

Abstract
Background
Enhancing flag leaf nitrogen use efficiency (NUE) in wheat production can substantially increase crop productivity while minimizing nitrogen application. Quantitative trait loci (QTLs) for NUE-related have been rarely reported in wheat flag leaf traits.
Results
In this study, a natural population of 243 varieties and an RIL population of 123 F7 recombinants were subjected to different nitrogen treatments. A genome-wide association study (GWAS) and linkage analysis were performed for four agronomic traits in terms of flag leaf length, flag leaf width, flag leaf area, and SPAD (chlorophyll content) under low and normal nitrogen conditions. Through GWAS, 1,016 significant SNP loci were identified and clustered into 290 QTLs, including 11 stably mapped QTLs (stable detection in multiple environments). Additionally, an AC population was established to verify the GWAS results and identify reliable QTL intervals. Three stable loci, namely, QFLLR6D.3 QFLWR6A.6, and QSPADR5B.3, were validated in the AC population, located 1.34 Mb, 2.84 Mb, and 5 Mb away from linkage mapping significant QTL, respectively. Through further transcriptome analysis of Chilero leaves at the jointing, anthesis and grain filling stages, four DEGs were identified within QSPADR5B.3. Among them, TraesCS5B02G394300, TraesCS5B02G394200, and TraesCS5B02G39390 encode beta-glucosidases, and TraesCS5B02G396400 encodes a potassium channel.
Conclusions
These findings offer potential candidate genes for wheat breeding, and provide a foundation for exploring the molecular targets underlying wheat NUE.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12864-025-12025-7.
Keywords: Wheat, Nitrogen use efficiency, Flag leaf, GWAS, QTL mapping, RNA-seq, Candidate genes
Background
Photosynthesis in plant leaves is the foundational process for energy conversion and the production of essential nutrients, and it serves as a critical indicator for assessing plant growth status [1–3]. In wheat, the flag leaf, which is the uppermost functional leaf of the plant, plays a decisive role in the yield formation of wheat. Through mechanisms that regulate photosynthetic efficiency, material transport, and stress responses, the flag leaf directly influences the accumulation of organic matter in the grains and the final yield [4]. Given its pivotal role, the flag leaf is often considered a key determinant of wheat productivity under various environmental conditions [5, 6].
Nitrogen is one of the essential macronutrients required for crop growth, and the amount of nitrogen fertilizer applied to the field can significantly influence crop yield and quality [7, 8]. Previous studies have demonstrated that under nitrogen stress conditions, varieties with relatively high chlorophyll contents can maintain relatively strong photosynthetic capacity, which is conducive to stable yield formation [9–12]. These findings suggest that the chlorophyll content, as an indicator of photosynthetic potential, is crucial for maintaining productivity under adverse conditions. The traits of flag leaf, such as length, width, area and chlorophyll content are closely related to yield, and they are important phenotypic indicators in breeding programs [3, 5]. Therefore, understanding the genetic basis of these traits is essential for developing high-yielding wheat varieties that can perform well under varying N conditions [13, 14].
To date, numerous studies have been conducted on quantitative trait locus (QTL) mapping of wheat flag leaf traits. These studies have provided valuable insights into the genetic architecture underlying flag leaf morphology and function [15–17]. However, relatively few reports on QTLs associated with the nitrogen response of wheat flag leaves in high and low nitrogen environments exist [18]. Given the importance of nitrogen management in agricultural practices, identifying QTLs that improved nitrogen use efficiency (NUE) and stress tolerance in wheat flag leaves could have significant implications for sustainable crop production. Future research should focus on elucidating the genetic mechanisms underlying these complex traits and exploring their potential applications in breeding programs aimed at enhancing wheat productivity under diverse environmental conditions [15].
At present, three main approaches are used to discover genes for complex quantitative trait genes. The first is to construct a genetic linkage map through hybridization and perform QTL mapping for agronomic, physiological, and biochemical traits related to nitrogen absorption and utilization [19–23]. The second approach is to use the GWAS method to rapidly construct a mapping population from natural populations and draw high-resolution genetic maps [24–27]. The third approach involves the use of transcriptomics, proteomics, and other studies to identify genes and proteins that respond to nitrogen and perform localization [25, 28–30]. These genes may be major genes in the nitrogen absorption and metabolism pathways or genes related to traits associated with the control of NUE. However, most of the previous studies have problems such as a small number of SNP markers, a small population size, and an experimental environment that deviates from production conditions. These issues result in a relatively low application value in wheat breeding and poor reproducibility [20, 31]. This study aims to combine these three main methods and, on the basis of the results of multi-year and multi-site experiments, identify QTL loci and candidate genes with greater stability and application value.
In this study, natural populations of 243 varieties derived from China’s Yellow-Huai River Valley (named as CH population detailed in Supplementary Table S1) and an RIL populations of 123 F7 recombinants derived from the cross between Avocet and Chilero (named as AC population) were utilized to investigate four agronomic traits associated with wheat flag leaf, namely FLL (flag leaf length), FLW (flag leaf width), FLA (flag leaf area), SPAD (chlorophyll content). Under two nitrogen treatments, normal nitrogen supply (NN) and low nitrogen stress (LN), genomic regions associated with nitrogen-stress tolerance and NUE were explored. The objectives of this study were as follows: (1) to study the low nitrogen tolerance indices (LNTI) of agronomic traits in response to nitrogen stress in the two populations; (2) to identify genomic regions related to NUE by combining linkage analysis and association mapping; (3) to predict the main candidate genes for flag leaf nitrogen-stress-related traits by integrating RNA-seq. This study aims to jointly anchor the genomic regions and candidate genes related to wheat flag leaf NUE through three methods, providing valuable information for the molecular breeding of wheat NUE.
Results
Phenotypic evaluation
As shown in Fig. 1(a, b) and Table S2, the four traits in both populations displayed approximately normal distributions, aligning with the polygenic inheritance patterns typical of continuously distributed quantitative traits. Pearson correlation analysis revealed strong positive correlations (p < 0.001) between FLA and both FLL and FLW in both populations. In the CH population, FLL and FLW also demonstrated a significant positive correlation (p < 0.001); however, there was no significant association between these two traits in the AC population. Notably, SPAD values showed a significant negative correlation with FLA in the AC population (p < 0.01 NN condition), whereas this relationship was not detected in the CH population.
Fig. 1.

The results of the phenotypic analysis of the four flag leaf traits, which present matrix scatter plots. a, b Frequency distribution histograms, and Pearson correlation analysis plots for the CH population and AC population. In these visualizations, blue denotes NN treatment, while yellow denotes LN treatment. c–f (CH population) and g–j (AC population) Present the results of student's t-tests comparing phenotypic traits. *, **, and*** indicate significant at p < 0.05, p < 0.01, and p < 0.001, respectively. FLL: flag leaf length; FLW: flag leaf width; FLA: flag leaf area; SPAD: chlorophyll content. NN: normal nitrogen treatment; LN: low nitrogen treatment. The 23XYT and 24XYT refer to the 2022–2023 and 2023–2024 field experiments in Xiyangtun, respectively, and 23ZC and 24ZC refer to the 2022–2023 and 2023–2024 field experiments in Zhaocun, respectively; BLUP: best linear unbiased predictions
The FLL, FLW, FLA, and SPAD in both wheat populations exhibited a significant reduction under LN treatment compared to NN treatment (p < 0.001). This indicated a pronounced inhibitory effect of LN stress on these traits. Specifically, the reduction in FLL ranged from 14 to 20% in the AC population, which was slightly larger than that observed in the CH population (13%–18%). Similarly, FLW showed a greater reduction in the AC population (12%–24%) compared to the CH population (11%–18%). The most substantial LN-induced reduction was observed in FLA, with the AC population displaying the highest sensitivity (30%–51%), followed by the CH population (29%–37%). In contrast, the CH population exhibited relatively minor reductions in SPAD (4%–12%) under the two N treatments, while the AC population showed moderate reductions in SPAD (11%–18%) under LN treatment. These results highlight population-specific variations in nitrogen stress tolerance, with the AC population demonstrating greater susceptibility in leaf morphology traits (FLL, FLW, FLA), whereas the CH population maintained comparatively stable chlorophyll (SPAD) retention under LN treatment (Fig. 1c–f, g–j).
Broad-sense heritability (H2) analysis revealed that all four flag leaf traits exhibited high heritability (H2 > 0.5) under NN treatment. Under LN treatment, however, with the exception of the low heritability observed for FLW in both populations (H2 = 0.22–0.35) and for FLA in the AC population (H2 = 0.34), all other flag leaf traits demonstrated moderate to high heritability (H2 > 0.5). However, heritability diverged significantly between the AC population and the CH population, with a further influence under LN stress. For instance, the CH population displayed high heritability for FLA under NN treatment (H2 = 0.91), whereas the AC population exhibited a markedly reduction in FLA heritability under NN treatment (H2 = 0.53) (Table 1).
Table 1.
Summary statistics of the BLUP of the agronomic traits of the CH population and AC population
| Population | Statistic analysis | Normal nitrogen | Low nitrogen | ||||||
|---|---|---|---|---|---|---|---|---|---|
| FLL (cm) | FLW (cm) | FLA (cm2) | SPAD | FLL (cm) | FLW (cm) | FLA (cm2) | SPAD | ||
| CH population | Mean | 20.89 | 2.1 | 36.54 | 58.02 | 18.06 | 1.84 | 27.7 | 53.34 |
| Min | 15.83 | 1.98 | 27.78 | 54.61 | 13.22 | 1.7 | 19.64 | 48.95 | |
| Max | 27.49 | 2.21 | 44.09 | 62.71 | 24.07 | 1.99 | 37.83 | 58.66 | |
| SD | 1.71 | 0.04 | 3.35 | 1.35 | 1.76 | 0.05 | 3.01 | 1.82 | |
| CV (%) | 8.19 | 1.91 | 9.15 | 2.33 | 9.76 | 2.98 | 10.85 | 3.42 | |
| Skewness | 0.27 | –0.06 | 0.07 | 0.09 | 0.03 | 0.3 | 0.25 | –0.07 | |
| Kurtosis | 0.82 | –0.09 | –0.4 | 0.21 | 0.27 | 0.08 | 0.24 | –0.29 | |
| H2 | 0.84 | 0.73 | 0.91 | 0.89 | 0.84 | 0.35 | 0.63 | 0.9 | |
| G | *** | *** | *** | *** | *** | *** | *** | *** | |
| S | *** | *** | *** | *** | *** | *** | *** | *** | |
| Y | *** | *** | *** | *** | *** | *** | *** | *** | |
| G × S | *** | ns | *** | ns | *** | *** | *** | ns | |
| G × Y | *** | ns | * | ns | *** | ns | *** | ns | |
| S × Y | *** | *** | *** | *** | *** | *** | *** | *** | |
| G × S × Y | *** | ns | *** | ns | *** | ns | *** | ns | |
| AC population | Mean | 24.7 | 2.01 | 41.44 | 54.1 | 20.9 | 1.73 | 30.35 | 49.32 |
| Min | 19.26 | 1.77 | 33.77 | 49.95 | 15.42 | 1.56 | 25.31 | 41.53 | |
| Max | 29.35 | 2.2 | 51.21 | 58.56 | 25.11 | 1.95 | 37.59 | 53.71 | |
| SD | 1.77 | 0.09 | 3.12 | 1.67 | 1.86 | 0.08 | 2.62 | 1.6 | |
| CV (%) | 7.16 | 4.72 | 7.53 | 3.08 | 8.88 | 4.34 | 8.63 | 3.25 | |
| Skewness | –0.06 | –0.21 | 0.14 | 0.16 | 0.04 | 0.3 | 0.44 | –0.63 | |
| Kurtosis | 0.32 | –0.66 | –0.09 | 0.15 | –0.27 | 0.23 | –0.2 | 4.17 | |
| H2 | 0.81 | 0.5 | 0.53 | 0.81 | 0.84 | 0.22 | 0.34 | 0.85 | |
| G | *** | *** | *** | *** | *** | *** | *** | *** | |
| S | *** | *** | *** | *** | *** | *** | *** | * | |
| Y | *** | ns | *** | *** | *** | *** | *** | ns | |
| G × S | *** | *** | *** | ns | *** | *** | *** | * | |
| G × Y | *** | ns | * | ns | *** | *** | *** | ns | |
| Y × S | *** | *** | *** | ns | * | *** | *** | ns | |
| Y × S × G | *** | * | *** | ns | *** | *** | *** | ns | |
FLL flag leaf length, FLW flag leaf width, FLA flag leaf area, SPAD chlorophyll content, BLUP best linear unbiased predictions, Min minimum, Max maximum, SD standard deviation, CV coefficient of variation, H2 broad-sense heritability, G genotype, S site, Y year
Significance level: *** p < 0.001, ** p < 0.01, * p < 0.05, and ns p ≥ 0.05
Genotype (G), site (S), year (Y) and their interaction significantly affected the four traits of flag leaf in most cases (Table 1). The exceptions included the FLW in the AC population under NN treatment and the SPAD value under LN treatment displayed no significant year-specific differences. The interaction effects on the FLW and SPAD were not significant (Table 1).
Genotype data, Molecular markers, population structure, and LD decay
Following rigorous quality control, a total of 395,783 high-quality polymorphic SNP Markers were retained for GWAS. These Markers exhibited comprehensive coverage across all 21 wheat chromosomes. Analysis of linkage disequilibrium (LD) decay patterns revealed sub-genome-specific differences: the LD half-decay distance (r2 = 0.5) was 3 Mb for Both the A and D sub-genomes, whereas the B sub-genome displayed an extended decay distance of 10 Mb (Fig. 2a). Neighbor-joining clustering of the 243 accessions supported a primary population structure with three distinct subpopulations (Fig. 2b). Furthermore, cross-validation error minimization criteria identified k = 12 as the optimal subgroup number, evidenced by the trough position in the error rate curve (Fig. 2c, d).
Fig. 2.

Characterizing population stratification and LD decay in a collection of CH population. a LD decay for three sub-genomes. b Plot of kinship; c Plot of cross. validation. Error. Phenotypic variation explained; d Stacked bar plot of ancestry relationship. r2: squared correlation coefficient; Distance (Kb): attenuation distance; k: number of ancestral clusters
Marker trait associations (MTAs) revealed by association mapping
GWAS analysis using phenotypic data from four environments and best linear unbiased predictions (BLUP) values identified 1,016 MTAs for LNTI across four flag leaf traits. Manhattan/QQ plots (Fig. 3; Table S3) visualized these associations. LD decay analysis (The LD decay distances of genomes subgenomes A 3 Mb, B 10 Mb, D 3 Mb) showed that MTAs were condensed into 290 QTLs: 58 FLLR, 110 FLWR, 72 FLAR, 50 SPADR. Among them, the PVE of FLLR ranged from 8.1% to 16.5%, with 22 major QTLs (PVE ≥ 10%). The PVE of FLWR ranged from 7.7% to 15.5%, with 71 Major QTLs. The PVE of FLAR ranged from 7.3% to 13.2%, with 48 Major QTLs. The PVE of SPADR ranged from 6.9% to 11.8%, with one major QTL. QTLs detected in at least one environment and localized through BLUP analysis were classified as stably inherited loci. Eleven loci were prioritized as stable QTLs (Table 2). QSPADR5B.3 emerged as a Major stable QTL, Mapped across two environments and BLUP value, accounting for 7.7%–11.8% of phenotypic variation. Similarly, QFLAR6D.3 demonstrated stability through two environment and BLUP value accounting for 7.3%–9.4% of phenotypic variation. Remaining loci (QFLWR2B.5, QSPADR3B.4, QFLWR3A.3, QFLWR6A.11, QFLWR3A.2, QFLWR6A.6, QSPADR6B.1, QFLWR7B.9, QFLWR7D.1) showed moderate stability with single-environment and BLUP value. The comparison with prior research on NUE revealed that the majority of identified loci overlapped or showed similarity to those reported previously, validating the reliability of the detected QTL loci in this study (Table 2). Among them, there were still two loci (QFLAR6D.3, QSPADR3B.4) that have not been found to overlap with previous loci, which may be new loci.
Fig. 3.

Manhattan plots for low nitrogen tolerance indices of studied traits under multiple environments from GWAS. The black solid horizontal lines in the Manhattan plots refer to the thresholds for significance at –log10(p) = 4.0. FLL: flag leaf length; FLW: flag leaf width; FLA: flag leaf area; SPAD: chlorophyll content; BLUP: best Linear unbiased predictions. The 23XYT and 24XYT refer to the 2022–2023 and 2023–2024 field experiments in Xiyangtun, respectively, and 23ZC and 24ZC refer to the 2022–2023 and 2023–2024 field experiments in Zhaocun, respectively
Table 2.
Most stable and major QTLs for NUE-related traits mapped in this study
| QTL | chr | Representative Marker | Environmenta | PVE% | Position (Mb) | References Marker | Position (Mb) | References |
|---|---|---|---|---|---|---|---|---|
| QSPADR5B.3 | 5B | AX-94551404 | 3, 4, 5 | 7.7–11.8 | 542.09–548.86 | AX-95685141 | 545.15 | Shi et al. (2022) [32] |
| QFLAR6D.3 | 6D | AX-109030772 | 1, 4, 5 | 7.3–9.4 | 359.62–365.62 | |||
| QFLWR2B.5 | 2B | AX-109949037, AX-109338641 | 4, 5 | 9.–12.7 | 746.47–756.42 | AX-108916587 | 731.87 | Zhang et al. (2024) [25] |
| QSPADR3B.4 | 3B | AX-111830686, AX-108956728 | 4, 5 | 7.2–7.8 | 687.76–687.85 | |||
| QFLWR3A.3 | 3A | AX-108797523, AX-94926374 | 3, 5 | 8.6–8.9 | 683.09–683.33 | AX-111488546 | 683.57 | Shi et al. (2022) [32] |
| QFLWR6A.11 | 6A | AX-110590286, AX-111823265 | 3, 5 | 8.7–8.8 | 388.22–393 | |||
| QFLWR3A.2 | 3A | AX-110366503, AX-110052451 | 2, 5 | 9.3–11.2 | 623.08–623.54 | AX-110542396 | 637.16 | Shi et al. (2022) [32] |
| QFLWR6A.6 | 6A | AX-110669543, AX-110987033 | 2, 5 | 8.8–14.2 | 227.45–237.33 | Xcn138 | 236 | Cui et al. (2014) [33] |
| QSPADR6B.1 | 6B | AX-94446754 | 2, 5 | 7.1–7.2 | 27.36–44.59 | AX-109395836 | 27.37 | Xiong et al. (2019) [38] |
| QFLWR7B.9 | 7B | AX-110390611, AX-109866042 | 2, 5 | 9.9–10 | 697.18–706.06 | AX-111644876 | 698.52 | Shi et al. (2022) [32] |
| QFLWR7D.1 | 7D | AX-94823348, AX-110282815 | 2, 5 | 8.2–10.8 | 2.14–3.61 | gwm635a | 3.2 | Habash et al. (2007) [34] |
aThe five environments of 2023XYT, 2023ZC, 2024XYT, 2024ZC, and BLUP are Marked as 1 to 5
QTLs identified by linkage analysis
Linkage analysis of the AC population identified 65 QTLs associated with the LNTI of flag leaf traits (Table S4). Among these, 15 FLLR-related loci were distributed across 8 chromosomes (2A, 3B, 4D, 5 A, 6 A, 6B, 6D, 7 A), including 4 major loci (QFLLR6A.3, PVE = 10.8%; QFLLR3B.2, PVE = 10.9%; QFLLR6A.4, PVE = 12.2%; QFLLR2A.5, PVE = 16.6%), all detected in single environments without stable inheritance (Fig. 4; Table S4).
Fig. 4.

Major QTLs of flag leaf traits in AC population. The red font represents the flanking markers of the main-effect QTL. FLL: flag leaf length; FLW: flag leaf width; FLA: flag leaf area; SPAD: chlorophyll content; BLUP: best Linear unbiased predictions. The 23XYT and 24XYT refer to the 2022–2023 and 2023–2024 field experiments in Xiyangtun, respectively, and 23ZC and 24ZC refer to the 2022–2023 and 2023–2024 field experiments in Zhaocun, respectively. The distances shown in the figure represent genetic distances
For FLWR traits, 15 loci were Mapped to 5 chromosomes (2B, 3 A, 5 A, 5B, 6 A), with 3 major loci (QFLWR2B.7, PVE = 11.7%; QFLWR3A.5, PVE = 12.4%; and QFLWR6A.16, PVE = 14.7%). Notably, QFLWR3A.5 showed consistent detection across four environments (23XYT, 23ZC, 24XYT, 24ZC) with PVE ranging from 4.3% to 9.7% (Fig. 4; Table S4).
The analysis revealed 21 FLAR-related loci spanning 9 chromosomes (1A, 3 A, 3D, 4D, 5B, 6 A, 6D, 7 A, 7D), including 5 major loci (QFLAR1A.8, PVE = 10.5%; QFLAR4D.6, PVE = 12.9%; QFLAR3A.4, PVE = 14.2%; QFLAR3A.5, PVE = 12.8%; QFLAR6D.6, PVE = 16.9%). QFLAR3A.5 was detected in four environments (23ZC, 23XYT, 24XYT, BLUP; PVE = 4.6–12.8%), while QFLAR4D.6 (24XYT, 24ZC; PVE = 8.1–12.9%) and QFLAR6D.6 (23XYT, 24ZC; PVE = 15.1–16.9%) were each identified in two environments (Fig. 4; Table S4).
Additionally, 14 SPAD-related loci were found on 9 chromosomes (2A, 2D, 3 A, 3B, 4 A, 5B, 7 A, 7B, 7D), with 5 major loci (QSPADR2A.2, PVE = 10.2%; QSPADR3B.6, PVE = 12.4%; QSPADR2A.3, PVE = 13.2%; QSPADR4A.5, PVE = 15.2%; QSPADR5B.6, PVE = 10.9%). QSPADR2A.2 (23ZC, 24XYT; PVE = 15.1–16.9%), QSPADR5B.6 (24XYT, BLUP; PVE = 7.2–10.9%), QSPADR2A.2 (23ZC, 24XYT; PVE = 10.2–12.4%) and QSPADR7A.4 (24XYT, BLUP; PVE = 9.4–9.8%) were detected in two environments (Fig. 4; Table S4).
Figure 5 showed that Chelio is enriched with more favorable alleles in nitrogen efficiency. In the future breeding, enhancing nitrogen use efficiency can be achieved by enriching Chelio alleles.
Fig. 5.

The source of additive effect in AC population. FLL: flag leaf length; FLW: flag leaf width; FLA: flag leaf area; SPAD: chlorophyll content. Avocet and Chilero are two parents of AC population. The numerical value represents the sum of additive effects
Genome segments discovered by association mapping and linkage analysis
To clarify the physical position of the marker, the DArT marker sequence was subjected to alignment with the whole genome database of Chinese Spring (https://wheat-urgi.versailles.inra.fr/) via the BLAST tool. Combined with the loci associated with Mapped by GWAS and Linkage analysis, 11 co-localized loci significantly were detected. Three of these loci were stable QTL detected by GWAS (QFLAR6D.3, QFLWR6A.6, QSPADR5B.3). QFLAR6D.3 (AX-109030772 physical position 362.56 Mb), a stable genetic locus identified in the CH population, was validated in the QFLAR6D.6 flanking marker DArT3937837 (physical position 359.72 Mb) of the AC population and further narrowed to a 2.84 Mb physical interval. The stable locus, QFLWR6A.6 (AX-110669543 physical position 224.45 Mb) was validated in the QFLWR6A.16 flanking marker DArT1031359 (physical position 226.11 Mb) of the AC population and further narrowed to a 1.34 Mb physical interval. The stable locus, QSPADR5B.3 (physical position 542.1 Mb–549.9 Mb) was validated in the QSPADR5B.6 flanking marker DArT3947164 (physical position 544.5 Mb–549.5 Mb) of the AC population and further narrowed to a 5 Mb physical interval (Fig. 6). Additional loci detected in specific environments of the CH population were also validated in the AC population, as summarized in Table S5.
Fig. 6.

A segment on chromosome 5B related to SPADR is jointly identified by GWAS and linkage analysis. a Fine mapping of QSPADR-5B.3 in wheat flag leaves. b Physical location and length of QSPADR-5B.3 in the CH population. c Physical location and length of QSPADR-5B.5 in AC population. d Co-localization interval the red box represents differentially expressed genes (FDR < 0.05, Log2 (FC) > 1)
Candidate gene analysis
We searched for candidate genes within these three critical intervals and identified a total of 128 annotated candidate genes. The interval QFLLR6D.3 contained 34 genes, that of QFLWR6A.6 contained 1 gene, and that of QSPADR5B.3 contained 94 genes. To further identify these candidate genes, we subjected the leaves of the low nitrogen tolerant parent Chileo to low nitrogen treatment at JS, AS and GS stages and identified DEGs (Fig. 7).
Fig. 7.

Heatmap of 127 candidate genes in leaves of low nitrogen tolerant parent Chileo. JS: jointing stage, AS: anthesis stage, GS: grain filling stage. Red, yellow, and green represent that a specific accession (gene) at a specific growth stage under LN were higher, close and lower than NN, respectively
According to the transcriptome sequencing results, there were no DEGs in QFLLR6D.3 and QFLWR6A.6, while four DEGs (FDR < 0.05, Log2 (FC) > 1) were found in QSPADR5B.3. Among these, TraesCS5B02G394300, TraesCS5B02G394200, and TraesCS5B02G39390 encode beta-glucosidase, and gene TraesCS5B02G396400 encodes a potassium channel (Figs. 6d and 7; Table S6). These genes were assigned unique numerical identifiers (Table S6).
Discussion
The integration of linkage analysis, GWAS, and transcriptome analysis for screening candidate genes
GWAS leverage high-density SNP markers (660 K) and linkage disequilibrium characteristics to achieve precise of genotype–phenotype associations across environments [32]. This study innovatively integrates the controlled genetic background advantage of linkage analysis with the fine mapping capability of GWAS to construct a hybrid analytical framework. This strategy has demonstrated exceptional performance in stress response trait studies, such as successfully mapping drought tolerance QTLs in wheat [24], and has been validated in studies of maize grain traits [35] and cucumber fruit development [36]. Specifically, in this study, GWAS based on 660 K SNP Markers identified 290 candidate QTLs, with 11 showing stable expression across environmental trials. Through validation via the AC population, 11 co-localized regions were identified, including loci corresponding to the three stable GWAS signals: QFLAR6D.3, QFLWR6A.6, and QSPADR5B.3.
Compared to traditional single-population analyses, the multi-system integration strategy developed in this study exhibits dual technical advantages: (1) reducing the false positive rate through an inter-population verification mechanism, significantly enhancing the reliability of QTL detection; and (2) incorporating high-density molecular markers to improve the resolution of genetic maps, thereby achieving a leap in mapping precision. This integrated approach provides a standardized solution for the genetic of complex traits, particularly in the identification of pleiotropic genes and the mining of minor-effect QTLs, offering significant application value.
Comparison of flag leaf NUE loci with reported loci in wheat
Previous studies on flag leaves have focused primarily on QTL mapping of flag leaf traits [15, 16, 37–39], with limited exploration of their responses to varying nitrogen treatments. In this study, leveraging the Chinese Spring physical map v1.0 as a reference, we compared the loci identified in this study with those reported in prior NUE research. In conjunction with prior Literature reviews on NUE in wheat, 8 out of the 11 QTLs that consistently emerged were closely associated with QTL loci or genes previously identified for traits related to wheat NUE. For instance, Shi et al. [32] performed GWAS of 389 wheat accessions under five environments, analyzed eight NUE-related agronomic traits, and identified 347 LNTI-associated QTLs [32]. In our study, we also employed the same method (LNTI value) for trait evaluation. The QTKWR2B.7 (identified in the study reported by Shi et al. [32]) detected via multi-trait and multi-environment analyses overlapped with the stable locus QFLWR2B.5, which was found that the QFLWR2B.5 locus has been reproducibly detected across studies and was identified as a Major QTL, with PVE ranging from 9.03% to 12.73%. QFLWR2B.5 is 3 Mb away from the previously reported QTKWR2B.7. The results suggest that QFLWR2B.5 and QTKWR2B.7 may represent the same QTL (Table 2). Moreover, a significant the NUE-related locus, QTNR3A.3 (identified in the study reported by Shi et al. [32]) overlaped with the stable locus QFLWR3A.3 in our trial, indicating that this region likely represents another critical genomic region associated with wheat NUE. Furthermore, QFLWR2B.5 is in close proximity, 0.8 Mb, to the previously identified AX-111559064 locus, which aligned with NUE-related QTL regions reported by Zhang et al. [25]. In addition, QFLWR3A.3, QFLWR2B.5, QSPADR6B.1, QFLWR7D.1, QFLWR6A.6 were also found to be consistent with or similar to those reported in previous studies [25, 33, 34, 40], however, QFLAR6D.3 and QSPADR3B.4 were two stable loci that did not overlap or have similar QTLs to those identified in previous studies on NUE, which may be two new loci (Table 2).
The loci identified in this study overlapped with multiple known NUE-related genes. For instance, the major QTL QFLWR2A.6 (PVE = 11.3%–11.8%) in the CH population co-localized with the nitrogen assimilation gene GS2-2A (TraesCS2A02G500400), while GS2-2B (TraesCS2B02G528300) was mapped within QSPADR2B.6. Additionally, the major QTL QFLWR4B.1 (PVE = 11.2%) and QFLWR6A.13 (PVE = 11.9%) in the CH population harbored the nitrate transporter gene NPF2.4-4B (TraesCS4B02G029600) and nitrite reductase gene NiR-6A (TraesCS6A02G333900), respectively. In summary, these results showed that these four flag leaf traits can be taken as the indicators for screening NUE-related genes.
Prediction of flag leaf NUE candidate genes
Combined with the loci associated with Mapped by GWAS and Linkage analysis, 11 co-localized loci significantly were detected. Three of these loci were stable QTL detected by GWAS (QFLAR6D.3, QFLWR6A.6, QSPADR5B.3). We searched for candidate genes within these three critical intervals and identified a total of 128 candidate genes. According to the transcriptome results, there were no DEGs in QFLLR6D.3 and QFLWR6A.6, while four DEGs were found in QSPADR5B.3 (the AC population name is QSPADR5B.6, and its additive effect originates from Chilero). Among them, TraesCS5B02G394300, TraesCS5B02G394200, and TraesCS5B02G39390 encode beta-glucosidase, and one gene TraesCS5B02G396400 encodes a potassium channel. However, no DEGs were found within the two important intervals QFLAR6D.3 and QFLWR6A.6. On the one hand, this might be because the functional genes within these two intervals involve multiple genes and different regulatory levels (including post-transcriptional regulation, translational regulation, etc.), and these complex regulatory mechanisms may be difficult to be comprehensively revealed in conventional transcriptome analysis [41]. On the other hand, the functional genes within these two intervals have spatiotemporal expression specificity and are expressed only at specific developmental stages, requiring spatiotemporal expression analysis of each period and each part [42].
In plants, beta-glucosidas are involved in various physiological processes such as lignification of cell walls, hormone metabolism, and stress defense responses. It is hypothesized that beta-glucosidase genes may participate in cell wall remodeling (e.g., lignin biosynthesis), thereby affecting root system development and nutrient uptake capacity [43]. Additionally, certain β-glucosidases contribute to the activation of plant hormones (such as cytokinins) and may regulate the expression of nitrogen metabolism-related genes [43–46]. In this study, three candidate DEGs TraesCS5B02G394300, TraesCS5B02G394200, and TraesCS5B02G39390 were encode to beta-glucosidas (Figs. 6 and 7). These genes exhibited downregulated expression during the AS and GS stages under LN treatment. These genes may serve as potential candidate genes associated with NUE in wheat.
The potassium channel protein family is known to play a pivotal role in plant responses to abiotic stress, as well as in promoting growth, regulating development, and controlling pollen tube elongation [47]. TraesCS5B02G396400 encodes a potassium channel, a crucial component in potassium uptake pathways in plants. TraesCS5B02G396400 has been identified nitrate transporter gene NRT1.5 integrate nitrate deficiency signaling with potassium homeostasis regulation as a key player in inhibiting leaf senescence [48]. Downregulated potassium transporter genes, reducing xylem potassium loading and resulting in leaf potassium deficiency, thereby inducing leaf senescence under nitrogen deficiency [48, 49]. In this trial, the gene TraesCS5B02G396400 showed downregulated expression under LN during the AS and GS stages (Fig. 7). Therefore, we postulate that TraesCS5B02G396400 may be an important candidate gene related to NUE. However, its specific function requires further analysis and validation.
Conclusion
In this study, we performed GWAS and QTL mapping on the flag leaf trait under two N conditions (LN and NN) via a natural population (CH population) and an a RIL population (AC population). We identified three stable co-localized loci (QFLAR6D.3, QFLWR6A.6, and QSPADR5B.3), which were located 1.34 Mb, 2.84 Mb, and 5 Mb away from GWAS-derived significant SNP loci, respectively. Differential expression analysis of genes within these loci revealed four DEGs in the QSPADR5B.3 region. Among them, TraesCS5B02G394300, TraesCS5B02G394200, and TraesCS5B02G393900 encode beta-glucosidase, while TraesCS5B02G396400 encodes a potassium channel. These findings provide a foundation for exploring molecular targets underlying wheat NUE.
Methods
Plant materials
This investigation employed two distinct wheat genetic resources for complementary analyses. The first cohort (CH population) consisted of 243 elite cultivars derived from China’s Yellow-Huai River Valley, serving as the foundation for genome-wide association mapping (detailed in Supplementary Table S1). The second genetic resource (AC population) involved a biparental population of 123 F7 RILs developed through crossing Avocet × Chilero from the International Maize and Wheat Improvement Center (CIMMYT, Mexico, Mexico).
Field trials for evaluation of phenotypic traits
The field trials were conducted across four distinct environments (year × site combinations) in Henan Province, China, including two experimental stations: Xiyangtun (34.64°N, 112.34°E) at Henan University of Science and Technology and Zhaocun (34.63°N, 112.45°E) at Luoyang Academy of Agriculture and Forestry Sciences, in the two consecutive growing seasons (2022–2023 and 2023–2024) evaluated at each location, designated as 23XYT, 24XYT, 23ZC, and 24ZC, respectively. A completely randomized block design with triplicate plots was implemented, each containing four rows (0.8 m × 0.25 m). The planting density was standardized to 45 plants per square meter (45 plants m⁻2) following thinning and the final seedling establishment fertilization regimes comprised two nitrogen levels: normal nitrogen (NN: 240 kg N ha⁻1, 90 kg P2O5 ha⁻1, 60 kg K2O ha⁻1) and low nitrogen (LN: 0 kg N ha⁻1 with equivalent P and K applications).
The soil nutrient status of the 0–20 cm soil layer in the experimental field before sowing is shown in Table S7. All nitrogen fertilizers were applied in two steps. First, the base fertilizer was applied, and then top-dressing was carried out during the jointing stage. The ratio of the base fertilizer to the top-dressing was 1:1.
Phenotypic measurements were conducted following standardized procedures. At 15 days post anthesis, five uniformly grown individual plants were selected to measure the flag leaf length and width of the main stem. The maximum length and widest portion of the flag leaves were recorded. FLA (cm2) was calculated as length × width × 0.83. The chlorophyll content of the flag leaves was determined using a SPAD-502 chlorophyll meter (Konica-Minolta, Japan). The final values represent the means of measurements from five plants.
Statistical analysis of phenotypic data
For each genotype, BLUP of all traits under LN and NN treatments (denoted as trait-LN-BLUP or trait-NN-BLUP) were calculated across four experimental environments (23XYT, 23ZC, 24XYT, 24ZC). A linear mixed-effects model with random variance components was implemented via the lme4 package (v1.1–34) in R 4.3.0, following the methodological framework established by Shi et al. [32].
The LNTI for each trait across environments were the relative ratio of LN to NN. For phenotypic values, such as the flag leaf length ratio (FLLR = FLL LN/FLL NN) were derived. To account for genetic potential, BLUP-based LNTI (BLUPR) was calculated by the relative ratios (BLUP values) of LN to NN (e.g., FLL-LNTI = FLL-LN-BLUP/FLL-NN-BLUP), where trait-LN-BLUP and trait-NN-BLUP represent the BLUP for each trait under the respective nitrogen regimes. H2 was estimated across multiple environment trials via the variance component method [50].
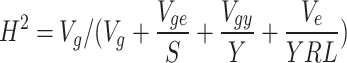 |
where Vg is the genotypic variance, Vge is the genotype by location variance, Vgy is the genotype by year variance Ve is the residual variance, S is the number of sites, Y is the number of years, and R is the number of replicates.
Data collation was carried out via Microsoft Excel 2016 software. ANOVA was used to divide the variation into genotype (G), site (S), year (Y). The contributions of G, S, Y, and their interactions to phenotypic variation were quantified through athree-way factorial analysis of variance (ANOVA) under a general linear model framework. This univariate ANOVA was implemented in SPSS Statistics (v26.0, IBM Corp., Armonk, NY, USA), with G, S, and Y as fixed factors and all possible interaction terms (G × S, G × Y, S × Y, and G × S × Y) included. Graphical outputs (e.g., interaction effect plots, correlation matrixes) were generated using OriginPro (v2024, Origin Lab Corp., Northampton, MA, USA), with adherence to scientific visualization standards. Post-hoc refinements to layout, labeling, and color schemes were applied in Microsoft PowerPoint (v2016, Microsoft Corp., Redmond, WA, USA) to enhance graphical clarity and audience interpretability, without altering raw data or statistical conclusions.
Linkage disequilibrium (LD) of the CH population
LD was quantified via the squared correlation coefficient (r2) of allele frequencies between SNP pairs, which was calculated via the r2 algorithm implemented in the RTM-GWAS software package (v1.1; Institute of Crop Science, CAAS, Beijing). The LD decay rate was determined by plotting the genome-wide average r2 values against physical distance. A critical threshold for LD decay was defined as the chromosomal distance at which the smoothed r2 curve declined to 50% of its maximum initial value [51].
Population structure analysis
Population structure analysis was performed via ADMIXTURE (v1.3.0) with a set of unlinked markers pruned for LD at r2 < 0.1 via an iterative pruning approach. The optimal number of genetic clusters (K) was inferred by testing a putative range of K = 1 to K = 20. For each K, the analysis employed a burn in period of 1,000 iterations to stabilize parameter estimates. The final K value was selected on the basis of the cross-validation error minimization criterion, as implemented in the ADMIXTURE framework [52].
Genome wide association analysis
The natural population was genotyped via the Illumina Wheat 660 K SNP array (CapitalBio Technology Inc., Beijing, China; https://www.capitalbio.com/). The raw SNP data were subjected to stringent quality control procedures in PLINK v1.9 [53] by applying the following exclusion thresholds: minor allele frequency (MAF) < 0.02 and per-marker missing rate >10%. After filtering, 395,782 high-quality SNPs were retained for subsequent analyses. Association mapping was performed in TASSEL v5.0 [54–56] via a mixed linear model (MLM) that accounts for population structure (Q matrix) and kinship (K matrix). The model structure was defined as follows:
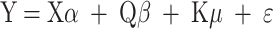 |
where Y is the vector of the phenotype; X is the vector of marker genotypes; Q is the population structure matrix; K is the relative kinship matrix; α, β and μ are the corresponding effects; and ε is a matrix of residual effects. The X and Q matrixes are considered fixed effects, and the K and ε matrixes are considered as random effects.
The associations between flag leaf traits and genetic markers in the natural population was investigated via the MLM framework in TASSEL v5.0. Population structure (Q matrix) and kinship (K matrix) were incorporated as covariates to mitigate confounding effects of genetic relatedness and population stratification. Genomewide significance was defined at a threshold of − log10(p) ≥ 4 (equivalent to p < 1 × 10−4). Significant loci were visualized through Manhattan plots and quantile‒quantile (Q‒Q) plots were generated using via the CMplot package.
Linkage analysis
The RIL population and parental lines were genotyped via the Diversity Arrays Technology (DArT) platform, as described by Zhao et al. [57]. A high-density genetic linkage Map, comprising 3,627 DArT Markers distributed across all 21 chromosomes, spanning a total genetic distance of 8,202.1 centimorgans (cM) with an average Marker interval of 2.26 cM, was constructed. QTL mapping was performed via QTL IciMapping v4.2 with the inclusive composite interval mapping (ICIM) algorithm. The analysis parameters were configured as follows: a step size of 1 cM for genome scanning, a probability for variable entry of 0.001, and a significance threshold of logarithm of odds (LOD) ≥ 2.5, as established in previous studies [24, 57]. Genetic map visualization and QTL positional mapping were executed in MapChart v2.32, with linkage groups annotated to reflect chromosomal positions and marker intervals [58].
RNA sequencing (RNA-seq)
Wheat leaves (the fully expanded penultimate leaves at JS, and flag leaves at AS and GS) of Chilero (a low-nitrogen-tolerant parental line) were sampled under the NN and LN regimes with three biological replicates per treatment, yielding 18 samples (3 stages × 2 treatments × 3 replicates). Eight flag leaves per plot were randomly collected, flash-frozen in liquid nitrogen, and stored at − 80 °C until RNA extraction. Total RNA was isolated via TRIzol reagent, and mRNA libraries were prepared via poly-A selection and strand-specific protocols by Biomarker Technologies Co., Ltd. (Beijing, China).
Library quality was assessed via Qubit 2.0 fluorometer (Thermo Fisher Scientific) for cDNA quantification and Agilent 2100 Bioanalyzer for insert size validation. Quantitative PCR (qPCR) was employed to normalize library concentrations, with libraries ≥ 2 nM pooled proportionally for sequencing on the Illumina NovaSeq 6000 platform (150 bp paired-end reads). After sequencing, the raw data underwent stringent quality control via FastQC (v0.11.9), yielding 196.36 Gb of high-quality clean data (Q30 ≥ 95.48% across all the samples). Differential gene expression analysis was performed on the OmicsMart platform (https://www.omicsmart.com), with transcript abundance quantified as FPKM (Fragments Per Kilobase of transcript per Million mapped reads). DEGs were identified using thresholds of false discovery rate (FDR) ≤ 0.05 and absolute |log2 (fold change)|≥ 1. Heatmaps visualizing expression patterns were generated using Chiplot (https://www.chiplot.online), with hierarchical clustering based on euclidean distance and complete linkage.
Determination of co-localization interval and analysis of candidate genes
To reveal candidate genes detected via GWAS and linkage analysis, BLAST search was performed on the based of the IWGSC Chinese spring reference genome v1.0 (http://plants.ensembl.org/index.html) to determine the physical spacing between identified loci and QTLs [59]. The high confidence genes for these regions were extracted from WheatOmics 1.0 (http://wheatomics.sdau.edu.cn/) [41]. When DEGs were screened through transcriptome analysis at co-localization sites, QTLs with additive effects originating from Chilero were selected for screening. The naming of co-located QTLs is based on the names of the QTLs in the CH population.
Supplementary Information
Authors’ contributions
CW, MH, and YL designed the study. YJ, NX, and JW performed the experiments and participated in field trials. YJ, and MH analyzed experimental results and wrote the manuscript. CW and YL, revised the manuscript, and all authors reviewed and com-mented on the manuscript.
Funding
This study was financially supported by National Key Research and Development Program of China (2022YFD2300800); National Natural Science Foundation of China (32401870); Key Research Project of the Shennong Laboratory (SN01–2022–01); Henan Province Science and Technology Research Project (232102111009); Henan Province Major Science and Technology Project (231100110300).
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2024), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: accession number CRA024358) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. All data generated or analyzed in this study are available.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ming Huang, Email: huangming_2003@126.com.
Youjun Li, Email: lyj@haust.edu.cn.
References
- 1.Reynolds M, Foulkes J, Furbank R, Griffiths S, King J, Murchie E, et al. Achieving yield gains in wheat. Plant Cell Environ. 2012;35(10):1799–823. 10.1111/j.1365-3040.2012.02588.x. [DOI] [PubMed] [Google Scholar]
- 2.Ullah A, Nadeem F, Nawaz A, Siddique KH, Farooq M. Heat stress effects on the reproductive physiology and yield of wheat. J Agron Crop Sci. 2022;208(1):1–17. 10.1111/jac.12572. [Google Scholar]
- 3.Kang J, Chu Y, Ma G, Zhang Y, Zhang X, Wang M, et al. Physiological mechanisms underlying reduced photosynthesis in wheat leaves grown in the field under conditions of nitrogen and water deficiency. Crop J. 2023;11(2):638–50. 10.1016/j.cj.2022.06.010. [Google Scholar]
- 4.Evans JR. Nitrogen and photosynthesis in the flag leaf of wheat (Triticum aestivum L.). Plant Physiol. 1983;72(2):297–302. 10.1104/pp.72.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Qiao L, Yang C, Li X, Zhao J, Wu B, et al. Identification of genetic loci for flag-leaf-related traits in wheat (Triticum aestivum L.) and their effects on grain yield. Front Plant Sci. 2022;13:990287. 10.21203/rs.3.rs-1408600/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zanella CM, Rotondo M, McCormick Barnes C, Mellers G, Corsi B, Berry S, et al. Longer epidermal cells underlie a quantitative source of variation in wheat flag leaf size. New Phytol. 2023;237(5):1558–73. 10.1111/nph.18676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lassaletta L, Billen G, Garnier J, Bouwman L, Velazquez E, Mueller ND, et al. Nitrogen use in the global food system: past trends and future trajectories of agronomic performance, pollution, trade, and dietary demand. Environ Res Lett. 2016;11(9):95007. 10.1088/1748-9326/11/9/095007. [Google Scholar]
- 8.Moklyachuk L, Furdychko O, Pinchuk V, Mokliachuk O, Draga M. Nitrogen balance of crop production in Ukraine. J Environ Manage. 2019;246:860–7. 10.1016/j.jenvman.2019.05.108. [DOI] [PubMed] [Google Scholar]
- 9.Vijayalakshmi K, Fritz AK, Paulsen GM, Bai G, Pandravada S, Gill BS. Modeling and mapping QTL for senescence-related traits in winter wheat under high temperature. Mol Breed. 2010;26:163–75. 10.1007/s11032-009-9366-8. [Google Scholar]
- 10.Kumar S, Sehgal SK, Kumar U, Prasad PV, Joshi AK, Gill BS. Genomic characterization of drought tolerance-related traits in spring wheat. Euphytica. 2012;186:265–76. 10.1007/s10681-012-0675-3. [Google Scholar]
- 11.Ilyas M, Ilyas N, Arshad M, Kazi AG, Kazi AM, Waheed A. Qtl mapping of wheat doubled haploids for chlorophyll content and chlorophyll fluorescence kinetics under drought stress imposed at anthesis stage. Pak J Bot. 2014;46(5):1889–97. [Google Scholar]
- 12.Talukder SK, Babar MA, Vijayalakshmi K, Poland J, Prasad PVV, Bowden R, et al. Mapping QTL for the traits associated with heat tolerance in wheat (Triticum aestivum L.). BMC Genet. 2014;15:1–13. 10.1186/s12863-014-0097-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avenson TJ, Cruz JA, Kanazawa A, Kramer DM. Regulating the proton budget of higher plant photosynthesis. Proc Natl Acad Sci U S A. 2005;102(27):9709–13. 10.1073/pnas.0503952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao C, Ma G, Zhou L, Zhang S, Su L, Sun X, et al. Effects of nitrogen levels on gene expression and amino acid metabolism in Welsh onion. BMC Genomics. 2021;22:1–10. 10.1186/s12864-021-08130-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu K, Xu H, Liu G, Guan P, Zhou X, Peng H, et al. QTL mapping of flag leaf-related traits in wheat (Triticum aestivum L.). Theor Appl Genet. 2018;131:839–49. 10.1007/s00122-017-3040-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farokhzadeh S, Fakheri BA, Nezhad NM, Tahmasebi S, Mirsoleimani A. Mapping QTLs of flag leaf morphological and physiological traits related to aluminum tolerance in wheat (Triticum aestivum L.). Physiol Mol Biol Plants. 2019;25:975–90. 10.1007/s12298-019-00670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Tao Y, Wang Z, Guo Q, Wu F, Yang X, et al. Identification of QTL for flag leaf length in common wheat and their pleiotropic effects. Mol Breed. 2018;38:1–11. 10.1007/s11032-017-0766-x. [Google Scholar]
- 18.Tian Z, Chai H, Guo H, Lu Y, Yang S, Liu X, et al. Genetic improvement of photosynthetic nitrogen use efficiency of winter wheat in the Yangtze River Basin of China. Field Crops Res. 2024;305:109199. 10.1016/j.fcr.2023.109199. [Google Scholar]
- 19.Sun J, Guo Y, Zhang G, Gao M, Zhang G, Kong F, et al. QTL mapping for seedling traits under different nitrogen forms in wheat. Euphytica. 2013;191:317–31. 10.1007/s10681-012-0834-6. [Google Scholar]
- 20.Cui F, Fan X, Chen M, Zhang N, Zhao C, Zhang W, et al. Qtl detection for wheat kernel size and quality and the responses of these traits to low nitrogen stress. Theor Appl Genet. 2016;129:469–84. 10.1007/s00122-015-2641-7. [DOI] [PubMed] [Google Scholar]
- 21.Fan X, Zhang W, Zhang N, Chen M, Zheng S, Zhao C, et al. Identification of QTL regions for seedling root traits and their effect on nitrogen use efficiency in wheat (Triticum aestivum L.). Theor Appl Genet. 2018;131:2677–98. 10.1007/s00122-018-3183-6. [DOI] [PubMed] [Google Scholar]
- 22.Zhang M, Gao M, Zheng H, Yuan Y, Zhou X, Guo Y, et al. QTL mapping for nitrogen use efficiency and agronomic traits at the seedling and maturity stages in wheat. Mol Breed. 2019;39:1–17. 10.1007/s11032-019-0965-8. [Google Scholar]
- 23.Mourad AM, Belamkar V, Baenziger PS. Molecular genetic analysis of spring wheat core collection using genetic diversity, population structure, and linkage disequilibrium. BMC Genomics. 2020;21:1–12. 10.1186/s12864-020-06835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo J, Guo J, Li L, Bai X, Huo X, Shi W, et al. Combined linkage analysis and association mapping identifies genomic regions associated with yield-related and drought-tolerance traits in wheat (Triticum aestivum L.). Theor Appl Genet. 2023;136(12):250. 10.1007/s00122-023-04494-9. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Peng C, Xu W, Li Y, Qi X, Zhao M. Genome-wide association study of agronomic traits related to nitrogen use efficiency in Henan wheat. BMC Genomics. 2024;25(1):7. 10.1007/s00122-022-04218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pace J, Gardner C, Romay C, Ganapathysubramanian B, Lübberstedt T. Genome-wide association analysis of seedling root development in maize (Zea mays L.). BMC Genomics. 2015;16:1–12. 10.1186/s12864-015-1226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delfan S, Bihamta MR, Dadrezaei ST, Abbasi A, Alipour H. Exploring genomic regions involved in bread wheat resistance to leaf rust at seedling/adult stages by using GWAS analysis. BMC Genomics. 2023;24(1):83. 10.1186/s12864-022-09096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi H, Wang W, Gao L, Wu J, Hu C, Yan H, et al. Genome-wide association study of seedling nitrogen-use efficiency-associated traits in common wheat (Triticum aestivum L.). Crop J. 2024;12(1):222–31. 10.1016/j.cj.2023.10.014. [Google Scholar]
- 29.Kaur S, Shamshad M, Jindal S, Kaur A, Singh S, Sharma A. RNA-seq-based transcriptomics study to investigate the genes governing nitrogen use efficiency in Indian wheat cultivars. Front Genet. 2022;13:853910. 10.3389/fgene.2022.853910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng X, Wang X, Zhang Z, Xiong S, Wei Y, Guo J, et al. Transcriptomic, proteomic, and physiological studies reveal key players in wheat nitrogen use efficiency under both high and low nitrogen supply. J Exp Bot. 2021;72(12):4435–56. 10.1093/jxb/erab153. [DOI] [PubMed] [Google Scholar]
- 31.Le TD, Gathignol F, Vu HT, Nguyen KL, Tran LH, Vu HTT, et al. Genome-wide association mapping of salinity tolerance at the seedling stage in a panel of Vietnamese landraces reveals new valuable QTLs for salinity stress tolerance breeding in rice. Plants. 2021;10(6):1088. 10.3390/plants10061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi H, Chen M, Gao L, Wang Y, Bai Y, Yan H, et al. Genome-wide association study of agronomic traits related to nitrogen use efficiency in wheat. Theor Appl Genet. 2022;135(12):4289–302. 10.1007/s00122-022-04218-5. [DOI] [PubMed] [Google Scholar]
- 33.Cui F, Fan X, Zhao C, Zhang W, Chen M, Ji J, et al. A novel genetic map of wheat: utility for mapping QTL for yield under different nitrogen treatments. BMC Genet. 2014;15:1–17. 10.1186/1471-2156-15-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Habash DZ, Bernard S, Schondelmaier J, Weyen J, Quarrie SA. The genetics of nitrogen use in hexaploid wheat: N utilisation, development and yield. Theor Appl Genet. 2007;114:403–19. 10.1007/s00122-006-0429-5. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Zhou Z, Ding J, Wu Y, Zhou B, Wang R, et al. Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize. Front Plant Sci. 2016;7:833. 10.3389/fpls.2016.00833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bo K, Wei S, Wang W, Miao H, Dong S, Zhang S, et al. QTL mapping and genome-wide association study reveal two novel loci associated with green flesh color in cucumber. BMC Plant Biol. 2019;19(1):1–13. 10.1186/s12870-019-1835-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hussain W, Baenziger PS, Belamkar V, Guttieri MJ, Venegas JP, Easterly A, et al. Genotyping-by-sequencing derived high-density linkage map and its application to QTL mapping of flag leaf traits in bread wheat. Sci Rep. 2017;7(1):16394. 10.1038/s41598-017-16006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Francki MG, Walker E, Li DA, Forrest K. High-density SNP mapping reveals closely linked QTL for resistance to Stagonospora nodorum blotch (SNB) in flag leaf and glume of hexaploid wheat. Genome. 2018;61(2):145–9. 10.1139/gen-2017-0203. [DOI] [PubMed] [Google Scholar]
- 39.Ma J, Tu Y, Zhu J, Luo W, Liu H, Li C, et al. Flag leaf size and posture of bread wheat: genetic dissection, QTL validation and their relationships with yield-related traits. Theor Appl Genet. 2020;133:297–315. 10.1007/s00122-019-03458-2. [DOI] [PubMed] [Google Scholar]
- 40.Xiong H, Guo H, Zhou C, Guo X, Xie Y, Zhao L, et al. A combined association mapping and t-test analysis of SNP loci and candidate genes involving in resistance to low nitrogen traits by a wheat mutant population. PLoS One. 2019;14(1):e211492. 10.1371/journal.pone.0211492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mauceri A, Abenavoli MR, Toppino L, Panda S, Mercati F, Aci MM, et al. Transcriptomics reveal new insights into molecular regulation of nitrogen use efficiency in Solanum melongena. J Exp Bot. 2021;72(12):4237–53. 10.1093/jxb/erab121. [DOI] [PubMed] [Google Scholar]
- 42.Shi X, Cui F, Han X, He Y, Zhao L, Zhang N, et al. Comparative genomic and transcriptomic analyses uncover the molecular basis of high nitrogen-use efficiency in the wheat cultivar Kenong 9204. Mol Plant. 2022;15(9):1440–56. 10.1016/j.molp.2022.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Malboobi MA, Lefebvre DD. A phosphate-starvation inducible β-glucosidase gene (psr3. 2) isolated from Arabidopsis thaliana is a member of a distinct subfamily of the BGA family. Plant Mol Biol. 1997;34:57–68. 10.1023/A:1005865406382. [DOI] [PubMed] [Google Scholar]
- 44.van de Ven WT, LeVesque CS, Perring TM, Walling LL. Local and systemic changes in squash gene expression in response to silverleaf whitefly feeding. Plant Cell. 2000;12(8):1409–23. 10.1105/tpc.12.8.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawasaki S, Borchert C, Deyholos M, Wang H, Brazille S, Kawai K, et al. Gene expression profiles during the initial phase of salt stress in rice. Plant Cell. 2001;13(4):889–905. 10.1105/tpc.13.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lipka V, Dittgen J, Bednarek P, Bhat R, Wiermer M, Stein M, et al. Pre-and postinvasion defenses both contribute to nonhost resistance in Arabidopsis. Sci. 2005;310(5751):1180–3. 10.1126/science.1119409. [DOI] [PubMed] [Google Scholar]
- 47.Huertas R, Rubio L, Cagnac O, García Sánchez MJ, Alché JDD, Venema K, et al. The K+/H+ antiporter LeNHX2 increases salt tolerance by improving K+ homeostasis in transgenic tomato. Plant, Cell Environ. 2013;36(12):2135–49. 10.1111/pce.12109. [DOI] [PubMed] [Google Scholar]
- 48.Drechsler N, Zheng Y, Bohner A, Nobmann B, von Wirén N, Kunze R, et al. Nitrate-dependent control of shoot K homeostasis by the nitrate transporter1/peptide transporter family member NPF7. 3/NRT1. 5 and the stelar K+ outward rectifier SKOR in Arabidopsis. Plant Physiol. 2015;169(4):2832–47. 10.1080/15592324.2016.1176819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meng S, Peng J, He Y, Zhang G, Yi H, Fu Y, et al. Arabidopsis NRT1.5 mediates the suppression of nitrate starvation-induced leaf senescence by modulating foliar potassium level. Mol Plant. 2016;9(3):461–70. 10.1016/j.molp.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 50.Piepho H, Möhring J. Computing heritability and selection response from unbalanced plant breeding trials. Genetics. 2007;177(3):1881–8. 10.1534/genetics.107.074229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang X, Wei X, Sang T, Zhao Q, Feng QI, Zhao Y, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42(11):961–7. 10.1038/ng.695. [DOI] [PubMed] [Google Scholar]
- 52.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–59. 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basnet BR, Singh RP, Ibrahim A, Herrera-Foessel SA, Huerta-Espino J, Lan C, et al. Characterization of Yr54 and other genes associated with adult plant resistance to yellow rust and leaf rust in common wheat Quaiu 3. Mol Breed. 2014;33:385–99. 10.1007/s11032-013-9957-2. [Google Scholar]
- 55.Yang J, Wang M, Li W, He X, Teng W, Ma W, et al. Reducing expression of a nitrate-responsive bZIP transcription factor increases grain yield and N use in wheat. Plant Biotechnol J. 2019;17(9):1823–33. 10.1111/pbi.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niaz M, Zhang L, Lv G, Hu H, Yang X, Cheng Y, et al. Identification of TaGL1-B1 gene controlling grain length through regulation of jasmonic acid in common wheat. Plant Biotechnol J. 2023;21(5):979–89. 10.1111/pbi.14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao Y, Yan X, Zeng Z, Zhao D, Chen P, Wang Y, et al. Integrated genome-wide association study and QTL mapping reveals qSa-3A associated with English grain aphid, Sitobion avenae (Fabricius) resistance in wheat. Pest Manag Sci. 2023. 10.1002/ps.7598. [DOI] [PubMed] [Google Scholar]
- 58.Mapchart VE. Software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93(1):77r–8r. 10.1093/jhered/93.1. [DOI] [PubMed] [Google Scholar]
- 59.Appels R, Eversole K, Stein N, Feuillet C, Keller B, Rogers J, et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science. 2018;361(6403):r7191. 10.1126/science.aar7191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2024), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: accession number CRA024358) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. All data generated or analyzed in this study are available.