV molecular clone in transfection assays. The His13→L substitution was introduced into the tat gene of an infectious molecular clone of HIV-1 (pILIC). Jurkat cells were transfected either with wt proviral DNA (Fig. 4A) or with the TatH13→L mutated provirus (Fig. 4B) by electroporation. Cells were grown under standard conditions (⧫) or treated with TNF-α (800 U/ml; Genzyme) at day 3 (▪), day 10 (▴), or day 16 (×) after transfection. Virus replication was monitored at different intervals (2 to 3 days) by measuring supernatant RT activity.
To confirm these findings in the context of an HIV infection, virus stocks of both wt and mutated HIV-TatH13→L were generated after transfection of their DNAs into Jurkat-Tat72 cells. The presence of the tat mutation in these stocks was confirmed by sequence analysis of RT-PCR-amplified tat cDNA by using genomic viral RNA from ultracentrifuged virus as previously described (24). Normalized amounts of each virus stock were used to infect both Jurkat control and Jurkat-Tat101 cells. The growth kinetics of the HIV-Tat wt virus in Jurkat control cells showed a peak of replication at day 18 (Fig. 5A). In contrast, infection of Jurkat cells with HIV-TatH13→L produced low to undetectable amounts of virus (Fig. 5A). A low level of virus replication was detected at later time points (days 20 to 30). This low-level replication is likely to result from the presence of revertants in the virus population, since RT-PCR analysis of Tat showed that 10% of cDNAs had lost the original mutation that was introduced into the tat open reading frame. When the two virus stocks were used to infect Jurkat-Tat101 cells, comparable kinetics of replication were observed for the two viruses (Fig. 5B), with a peak of replication at day 12 for wt virus and at day 17 for HIV-TatH13→L. These data indicated that HIV-TatH13→L was defective for replication in Jurkat control cells and that this defective phenotype could be corrected by providing the wt Tat protein in trans.
FIG. 5.
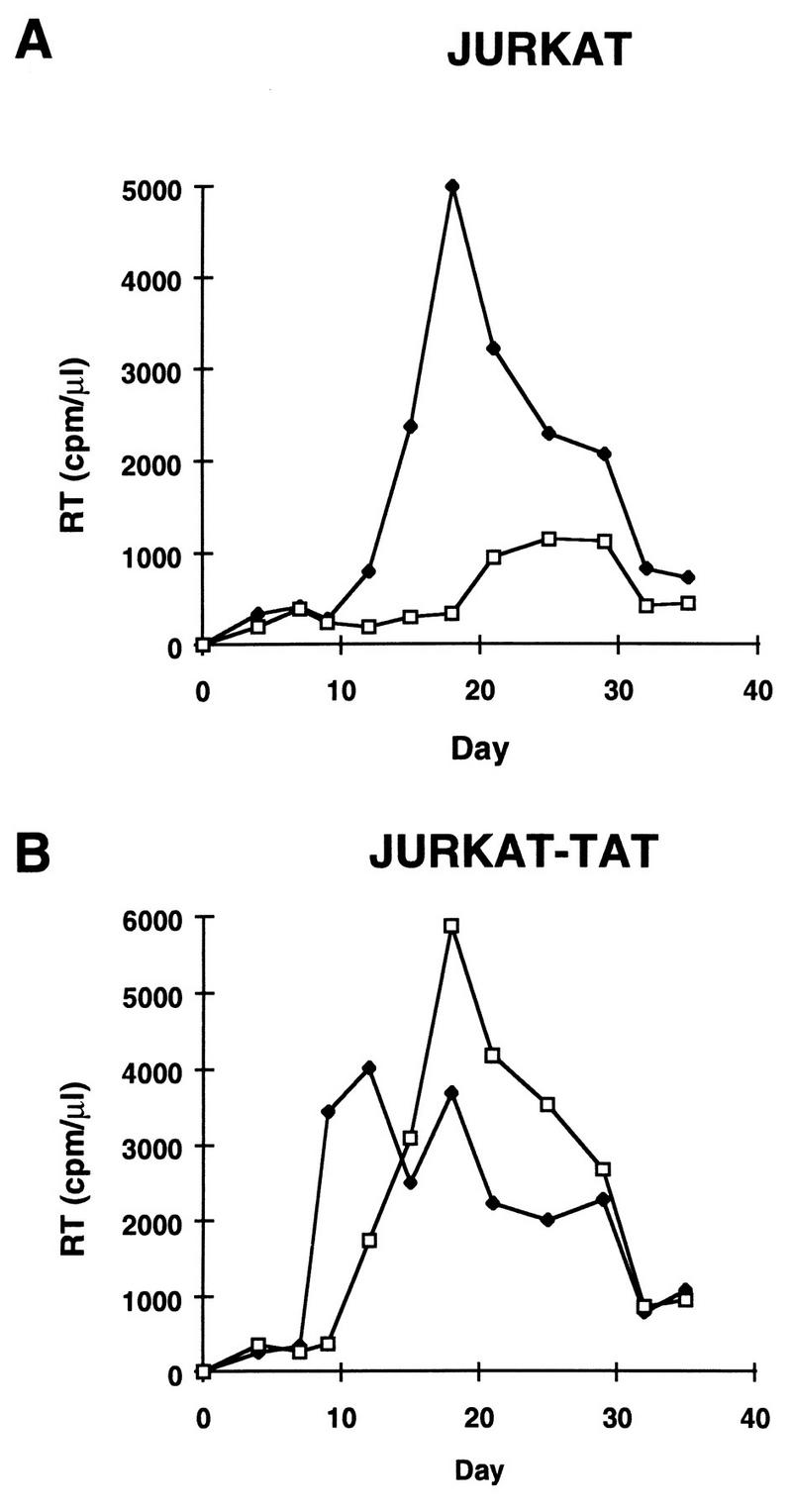
Replacement of histidine 13 with leucine in the tat open reading frame confers a latent phenotype to HIV in infection assays. Infectious stocks of wt (HIV-Tatwt) and mutated (HIV-TatH13→L) virus were generated following transfection of HIV-TatH13→L DNA into Jurkat-Tat72 cells. Infection of Jurkat (Fig. 5A) or Jurkat-Tat101 cells (Fig. 5B) was carried out by incubating 106 cells with 500,000 cpm of RT activity of HIV-Tatwt (⧫) or HIV-TatH13→L (□) for 2 h at 37°C in 100 μl. Virus replication was monitored at different intervals (2 to 3 days) by measuring RT activity in supernatants. A representative experiment out of four independent experiments is shown.
We have shown here that a defect in the Tat-TAR axis is involved in the latent phenotype of the U1 cell line, a model for postintegration latency. Determination of the sequences of the two tat genes encoded by the HIV-1 proviruses integrated into the U1 showed that both proviruses harbor a mutation in their tat open reading frames. Mutation of the start codon in the tat1U1 gene completely abolished the ability of the Tat1U1 expression vector to transactivate the HIV-1 LTR in transient-transfection experiments because of a translation defect. The Tat2U1 protein, which contains a single amino acid substitution (H13→L), exhibited reduced transactivating activity on the HIV-1 LTR. When introduced into the tat gene of an infectious HIV-1 clone, this substitution (H13→L) markedly impaired virus replication. This defect was compensated for by the constitutive expression of an active Tat protein in trans or by TNF-α treatment, faithfully reproducing the phenotype of the U1 provirus.
The H13→L substitution in the N-terminal region of tat modifies a domain that contains several acidic residues and exhibits potential amphipathic helicity (23). Examination of tat sequences from several HIV-1 isolates reveals that the histidine residue at position 13 is conserved among all virus strains (18), indicating the critical nature of this amino acid in Tat function and presumably in HIV replication. Further study will be required to determine how this mutation affects Tat function. However, its proximity to the Tat activation domain suggests that His13 plays an important role either in the structure of the activation domain or in its ability to interact with a cofactor(s) critical for Tat activity.
These observations and our previously reported identification of a TAR mutation in the ACH2 cell line (9) define the Tat-TAR axis as a critical target in the establishment of postintegration latency. The fact that HIV-infected latent cell lines contain defective virus genomes raises the question of whether replication-competent HIV can establish a true state of latent infection. True latency is indeed defined as a stable nonproductive interaction between a fully infectious virus and a host cell that is capable of being reversed to allow production of infectious virus. True latency is observed with herpesviruses and many endogenous retroviruses. Clearly, what has previously been called HIV postintegration latency does not respond to these criteria and should probably be relabeled as nonproductive defective infection. However, these observations do not exclude the possibility that a true state of latency can be achieved by HIV in its natural host. Further study of HIV-infected individuals should clarify this point. A high proportion of the tat genes amplified from the blood of infected individuals encode transactivation-defective Tat proteins (6). The presence of a disabling tat mutation in a provirus results in low levels of virus RNAs and proteins, as shown here for the U1 cells. Low or absent virus protein expression could allow the infected cell to escape immune surveillance and should therefore provide a selective advantage to cells infected by tat-defective viruses in comparison to cells infected by wt virus (5). While the role of defective viruses in HIV pathogenesis has not been clearly established, the selective survival of cells infected with tat-defective viruses might be one of the mechanisms used by the virus to persist in its host. Current efforts aimed at curing HIV infection by long-term treatment with antivirals will have to deal with this population of nonproductively infected cells as a possible source of infection reactivation after therapy has been terminated.
Acknowledgments
We thank the AIDS Research and Reference Reagent Program (NIAID, NIH), Anthony Fauci, and Guido Poli (NIAID, NIH) for providing the U1 cell line and for discussions. We thank Arnold Rabson and Malcolm Martin for providing the pILIC HIV molecular clone.
Carine Van Lint is a Chercheur Qualifié of the Fonds National de la Recherche Scientifique (FNRS, Belgium). This work was supported in part by a grant from the NIH of the U.S. Public Health Service (R01 AI 40847-01A1 ARRA) and by institutional funds from The Picower Institute for Medical Research.
REFERENCES
- 1.Adams M, Sharmeen L, Kimpton J, Romeo J M, Garcia J V, Peterlin B M, Groudine M, Emerman M. Cellular latency in human immunodeficiency virus-infected individuals with high CD4 levels can be detected by the presence of promoter-proximal transcripts. Proc Natl Acad Sci USA. 1994;91:3862–3866. doi: 10.1073/pnas.91.9.3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cannon P, Kim S H, Ulich C, Kim S. Analysis of Tat function in human immunodeficiency virus type 1-infected low-level-expression cell lines U1 and ACH-2. J Virol. 1994;68:1993–1997. doi: 10.1128/jvi.68.3.1993-1997.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen B K, Saksela K, Andino R, Baltimore D. Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by superinfection of nonproductively infected cell lines with recombinant luciferase-encoding viruses. J Virol. 1994;68:654–660. doi: 10.1128/jvi.68.2.654-660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chun T W, Carruth L, Finzi D, Shen X, Digiuseppe J A, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn T C, Kuo Y-H, Brookmeyer R, Zeiger M A, Barditch-Crovo P, Siliciano R F. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 5.Coffin J M. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 6.Delassus S, Meyerhans A, Cheynier R, Wain-Hobson S. Absence of selection of HIV-1 variants in vivo based on transcription/transactivation during progression to AIDS. Virology. 1992;188:811–818. doi: 10.1016/0042-6822(92)90536-x. [DOI] [PubMed] [Google Scholar]
- 7.Duan L, Oakes J W, Ferraro A, Bagasra O, Pomerantz R J. Tat and rev differentially affect restricted replication of human immunodeficiency virus type 1 in various cells. Virology. 1994;199:474–478. doi: 10.1006/viro.1994.1148. [DOI] [PubMed] [Google Scholar]
- 8.Embretson J, Zupancic M, Ribas J L, Burke A, Racz P, Tenner-Racz K, Haase A T. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- 9.Emiliani S, Van Lint C, Fischle W, Paras P, Ott M, Brady J, Verdin E. A point mutation in the HIV-1 tat responsive element is associated with post-integration latency. Proc Natl Acad Sci USA. 1996;93:6377–6381. doi: 10.1073/pnas.93.13.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feinberg M B, Baltimore D, Frankel A D. The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc Natl Acad Sci USA. 1991;88:4045–4049. doi: 10.1073/pnas.88.9.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Folks T, Justement J, Kinter A, Dinarello C, Fauci A. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science. 1987;238:800–802. doi: 10.1126/science.3313729. [DOI] [PubMed] [Google Scholar]
- 12.Folks T, Justement J, Kinter A, Schnittman S, Orenstein J, Poli G, Fauci A. Characterization of a promonocytic clone chronically infected with HIV and inducible by 13-phorbol-12-myristate acetate. J Immunol. 1988;140:1117–1122. [PubMed] [Google Scholar]
- 13.Griffin G E, Leung K, Folks T M, Kunkel S, Nabel G J. Activation of HIV gene expression during monocyte differentiation by induction of NF-kappa B. Nature. 1989;339:70–73. doi: 10.1038/339070a0. [DOI] [PubMed] [Google Scholar]
- 14.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 15.Kinter A, Poli G, Maury W, Folks T, Fauci A. Direct and cytokine-mediated activation of protein kinase C induces human immunodeficiency virus expression in chronically infected promonocytic cells. J Virol. 1990;64:4306–4312. doi: 10.1128/jvi.64.9.4306-4312.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leonard J, Parrott C, Buckler-White A J, Turner W, Ross E K, Martin M A, Rabson A B. The NF-κB binding sites in the human immunodeficiency virus type 1 long terminal repeat are not required for virus infectivity. J Virol. 1989;63:4919–4924. doi: 10.1128/jvi.63.11.4919-4924.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonard J M, Parrott C M, Buckler-White A, Martin M A, Rabson A B. Characterization of the infectivity of HIV proviruses containing mutations in LTR regulatory sequences. Int Conf AIDS. 1989;5:582. [Google Scholar]
- 18.Myers G, Berzofsky J A, Korber B, Smith R F, Pavlakis G N, editors. Human retroviruses and AIDS. A compilation and analysis of nucleic acid and amino acid sequences. Los Alamos, N.Mex: Los Alamos National Laboratory; 1995. [Google Scholar]
- 19.Ott M, Emiliani S, Van Lint C, Herbein G, Lovett J, Chirmule N, McCloskey T, Pahwa S, Verdin E. Immune hyperactivation of HIV-1 infected T cells mediated by Tat and the CD28 pathway. Science. 1997;275:1481–1485. doi: 10.1126/science.275.5305.1481. [DOI] [PubMed] [Google Scholar]
- 20.Pantaleo G, Graziosi C, Demarest J F, Butini L, Montroni M, Fox C H, Orenstein J M, Kotler D P, Fauci A S. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 21.Perelson A S, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho D D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 22.Pomerantz R J, Trono D, Feinberg M B, Baltimore D. Cells nonproductively infected with HIV-1 exhibit an aberrant pattern of viral RNA expression: a molecular model for latency. Cell. 1990;61:1271–1276. doi: 10.1016/0092-8674(90)90691-7. [DOI] [PubMed] [Google Scholar]
- 23.Rappaport J, Lee S-J, Khalili K, Wong-Staal F. The acidic amino-terminal region of the HIV-1 Tat protein constitutes an essential activating domain. New Biol. 1989;1:101–110. [PubMed] [Google Scholar]
- 24.Van Lint C, Amella C A, Emiliani S, John M, Jie T, Verdin E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J Virol. 1997;71:6113–6127. doi: 10.1128/jvi.71.8.6113-6127.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Lint C, Ghysdael J, Paras P, Jr, Burny A, Verdin E. A transcriptional regulatory element is associated with a nuclease-hypersensitive site in the pol gene of human immunodeficiency virus type 1. J Virol. 1994;68:2632–2648. doi: 10.1128/jvi.68.4.2632-2648.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, Saag M S, Shaw G M. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]