

Abstract
Reported here is the production of recombinant human rhinovirus 14 (HRV14) 2A protease from bacterial cells transformed with a heat-inducible plasmid containing the HRV14 2A cDNA sequence. Overexpressed 2A protein partitioned into the inclusion bodies was solubilized in urea and then refolded in the presence of Zn2+. Transition metals were required for the restoration of 2A protease activity as a structural component, but appeared to be inhibitory if added exogenously once the enzyme was refolded. Based on the cleavage specificity studies, a colorimetric assay was developed for the highly purified HRV14 2A protease. A peptide with the sequence RKGDIKSY–p-nitroanilide was found to be cleaved by the 2A protease with a kcat/Km ratio of ∼335 M−1s−1, which allows its activity to be measured continuously with a spectrophotometer or a microplate reader.
Human rhinoviruses (HRVs), the major etiologic agents of the common cold in humans, contain over a hundred distinct serotypes and belong to the picornavirus family (5). These small plus-strand RNA viruses encode a single open reading frame which is translated into a single large polyprotein with a size of 220 kDa (19, 20). Maturation cleavage of the polyprotein to generate functional viral proteins is mainly performed by two virally encoded proteases, designated 2A and 3C (19, 20). The first cleavage of the polyprotein is believed to be catalyzed by the 2A protease as a cotranslational event (19, 20). This cleavage, which takes place at the junction of capsid protein VP1 and the N terminus of the 2A protease itself, separates the viral capsid from the nonstructural proteins (19, 20). Most of the remaining cleavages are further processed by either the 3C protease or its precursor 3CD enzyme. In addition, it has been shown that these viral proteases are responsible for the cleavage of several important cellular proteins, including eukaryotic initiation factor eIF4G (p220), which may have a significant impact on host cell protein synthesis (2, 3, 7, 12, 13).
From a structural point of view, rhinovirus 2A and 3C proteins display a strong similarity to trypsin-like serine proteases, although both of them contain a cysteine as the active site nucleophile (16, 17, 21). The X-ray crystal structures of 3C proteases from both hepatitis A virus and HRV14 have been solved, revealing their structural similarity to the typical serine proteases (16, 17). However, no such study has been reported for the 2A enzymes. In spite of the similarities to the 3C enzymes, the HRV 2A protease has been proposed to be a zinc-binding protein. Sommergruber and his colleagues have demonstrated that zinc is essential for the structural integrity of the HRV2 2A protease (22, 23). Interestingly, the NS3 serine protease from hepatitis C virus (HCV) has been reported to contain a zinc-binding site comprising three cysteines and a water-histidine moiety (9, 14). Since the HRV 2A proteins contain such a conserved motif similar to the zinc-binding site of the HCV NS3 protease (6), it might be postulated that the viral 2A protease binds to the metal ion in approximately the same coordination geometry as that seen with the HCV NS3 protease (9, 14).
Great efforts have been made in the purification and characterization of the viral 2A proteases, especially the 2A enzyme from HRV2 (12, 13, 15, 18, 21–23). However, considerably less is known about the 2A proteases from other HRV strains. On the basis of sequence homology, HRV2 and HRV14 have been classified into two groups (19). Actually, HRV14 is more a polio-related enterovirus than a typical HRV strain. It is known that 2A proteases from polioviruses could not be purified with such a yield and purity as those reported for typical HRV 2A proteases (10, 13). In the present study, we describe the overexpression of the HRV14 2A protease in bacteria and the successful refolding of the enzyme in the presence of certain transition metals. The enzymatic parameters of the highly purified HRV14 2A protease were compared to those of its counterpart from HRV2. Furthermore, a simple colorimetric assay for the 2A protease was developed on the basis of its peptide substrate specificity, which should prove useful for the 2A enzyme characterization and high-throughput screening of 2A protease inhibitors.
Requirement of divalent cation for generation of active HRV14 2A protease expressed in Escherichia coli.
To produce a large quantity of recombinant 2A protein in E. coli, the gene encoding the HRV14 2A protease, derived from a biologically active cDNA clone of the virus pWR40, was amplified out of the cDNA with NdeI- and BamHI-modifying primers at the 5′ and 3′ ends, respectively. The coding sequence for the 2A protease was then inserted into a heat-inducible expression vector, pH10, described previously for the overexpression of human immunodeficiency virus type 1 protease and HRV14 3C protein in E. coli (1, 11). Since the 2A protease gene (total of 438 bp) was ligated into the vector at the 5′ NdeI and 3′ BamHI site, a start codon ATG from the NdeI recognition sequence was added in front of the 2A protease cDNA sequence. As a result, a methionine was expected at the N-terminal side of the recombinant 2A protein. The resulting construct, pH10/2A, was transformed into the competent E. coli strain RV308 cells, and the transformants were selected on tryptone-yeast extract (TY) plates containing tetracycline (10 mg/ml). A positive pH10/2A clone was selected and incubated at 30°C in 2× TY broth plus tetracycline (10 mg/ml) until the optical density at 600 nm reached approximately 0.7. The cell culture was induced to produce HRV14 2A protease by shifting the temperature to 42°C for 3 h. The cells were then harvested and analyzed for the presence of 2A protease by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). An overexpressed protein with an apparent molecular mass of ∼16 kDa was clearly seen in the crude extracts of transformed E. coli cells (see Fig. 2B, lane 1). This protein was not present in the control bacterial cells transformed with the vector with no insert (not shown). Further analysis showed that the 2A protein was present predominantly in the inclusion bodies as expected.
FIG. 2.
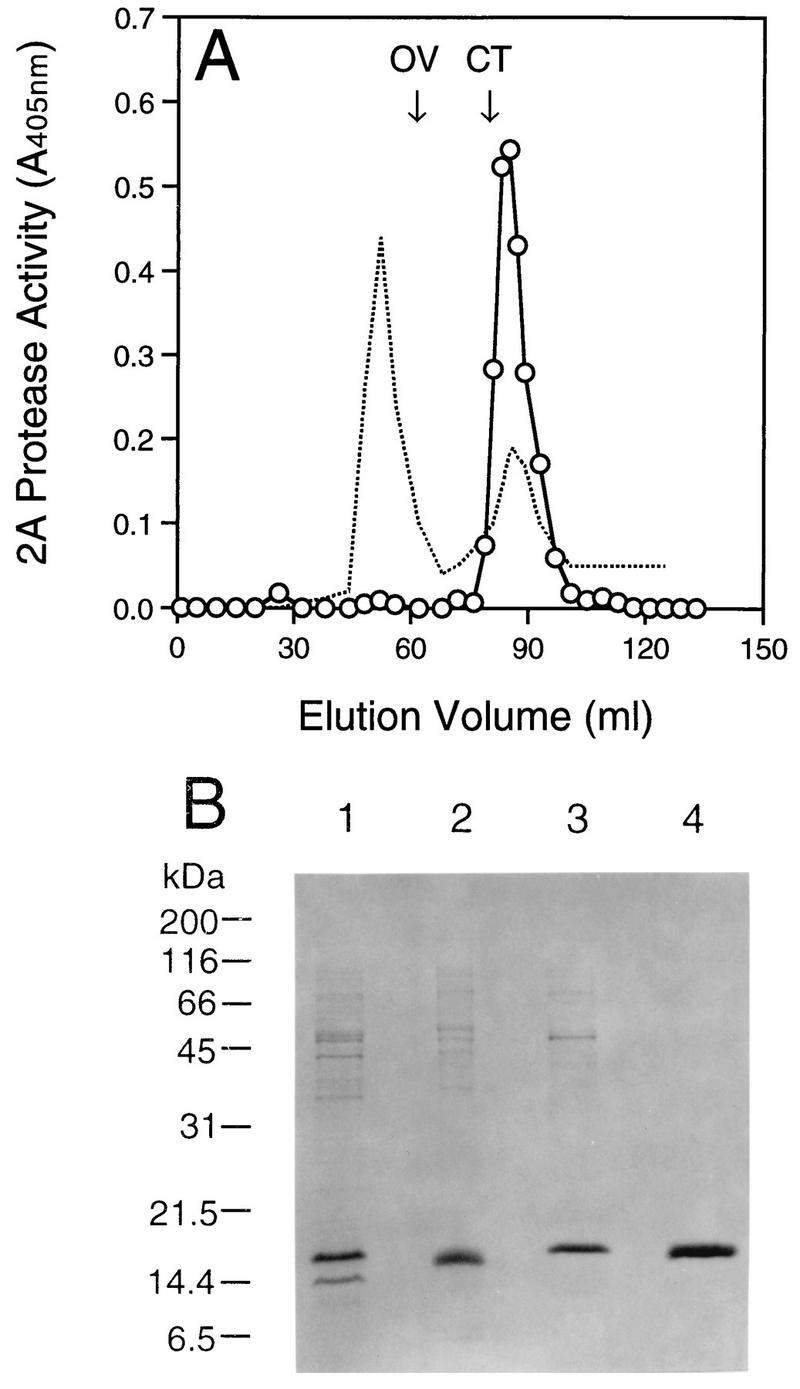
Purification of HRV14 2A protease. (A) Elution profile of HRV14 2A protease from Superdex-75 Hiload 16/60. Five milliliters of the 2A protein preparation or mixed gel filtration standards (1 mg/ml each) was loaded onto the column. Proteins were eluted at a flow rate of 1 ml/min with buffer B. Peaks were monitored at 280 nm (dashed line). The elution positions of the standards are labeled with OV (ovalbumin [43 kDa]) and CT (chymotrypsinogen A [19.5 kDa]). Fractions were examined for the pNA peptide cleavage activity as shown (○). (B) SDS-PAGE analysis of the purified HRV14 2A protein. Protein samples (∼1 μg) were separated by electrophoresis on a 16% gel and then stained with Coomassie blue. Lanes: 1, transformed bacterial cell lysate; 2, urea-solubilized inclusion bodies; 3 and 4, Mono Q and Superdex-75 column pooled fractions containing 2A protease activity, respectively.
To isolate the HRV14 2A protein, induced bacterial cells were collected from 2-liter cultures and resuspended in buffer A containing 25 mM HEPES (pH 8.0), 5 mM dithiothreitol (DTT), and 5% glycerol. The cells were treated with DNase I (1 U/ml) and lysozyme (0.5 μg/ml) for 60 min, followed by the addition of NaCl to 1 M. After lysis by sonication, cytoplasmic granules containing HRV14 2A protein were collected by centrifugation at 10,000 × g for 20 min. The pellet was washed first with 100 ml of 1 M NaCl, and then with 1 M urea, followed by water. Isolated inclusion bodies were solubilized overnight with 7 M urea in buffer A and then clarified by centrifugation at 10,000 × g for 30 min. The supernatant, containing the denatured 2A protein, was then diluted with the same buffer to a concentration of 0.1 mg/ml. To refold the urea-denatured 2A protein, the diluted sample was dialyzed overnight at 4°C against buffer B (25 mM HEPES [pH 8.0], 5% glycerol, 150 mM NaCl) in the presence of ZnCl2 or EDTA to see if metal ion was required for HRV14 2A protease refolding. As seen in Fig. 1A, maximal 2A protease activity was identified with the sample refolded in the presence of 0.1 mM Zn2+, although the control sample, refolded in the basic buffer, also displayed peptide cleavage activity, but to a lesser extent. In contrast, no active 2A enzyme was found in the sample refolded in the presence of 2 mM EDTA (Fig. 1A). No significant protein precipitation during dialysis was observed under these conditions (data not shown).
FIG. 1.
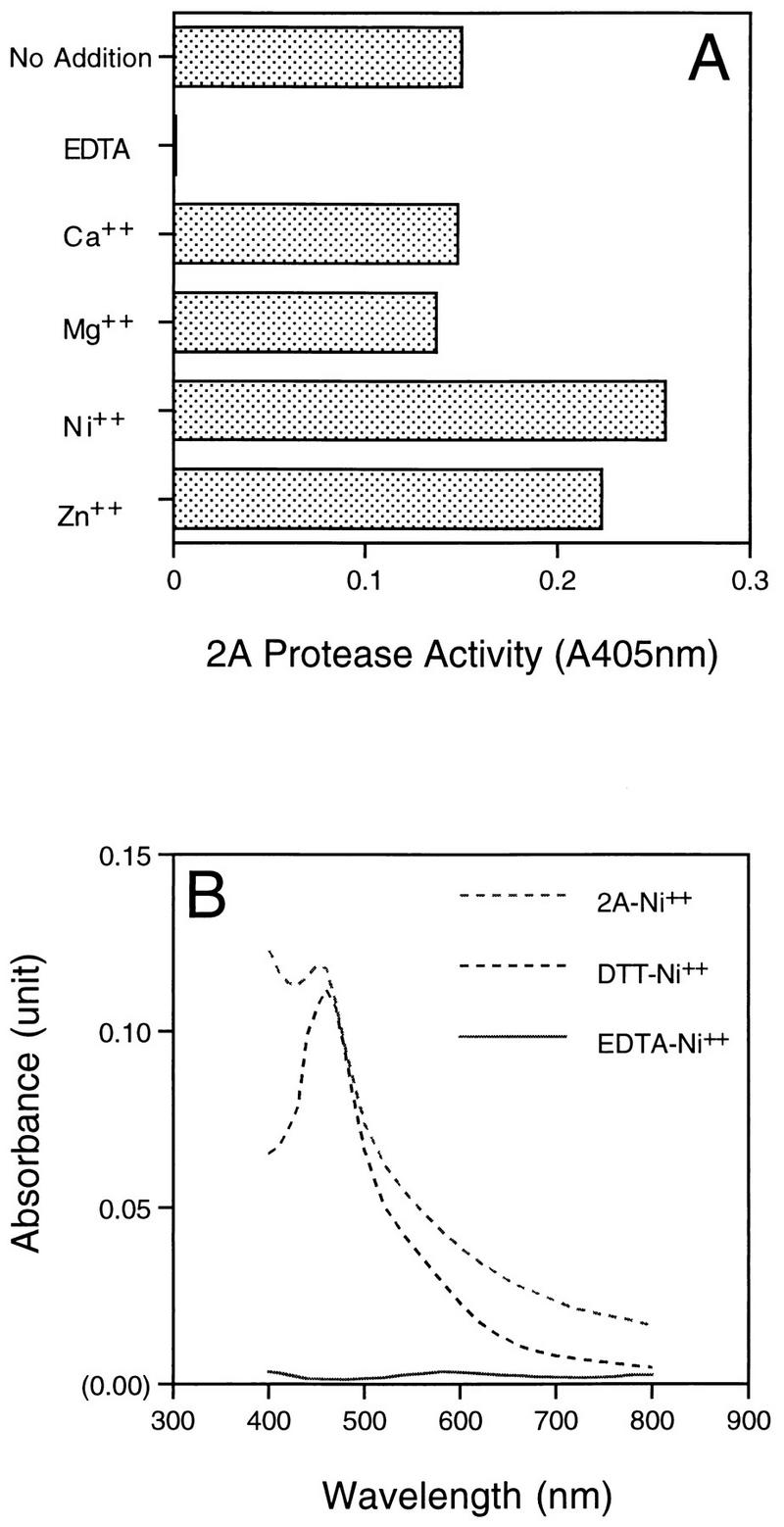
Metal requirement for generation of active HRV14 2A protease. (A) Refolding HRV14 2A protein. Urea-solubilized HRV14 2A protein was refolded through dialysis against buffer B with addition of EDTA (2 mM) or the specified divalent cations at 0.1 mM. After refolding, samples were all dialyzed against fresh buffer B to remove excess metals or chelator. The 2A protease activity was then detected with the pNA peptide RKGDIKSY-pNA as a substrate as described in the text. (B) Absorbance spectra of Ni2+ substituted 2A protease. Purified urea-denatured HRV14 2A protease (10 μM) was refolded with 0.1 mM Ni2+ in buffer B. DTT-Ni2+ and EDTA-Ni2+ complex were prepared freshly before the assay by mixing 15 μM DTT or 2 mM EDTA in buffer B with 0.1 mM Ni2+. Absorbance scanning of the samples was performed against a blank containing buffer B plus 0.1 mM Ni2+.
Active 2A protease could also be generated in the presence of a few other transition metals belonging to the thiophilic group. For example, the 2A protease refolded with Co2+ or Ni2+ displayed similar enzymatic activity to that with Zn2+ (Fig. 1A). On the other hand, Ca2+ and Mg2+ had no stimulatory effects on production of active 2A enzyme over the control sample (Fig. 1A). These data suggest that a divalent cation such as Zn2+ is required during protein folding and therefore is critical for the generation of active HRV14 2A protease. At the present time, it is unclear if HRV14 2A protein present in cells binds different metals, although HRV2 2A protease, purified as a recombinant protein from bacterial cells, has been shown to be complexed with zinc (24). In the case of NS3 protease from HCV, the enzyme complexes with Zn2+ via its conserved three cysteines and one water molecule forming a hydrogen bond with one histidine residue near the C terminus (9, 14). Sequence alignment of the HRV 2A proteins with the NS3 protease reveals that 2A protease also contains these four amino acids, and thus it may have a metal binding format similar to that seen in NS3 (6, 9, 14).
To verify that the 2A protein binds the metals via cysteine residues, we performed spectrophotometric analysis of the 2A protein-metal complex by using Ni2+-substituted 2A protease. Unlike zinc, thiolate-chelated Ni2+ such as the complex of DTT-Ni2+ has a rather characteristic absorption spectrum in the visible region (Fig. 1B). The 2A protease-Ni2+ complex was thus prepared by refolding the purified enzyme in the presence of Ni2+, and its absorbance spectrum was taken and compared to those of the DTT-Ni2+ and EDTA-Ni2+ complexes, in which the metal was ligated through sulfur and nitrogen-oxygen atoms, respectively. As seen in Fig. 1B, the 2A protease-Ni2+ complex displayed an absorption pattern similar to that of the DTT-Ni2+ complex, which was significantly different from that of the EDTA-Ni2+ complex, implying that the 2A protease might bind to the metal through its cysteines.
Cleavage of chromogenic peptides by purified HRV14 2A protease.
Purification of the 2A protein refolded in the presence of Zn2+ was achieved by chromatography on an ion-exchange column followed by size-exclusion separation. Briefly, the refolded proteins were loaded onto a Mono Q 5/5 column (Pharmacia) and then eluted with a linear gradient of 0.15 to 1 M NaCl in buffer A. Fractions containing the active 2A protease were identified by the colorimetric assay as described below, pooled, and loaded onto a Superdex-75 Hiload 26/60 column (Pharmacia). The proteins were then eluted with buffer B. The fractions containing the protease activity were identified, pooled, and used for further analyses. Most of the contaminants after the first column had sizes over 45 kDa, which could be readily separated from the 2A protease by gel filtration. Figure 2A shows the protein elution profile from the gel filtration column along with the peptide cleavage activity. The HRV14 2A protease eluted at the position corresponding to the calculated molecular mass of 16.1 kDa (Fig. 2A), which coeluted with the protease activity. Only one band was identified from the pooled fractions on a silver-stained gel (data not shown), and N-terminal amino acid analysis confirmed the presence of HRV14 2A protease in this sample (not shown). The purity of the isolated HRV14 2A was greater than 95%, as determined by high-performance liquid chromatography analysis and SDS-PAGE (Fig. 2B). The high-level expression of the HRV14 2A protease and its enrichment in inclusion bodies simplified its purification. Using the two-step purification protocol, we could obtain 5.7 mg of 2A protease per g of the transformed E. coli cells.
Similar to the HRV2 2A protease (26), we found that the minimal structure required for an efficient HRV14 2A protease cleavage was located at the N-terminal side of the scissile bond (data not shown). Thus, a chromogenic octapeptide (C2A14pNA) with a sequence of RKGDIKSY–p-nitroanilide (pNA) was tested as a substrate for the HRV14 2A protease. This peptide was designed on the basis of the viral protease native cleavage sites and was custom synthesized (American Peptide Co., Sunnyvale, Calif.). The N-terminal amino acids, corresponding to the 2A protease native cleavage site, were chemically linked to the chromophore pNA molecule riding at P1′. Since cleavage by the protease at the newly formed amide bond between P1 tyrosine and pNA would release free, yellow pNA, reactions could be monitored at a visible wavelength (405 nm) against a blank with either substrate or enzyme absent in the reaction mix (24, 26). A typical HRV14 2A protease assay was performed at 25°C for the time indicated in a 200-μl reaction mix containing 25 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM EDTA, and the HRV14 2A protease at the indicated concentrations. Reactions were directly performed in microtiter plate wells and monitored with a temperature-controlled microplate reader (Molecular Devices). To determine the kinetic parameters, reactions were monitored continuously at 405 nm to obtain the initial velocity of the cleavage reaction (prior to 10% substrate depletion). The kcat/Km values were determined with the equation v = (kcat/Km)[E][S], where [E] and [S] are enzyme and substrate concentrations, respectively. Kinetic parameters were calculated based on the assumption that all of the 2A protease present in the sample was active.
When peptide C2A14pNA was incubated with the purified HRV14 2A protease, increased A405 was detected, indicating that the enzyme could tolerate pNA located at P1′. Hydrolysis of this peptide by HRV14 2A protease was both time and enzyme concentration dependent (data not shown). HRV14 2A protease cleaved this peptide with a kcat/Km of ∼335 M−1 s−1. The Km for peptide C2A14pNA was determined as 0.96 mM. There was no measurable rate of peptide hydrolysis in the absence of HRV14 2A protease (result not shown). Both high-performance liquid chromatography and mass spectrometry analyses confirmed that cleavage of the C2A14pNA peptide by HRV14 2A protease occurred at the expected tyrosine-pNA scissile bond (results not shown).
HRV14 2A protease could also hydrolyze the peptide TRPIITTA-pNA designed for the HRV14 2A protease (26), but with approximately sixfold-less efficiency (Table 1). No detectable 2A protease activity was found when the pNA peptides derived from the HRV14 3C cleavage site (Table 1) were used, implying that the N-terminal residues were important for HRV14 2A protease substrate recognition. These results indicate that the peptides with a pNA directly linked to the scissile bond can be hydrolyzed by HRV14 2A protease, suggesting that the eight residues upstream of the scissile bond are sufficient for the 2A protease cleavage. Similar recognition features have also been described for the 2A protease from HRV2 and 3C protease from HRV14 (24, 26). It is possible that this characteristic is common for the 2A and 3C proteases from different HRV serotypes or even from other members in the picornavirus family. Since the pNA assay is convenient, quantitative, and can be performed with either a spectrophotometer or a microplate reader, it is expected that it will not only aid in the biochemical characterization of the 2A protease but also facilitate antiviral chemotherapeutic efforts.
TABLE 1.
Cleavage activity of HRV14 2A protease against chromogenic peptide substrates
| Codea | Peptide source | Peptide sequence | kcat/Km (M−1 s−1)b |
|---|---|---|---|
| C2A14pNA | HRV14 | RKGDIKSY-pNA | 335 ± 20 |
| C2A02pNA | HRV2 | TRPIITTA-pNA | 58.3 ± 7.5 |
| C2A02pNA2 | HRV2 | TRPIITTM-pNA | 25.0 |
| C3C14pNA8 | HRV14 | DSLETLFQ-pNA | NC |
| C3C14pNA5 | HRV14 | EALFQ-pNA | NC |
Peptides C3C14pNA8 and C3C14pNA5, representing the 2C/3A protease cleavage of HRV14 polyprotein, were originally designed for HRV14 3C protease (24). Peptide C2A02pNA is a specific substrate for HRV2 2A protease (26).
Kinetic parameters were determined as described in the text. Shown is the average of at least two independent experiments. The absence of products after a 3-h incubation with the enzyme (0.4 μM) is defined as no cleavage (NC).
Comparison of HRV14 2A protease with other HRV proteases.
With the assay described above, the enzymatic parameters of HRV14 2A protease were determined and compared to those of the 2A proteases from HRV2 and polioviruses. The purified HRV14 2A protease was very stable at 4°C for weeks without significant loss of activity at low concentrations. HRV14 2A protease activity was not significantly affected by high concentrations of NaCl, showing similar cleavage activity in the range of 15 mM to 1.5 M. Similar results were observed with the 2A protease from HRV2 (21). The HRV14 2A enzyme activity was slightly inhibited by organic solvents such as dimethyl sulfoxide and ethanol, even at low concentrations (5% [vol/vol]), being 27 and 25% less active than the control, respectively. HRV14 2A protease had an optimum pH range of 7 to 9, which was similar to that of the 2A enzyme from serotype 2. Inhibition studies of HRV14 2A protease with classic group-specific protease inhibitors gave results similar to those reported previously for the 3C protease from the same strain and the 2A protease from HRV2 (10, 21, 24, 25). For example, thiol alkylating reagents such as iodoacetamide and N-ethylmaleimide inactivated the enzyme, implying an important role for the cysteine residue in the 2A protease catalytic reaction. HRV14 2A protease was not sensitive to E-64, a specific inhibitor of papain-like cysteine proteases, even at a concentration of 0.1 mM (data not shown). However, the enzyme was found to be inhibited by elastatinal, a serine protease inhibitor, with a 50% inhibitory concentration of 90 μM (data not shown). Therefore, our inhibitor studies support the hypothesis that HRV 2A protease belongs to a novel class of cysteine proteases.
In contrast to HRV2 2A protease and the 3C protease from the same strain (4, 21), HRV14 2A protease demonstrated maximal enzymatic activity at low temperature, as shown in Fig. 3. HRV14 2A protease was sensitive to high temperature, while HRV2 2A protease demonstrated its highest cleavage activity at 40°C. Interestingly, Kuechler and his colleagues reported recently that several C-terminal residues, including phenylalanine 130 and histidines 135 and 137, are important for the HRV2 2A protein stability (15). Purified 2A enzymes carrying mutations at these sites demonstrate altered temperature tolerance (15). Sequence comparison results show that HRV14 2A protease contains the corresponding phenylalanine residue but not the two histidines. Thus, it is possible that the C-terminal amino acid difference between these proteins contributes to altered protein stability and integrity.
FIG. 3.

Effect of temperature on the protease activity of the 2A proteases from HRV2 and HRV14. The peptide substrates (250 μM) used for the HRV2 2A (□) and HRV14 2A (○) proteases are TRPIITTA-pNA and RKGDIKSY-pNA, respectively. Reactions were carried out at the indicated temperature for 30 min under the conditions described in the text, with no preincubation between the enzyme and the corresponding substrate involved.
In addition, HRV14 2A protease was eluted as a monomer from the gel filtration column as seen in Fig. 2A, while the HRV2 2A protease has been reported as a dimer (13). As mentioned above, HRV2 and HRV14 has been classified into different groups on the basis of amino acid sequence similarity (8). The 2A proteins encoded by HRV2 and HRV14 contain 142 and 146 amino acids, respectively, sharing only 40% identity and 57% similarity at the amino acid level. Interestingly, the 2A protease of coxsackievirus B4 also behaves as a monomer (13). These results together would support the notion that HRV14 is more closely related to enterovirus than to typical HRV strains.
Inhibition of refolded HRV14 2A protease activity by transition metals present in the reactions.
In contrast to their positive impact on generation of active 2A protease during the refolding process, metals, including Co2+, Cu2+, Ni2+, and Zn2+, were not required for its enzymatic activity. In fact, this group of metals strongly inhibited the 2A protease cleavage activity, even at low micromolar concentrations, as illustrated in Fig. 4. Interestingly, extensive dialysis of the 2A enzyme against EDTA or addition of chelating agents directly into the cleavage reactions did not significantly affect the 2A protease activity. For example, EDTA did not inhibit 2A protease cleavage activity at concentrations of ≤5 mM; 85% activity remained at the highest concentration tested (25 mM). Dialysis of the active 2A protease against the buffer containing 2 mM EDTA for 48 h did not affect the enzyme activity (not shown). Additionally, no detectable 2A enzyme inhibition was found when EGTA (up to 25 mM) was present in the reaction mixtures.
FIG. 4.

Effect of divalent cations on the HRV14 2A protease activity. Peptide RKGDIKSY-pNA at 250 μM was incubated at 22°C with the purified 2A protease (0.2 μM) for 30 min under the standard assay conditions. Different cations were included in the 2A cleavage reaction at three different concentrations. The effect of the divalent cations on the HRV14 2A activity was expressed as the percentage of activity of the control sample containing no metal ions. Shown is the average of two independent measurements with variation less than 3%.
These results suggest that the metal binds to the enzyme very tightly; the bound metal has only a structural role and is not required for the enzyme catalytic activity. Supporting evidence includes an efficient inhibition of 2A protease activity by zinc and other metals at low concentrations. Zinc inactivation of HRV14 2A protease has a 50% inhibitory concentration value of 0.5 μM, which is close to the 2A enzyme concentration used in the reaction. Unlike the NS3 protease, rhinovirus 2A protease is a cysteine protease requiring the presence of a free thiol group as the nucleophile during the hydrolytic reaction. Therefore, such an efficient 2A protease activity inhibition by Zn2+ could result from direct binding of the metal to the 2A protease active site cysteine residue. It should be noted that HRV14 2A protease contains six other cysteines and several histidines, which could also interact with the added metal ions. Nevertheless, these data strongly suggest that the transition metal bound to the 2A enzyme did not participate in the catalytic reaction directly. There is no doubt that crystallographic analyses of HRV 2A protease will generate a better understanding of the roles of the 2A protease-bound metal. Apparently, the availability of the highly purified HRV14 2A protease would facilitate this effort.
Acknowledgments
We thank John Richardson for performing mass spectrometry experiments, Joseph Colacino and Beverly Heinz for critical readings of the manuscript, and W. Sommergruber from Boehringer Ingelheim for providing us with the purified HRV2 2A protease and helpful suggestions. We are also grateful to Wai-Ming Lee and Roland Rueckert (University of Wisconsin, Madison) for providing the biologically active HRV14 cDNA clone.
REFERENCES
- 1.Birch G M, Black T, Malcolm S K, Lai M T, Zimmerman R E, Jaskunas S R. Purification of recombinant human rhinovirus 14 3C protease expressed in E. coli. Protein Expr Purif. 1995;6:609–618. doi: 10.1006/prep.1995.1080. [DOI] [PubMed] [Google Scholar]
- 2.Clark M E, Lieberman P M, Berk A J, Dasgupta A. Direct cleavage of human TATA-binding protein by poliovirus protease 3C in vivo and in vitro. Mol Cell Biol. 1993;13:1232–1237. doi: 10.1128/mcb.13.2.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark M E, Hämmerle T, Wimmer E, Dasgupta A. Poliovirus proteinase 3C converts an active form of transcription factor IIIC to an inactive form: a mechanism for inhibition of host cell polymerase III transcription by poliovirus. EMBO J. 1991;10:2941–2947. doi: 10.1002/j.1460-2075.1991.tb07844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cordingley M G, Callahan P L, Sardana V V, Garsky V M, Colonno R J. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J Biol Chem. 1990;265:9062–9065. [PubMed] [Google Scholar]
- 5.Couch R B. Rhinoviruses. In: Fields B N, Knipe D M, editors. Virology. New York, N.Y: Raven Press; 1990. pp. 607–629. [Google Scholar]
- 6.De Francesco R, Urbani A, Nardi M C, Tomei L, Steinkuhler C, Tramontano A. A zinc binding site in viral serine proteinase. Biochemistry. 1996;35:13282–13287. doi: 10.1021/bi9616458. [DOI] [PubMed] [Google Scholar]
- 7.Hellen C U T, Fäcke M, Kräusslich H-G, Lee C-K, Wimmer E. Characterization of poliovirus 2A proteinase by mutational analysis: residues required for autocatalytic activity are essential for induction of cleavage of eukaryotic initiation factor 4F polypeptide p220. J Virol. 1991;65:4226–4231. doi: 10.1128/jvi.65.8.4226-4231.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horsnell C, Gama R E, Hughes P J, Stanway G. Molecular relationships between 21 human rhinovirus serotypes. J Gen Virol. 1995;76:2549–2555. doi: 10.1099/0022-1317-76-10-2549. [DOI] [PubMed] [Google Scholar]
- 9.Kim J L, Morgenstern K A, Lin C, Fox T, Dwyer M D, Landro J A, Chambers S P, Markland W, Leper C A, O’Malley E T, Harbeson S L, Rice C M, Murcko M A, Caron P R, Thomson J A. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996;87:343–355. doi: 10.1016/s0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 10.König H, Rosenwirth B. Purification and partial characterization of poliovirus protease 2A by means of a functional assay. J Virol. 1988;62:1243–1250. doi: 10.1128/jvi.62.4.1243-1250.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai M T, Dee A G, Zervos P H, Heath W E, Scheetz M E. Biosynthesis and processing of the gag and pol polyproteins of human immunodeficiency virus type 1 in E. coli. In: Pearl L H, editor. Retroviral proteases: maturation and morphogenesis. London, United Kingdom: MacMillan; 1990. pp. 63–71. [Google Scholar]
- 12.Lamphear B J, Yan R, Yang F, Waters D, Liebig H-D, Klump H, Kuechler E, Skern T, Rhoads R E. Mapping the cleavage site in protein synthesis initiation factor eIF-4γ of the 2A proteases from human coxsackievirus and rhinovirus. J Biol Chem. 1993;268:19200–19203. [PubMed] [Google Scholar]
- 13.Liebig H-D, Ziegler E, Yan R, Hartmuth K, Klump H, Kowalski H, Blaas D, Sommergruber W, Frasel L, Lamphear B J, Rhoads R, Kuechler E, Skern T. Purification of two picornaviral 2A proteinases: interaction with eIF-4γ and influence on in vitro translation. Biochemistry. 1993;32:7581–7588. doi: 10.1021/bi00080a033. [DOI] [PubMed] [Google Scholar]
- 14.Love R A, Parge H E, Wichersham J A, Hostomsky Z, Habuka N, Moomaw E W, Adachi T, Hostomska Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell. 1997;87:331–342. doi: 10.1016/s0092-8674(00)81350-1. [DOI] [PubMed] [Google Scholar]
- 15.Luderer-Gmach M, Liebig H-D, Sommergruber W, Voss T, Fessl F, Skern T, Kuechler E. Human rhinovirus 2A proteinase mutant and its second-site revertants. Biochem J. 1996;318:213–218. doi: 10.1042/bj3180213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malcolm B A. The picornaviral 3C proteinases: cysteine nucleophiles in serine proteinase folds. Protein Sci. 1995;4:1439–1445. doi: 10.1002/pro.5560040801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matthews D A, Smith W W, Ferre R A, Condon B, Budahazi G, Sisson W, Villafranca J E, Janson C A, McElroy H E, Gribskov C L, Worland S. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell. 1994;77:761–771. doi: 10.1016/0092-8674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 18.Molla A, Hellen C U T, Wimmer E. Inhibition of proteolytic activity of poliovirus and rhinovirus 2A proteinases by elastase-specific inhibitors. J Virol. 1993;67:4688–4695. doi: 10.1128/jvi.67.8.4688-4695.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmenberg A C. Proteolytic procession of picornaviral polyprotein. Annu Rev Microbiol. 1990;44:603–623. doi: 10.1146/annurev.mi.44.100190.003131. [DOI] [PubMed] [Google Scholar]
- 20.Porter A G. Picornavirus nonstructural proteins: emerging roles in virus replication and inhibition of host cell functions. J Virol. 1993;67:6917–6921. doi: 10.1128/jvi.67.12.6917-6921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sommergruber W, Ahorn H, Zöphel A, Maurer-Fogy I, Fessl F, Schnorrenberg G, Liebig H-D, Blaas D, Kuechler E, Skern T. Cleavage specificity on synthetic peptide substrates of human rhinovirus 2 proteinase 2A. J Biol Chem. 1992;267:22639–22644. [PubMed] [Google Scholar]
- 22.Sommergruber W, Casari G, Fessl F, Seipelt J, Skern T. The 2A proteinase of human rhinovirus is a zinc containing enzyme. Virology. 1994;204:815–818. doi: 10.1006/viro.1994.1599. [DOI] [PubMed] [Google Scholar]
- 23.Voss T, Meyer R, Sommergruber W. Spectroscopic characterization of rhinoviral protease 2A: Zn is essential for the structural integrity. Protein Sci. 1995;4:2526–2531. doi: 10.1002/pro.5560041209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Q M, Johnson R B, Cox G A, Villarreal E C, Loncharich R J. A continuous colorimetric assay for rhinovirus-14 3C protease using peptide p-nitroanilides as substrates. Anal Biochem. 1997;252:238–245. doi: 10.1006/abio.1997.2315. [DOI] [PubMed] [Google Scholar]
- 25.Wang Q M, Johnson R B, Cohen J D, Voy G T, Richardson J M, Jungheim L N. Development of a continuous fluorescence assay for rhinovirus-14 3C protease using synthetic peptides. Antiviral Chem Chemother. 1997;8:303–310. [Google Scholar]
- 26.Wang Q M, Sommergruber W, Johnson R B. Cleavage specificity of human rhinovirus 2 2A protease for peptide substrates. Biochem Biophys Res Commun. 1997;235:562–566. doi: 10.1006/bbrc.1997.6830. [DOI] [PubMed] [Google Scholar]