

Abstract
Early detection of water blooms caused by potential toxin-producing cyanobacteria is important in environmental monitoring. We present a new nucleic acid-based method for detection of cyanobacteria in water that utilizes the same paramagnetic solid phase (beads) for both bacterial cell concentration and subsequent DNA purification. In the cell concentration step, the beads were attracted to a magnet after cell adsorption (in an alcohol- and salt-containing solution), and the supernatant was removed. For DNA purification, a buffer containing guanidine thiocyanate and Sarkosyl lysed the concentrated cells. The addition of alcohol precipitated the released DNA onto the same solid phase as was used for the cell concentration. Finally, to remove PCR inhibitors, the DNA was washed twice in alcohol while bound to the beads. All of the bead-DNA complex was used in the subsequent PCR amplification. The detection limit, as measured by 16S rDNA PCR amplification, was 50 cells in a 0.5-ml water sample, which is considerably lower than the limit (500 cells/ml) of toxic cyanobacteria tolerated in drinking water (New South Wales Blue-Green Algae Task Force, 1992). Testing of water from natural habitats showed a detection limit in the same range as that for the defined samples. The detection limits and the simplicity of the method (paramagnetic beads can be handled in automated systems) suggest that our method is suitable for routine environmental monitoring.
PCR allows high-sensitivity detection of bacteria in environmental water samples (11, 21). Isolation of bacteria and DNA purification are crucial steps for reproducibility and high sensitivity. Commonly, bacteria are isolated from water by centrifugation, filtration, or specific affinity binding to antibodies attached to a solid phase (5, 10, 22). Since many of the naturally occurring bacteria in natural waters contain gas vesicles (20), additional steps (e.g., heating to break the vesicles) are required for precipitation by centrifugation. Filtration procedures usually require resuspension of the cells on the filter for downstream applications. For antibody affinity binding, specific antibodies for each of the bacteria of interest have to be produced and tested. This limits the number of different bacterial species that can be detected by a particular cell concentration and DNA purification assay. All the above-mentioned cell concentration approaches require subsequent DNA preparation by using additional methods, often involving phenol-chloroform treatment (16), or commercially available DNA purification kits. However, for high-throughput and automated approaches, there is a need for simple assays (adaptable for several target organisms) which integrate cell concentration and DNA purification.
The aim of our work was to develop an integrated cell concentration and DNA purification assay for early detection of potential toxic water blooms formed by cyanobacteria in environmental water. We chose, as model organisms, cyanobacteria belonging to the genera Microcystis, Planktothrix, and Anabaena, which are commonly associated with toxic water blooms in both drinking water and recreational water (6). Previously, we showed that paramagnetic beads are suited for purification of PCR-ready DNA from cyanobacterial cultures (13). Here, we have developed an assay involving nonspecific adsorption of bacteria (by lowering the water activity by the addition of alcohol and salt) onto the beads. The DNA binding property of the beads was then used for DNA purification. Since paramagnetic beads are easy to manipulate both manually (13) and in automated systems (8), this integrated cell concentration and DNA purification method is suited for high-throughput assays. With the optimized protocol (combined with PCR amplification), as few as 50 cells could be detected in a 0.5-ml water sample. Health authorities (New South Wales Blue-Green Algae Task Force, 1992) have already adopted a system where the authorities are alerted when cell counts reach between 500 and 2,000 cells/ml for toxic cyanobacteria in drinking water (4), suggesting that our method is suited for early warning of potential toxic water blooms.
MATERIALS AND METHODS
Strains and strain cultivation.
Strains were cultivated in glass flasks containing 50 ml of medium Z8 in a constant-temperature room at 17 ± 2°C (15). Dilution series from 107 to 100 cells of the cyanobacteria Microcystis aeruginosa NIVA-CYA 43, Planktothrix agardhii NIVA-CYA 29 and Anabaena lemmermannii NIVA-CYA 83/1 per ml (18) were used in the development and optimization of the assays described in this work. The cells were counted by microscopy in a Fuchst-Rosenthal counting chamber (Karl Hecht, Sondheim, Germany).
Standard (reference) cell concentration and DNA purification protocols.
Bacterial cells were concentrated by centrifugation in a microcentrifuge (model 2231 M; Hermle GmbH, Goshe, Germany) at 3,000 × g for 10 min. Bacteria with gas vesicles were heated to 65°C for 2 min to break the vesicles before being pelleted. We tested two different filter types—a glass microfiber filter (GF/C; Whatman International Ltd.) and cellulose nitrate membrane (Sartorius Corp., Edgewood, N.Y.)—commonly used for concentration of cyanobacteria by filtration (17). For each water sample, 65 ml was filtered onto a 12.5-cm2 membrane. A 0.1-cm2 piece, corresponding to the filtration of 0.5 ml of water, was subsequently excised with a scalpel. The cells on the filter or on centrifugation pellets were resuspended in the respective lysis buffers by brief vortexing before DNA purification.
For reference DNA purification strategies, we used both a standard phenol-chloroform method (13, 16) and a solid-phase purification strategy (Dynabeads DNA DIRECT; Dynal A.S, Oslo, Norway) involving heating to 65°C (13). In the phenol-chloroform method, the cells were resuspended in 200 μl of buffer containing 10 mM EDTA (pH 8.0) and 12.5 mM TrisHCl (pH 8.0) and then homogenized for 5 min with 30 mg of alumina type A-5 (Sigma Chemical Co., St. Louis, Mo.) and a pestle (Kontes Scientific Instruments, Vineland, N.J.) to break the bacterial cell walls. Then 0.3 mg of lysozyme (Sigma Chemical Co.) and 0.1 mg of RNase A (Sigma Chemical Co.) were added, and the mixture was incubated at 37°C for 30 min. Sodium dodecyl sulfate was added to a final concentration of 0.5%, together with 0.1 mg of proteinase K (Boehringer GmbH, Mannheim, Germany), and the mixture was incubated at 65°C for 1 h. Cell debris and alumina were pelleted by a brief centrifugation, and the supernatant was transferred to a fresh tube. The supernatant was extracted twice with 200 μl of phenol-chloroform-isoamyl alcohol (25:24:1) and once with 100 μl of chloroform-isoamyl alcohol (24:1) with brief vortexing between the extractions. The DNA was precipitated at −20°C for 2 h with 2 volumes of ethanol and 0.1 volume of 3 M sodium acetate (pH 5.2) and pelleted by centrifugation at 15,000 × g in a microcentrifuge (model 2231 M) at 4°C for 30 min. The pellet was rinsed once with ice-cold 70% ethanol and dried in a vacuum centrifuge. Finally, the DNA was rehydrated in 40 μl of water with agitation for 2 h at room temperature. In the reference solid-phase method, 200 μl of Dynabeads DNA DIRECT (1 U) was added to the cell sample, and the mixture was incubated at 65°C for 15 min and stored at room temperature for 5 min. The beads bound to DNA were drawn to the side of a microcentrifuge tube with an MPC-E magnet (Dynal A.S). While bound to the beads, the DNA was washed twice with the washing buffer supplied with the kit. Finally, the bead-DNA complex was broken up by thorough resuspension in 40 μl of water. DNA for PCR amplification was also prepared by modifying a previously described protocol involving direct lysis of the cells without subsequent purification (9). In the modified protocol, the bacterial cells were resuspended in water, frozen at −70°C, and heated to 95°C for 5 min (to lyse the cells and to denature the DNases). The degree of cell lysis was determined by microscopic examination and agarose gel electrophoresis.
Combined solid-phase cell concentration and DNA purification protocols.
The cell detection limit with the standard Dynabeads DNA DIRECT buffer system is too high for the purpose of environmental monitoring (see Results and Discussion). An additional step involving ethanol and salt DNA precipitation of the DNA onto the beads was included to lower the detection limit in the optimized protocol. Incubation of the cells at 65°C in 4 M guanidine thiocyanate–1% Sarkosyl for 10 min resulted in complete cell lysis (determined by microscopic examination). Subsequent precipitation of the DNA onto 1 U of the beads (i.e., the beads in 200 μl of lysis buffer) in 2 volumes of ethanol led to reproducible detection of <50 cells (as determined by PCR amplification). One unit of beads was also the maximum amount tolerated in a 50-μl PCR mixture without enzyme inhibition (determined by titration experiments). The cell isolation conditions were accordingly optimized with 1 U of beads per sample test.
The bacteria tested have low affinity for the beads in water. To increase the this affinity, the water activity was lowered by the addition of alcohol and salt. Addition of 1 volume of ethanol increased the cell recovery considerably, but not at low cell concentrations (<104 cells/ml). This was improved by the addition of 1 volume of isopropanol instead, which increased the recovery about 10-fold at low cell concentrations. However, the optimal cell concentration protocol involved 1 volume of isopropanol and 0.1 volume of 7.5 M ammonium acetate for the cell adsorption step (the complex formed by P. agardhii NIVA-CYA 29 and the beads is shown in Fig. 1). Based on the optimized conditions, both large-scale and small-scale protocols (for 25 and 0.5 ml of water, respectively) were developed.
FIG. 1.
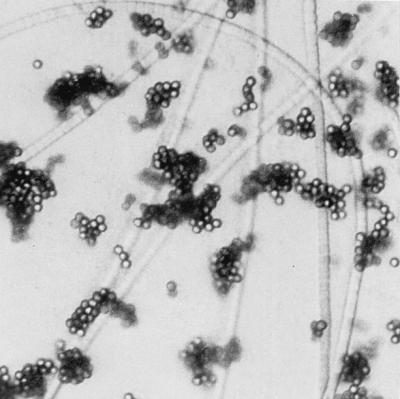
Micrograph showing adsorption of the filamentous cyanobacterium P. agardhii NIVA-CYA 29 to magnetic beads. Approximately 105 bacterial cells were mixed with the cell binding buffer, incubated at room temperature for 20 min, and concentrated with the MPC-E magnet for 2 min. Magnification, ×400, with interference contrast.
The protocols (large-scale values are indicated in brackets) involve mixing of 0.5-ml [25-ml] water sample with 1 U of beads (the beads in 200 μl of Dynabeads DNA DIRECT lysis buffer, prepared by removing the supernatant and washing the beads once with 200 μl of water), 50 μl [2.5 ml] of 7.5 M ammonium acetate, and 0.5 ml [25 ml] of isopropanol in a microcentrifuge tube [50-ml tube]. The samples were incubated at room temperature for 20 min and placed in an MPC-E [MPC-1] magnet for 2 min [5 min], and the supernatant was removed carefully. Then, 20 μl of 4 M GTC–1% Sarkosyl was added to the beads, and the mixture was incubated at 65°C for 10 min. To precipitate DNA onto the beads, 40 μl of 96% ethanol was added, and incubation was continued at room temperature for an additional 5 min. The beads were attracted to the tube wall with the magnet, the supernatant was removed, and the bead-DNA complex was washed twice with 200 μl of 70% ethanol. Finally, the complex was dried at 65°C for 5 min (it should be completely dried) and resuspended in 5 μl water. Maximum sensitivity was obtained by using all of the bead-DNA complex in the PCR.
PCR amplification.
For PCR amplification, the following 16S rDNA PCR primer pairs were used: 5′-AGCCAAGTCTGCCGTCAAATCA-3′ (CH) and 5′-ACCGCTACACTGGGAATTCCTG-3′ (CI) for amplifying M. aeruginosa, 5′-AAGGGTCCGCAGGTGGCAT-3′ (CL) and 5′-GCACAGCTCGGGTCGATACG-3′ (CM) for amplifying A. lemmermannii, and finally 5′-GGAAGGTTCTTGGATTGTCAACCC-3′ (CN) and 5′-TGCCTTTGCGAGGTTAAGCCT-3′ (CO) for amplifying P. agardhii. These primer sets are based on sequence information given by Rudi et al. (15). Amplifications were done with the GeneAmp 2400 PCR system (Perkin Elmer, Norwalk, Conn.) in 50-μl volumes containing 10 pmol of primers, 200 μM each deoxynucleoside triphosphate, 10 mM Tris-HCl (pH 8.8), 1.5 mM MgCl2, 50 mM KCl, 0.1% Triton X-100, 1 U DynaZyme thermostable DNA polymerase (Finnzymes Oy, Espoo, Finland), and 5 μl of bead-DNA complex. The PCR was initiated with a 4-min denaturation step at 94°C; this was followed by 40 cycles with the following denaturation, annealing, and synthesis parameters: 94, 58, and 72°C for 30 s each. An extension step for 7 min at 72°C was included at the end of the PCR.
RESULTS AND DISCUSSION
Testing of the combined solid-phase cell concentration and DNA purification protocols.
Based on the PCR results obtained, as visualized on ethidium bromide-stained agarose gels, we estimated a detection limit of 100 cells/ml by the small-scale protocol (Fig. 2A) and 10 cells/ml by the large-scale protocol (Fig. 2B). For the highest cell concentrations tested (106 and 107 cells/ml), we experienced difficulties in attracting the beads to the magnet in the cell concentration step. The purified DNA, however, gave reproducible PCR amplifications without enzyme inhibition (results not shown). Samples of water from natural habitats (containing Anabaena sp.) gave a detection limit of 250 cells/ml (Fig. 3), indicating that complex samples do not interfere with the analysis. The difference in the detection limit between diluted cultures and natural samples could be because the fast-growing organisms in culture contain more copies of the genome per cell than do the slow-growing organisms in nature. There were no significant differences in the sensitivity and specificity of the cell concentration and DNA purification assay between M. aeruginosa NIVA-CYA 43 and 228/1 (from 105 to 100 cells/ml) diluted in pure water, in water containing 105 cells of P. agardhii NIVA-CYA 29 per ml, in water containing 105 cells of A. lemmermannii NIVA-CYA 83/1 per ml, and in environmental water sampled from Lake Akersvatnet, Norway (results not shown). This indicates that the composition of the sample is not important for cell recovery, probably because there is an excess of bead surface for bacterial adsorption. The small-scale protocol has also successfully been used for detection of the gram-negative Escherichia coli NovaBlue and the gram-positive bacterium Bacillus cereus AH75. Although the protocol is not optimized for these species, the detection limits were in the same range as those of PCR directly on diluted cultures.
FIG. 2.

Detection limits for the small-scale (A) and large-scale (B) protocols. Cells and DNA were isolated from 0.5-ml (A) and 25-ml (B) cultures from the following dilution series: lane 1, 105 cells/ml; lane 2, 104 cells/ml; lane 3, 103 cells/ml; lane 4, 102 cells/ml; lane 5, 101 cells/ml; lane 6, 100 cells/ml. The agarose gel displays PCR products of 16S rDNA from the three species M. aeruginosa NIVA-CYA 43 amplified with primers CH and CI, A. lemmermannii NIVA-CYA 83/1 amplified with primers CL and CM, and P. agardhii NIVA-CYA 29 amplified with primers CN and CO. Twenty percent of the amplification products were loaded in each lane. mw, molecular weight standards. nt, fragment size (in nucleotides).
FIG. 3.

Water sample from Lake Fløtjønni, Smøla, Norway. The sample collected on 29 May 1996 contained approximately 2.5 × 104 cells of Anabaena sp. per ml (as determined by microscopic examination). Cells and DNA were isolated from 0.5 ml of water from the following dilution series: lane 1, 2.5 × 104 cells/ml; lane 2, 2.5 × 103 cells/ml; lane 3, 2.5 × 102 cells/ml; lane 4, 2.5 × 101 cells/ml; lane 5, 2.5 × 100 cells/ml. 16S rDNA was PCR amplified with primers CL and CM. The amplified bands were verified as Anabaena sp. by sequencing (98% sequence identity to A. lemmermannii NIVA-CYA 83/1 16S rDNA). Twenty percent of the amplification products were loaded in each lane.
Bacteria have been kept in the cell binding buffer for 1 week at room temperature with no detectable loss of sensitivity, as measured by PCR amplification. For field experiments, such stability may be crucial. Normally, the water sample has to be processed within a few hours after sampling because the cell composition can be altered with time and because DNA is unstable (with a half-life of 4 to 6 h) in environmental water (12).
Comparison of the combined solid-phase cell concentration and DNA purification protocols to standard (reference) protocols.
Commonly, nonspecific concentration of bacteria involves centrifugation or filtration. DNA is then purified in a second step, either by commercially available DNA purification kits or by phenol-chloroform-based methods (1–3, 19).
We evaluated the cell concentration step developed in this work by comparing the results with those of cell concentration by centrifugation or filtration. The centrifugation step resulted in a floating-cell fraction for organisms containing gas vesicles, e.g., A. lemmermannii NIVA-CYA 83/1. Pretreatment by heating to 65°C for 2 min was required to break these vesicles before cell pelleting. This caused precipitation of most of the cells in the centrifugation step. With the filtration approach, the cells on the filter were resuspended in the lysis buffers before being subjected to further processing. Both these approaches usually gave higher detection limits (for 0.5-ml water samples) and were less reproducible than the cell adsorption step developed in this work (Table 1). The lower reproducibility and higher detection limits for cell concentration by centrifugation or filtration compared than by the solid-phase strategy may partially be due to cell disruption (caused by mechanical shearing) and cell loss in the centrifugation and the filtration steps (7). For parameters which are not easily measured, such as the simplicity of the method and the risk of cross-contamination, the solid-phase cell concentration method also performed better than cell concentration by centrifugation or filtration. For example, the cell pellet formed by the filamentous cyanobacterium P. agardhii NIVA-CYA 29 was loose and difficult to handle, even after centrifugation at 15,000 × g for 30 min. Manual handling of the filters was difficult and increased the risk of cross-contamination. In the cell concentration protocol developed in this work, the cells were simply concentrated by being attracted to a magnet. The supernatant was then easily removed from the bead-cell complex (without extensive manual handling).
TABLE 1.
Comparison of cell concentration methods
| Straina | Cell detection limitb by:
|
||||
|---|---|---|---|---|---|
| Centrifugation | Heating and centrifugation | Filtration
|
Solid-phase cell concentration | ||
| Glass microfiber | Cellulose nitrate | ||||
| M. aeruginosa N-C 43 | ++ | NT | + | ++ | +++ |
| P. agardhii N-C 29 | +++ | NT | ++ | ++ | +++ |
| A. lemmermannii N-C 83/1 | + | ++ | ++ | ++ | +++ |
N-C, strain NIVA-CYA.
Cells from 0.5-ml water samples were recovered by the designated cell concentration protocols (see Materials and Methods). For all samples, the DNA was purified by the solid-phase DNA isolation strategy developed in this work. The cell detection limits were measured by PCR amplification and agarose gel electrophoresis of 10 μl of the amplification products. Symbols: +++, <100 cells; ++, 100 to 1,000 cells; +, 1,000 to 10,000 cells; NT, not tested.
Our DNA purification step was compared to DNA purification by the phenol-chloroform method or the commercially available Dynabeads DNA DIRECT method (the manufacturer recommends not to use more than 20% of the beads-DNA complex in each PCR for this buffer system). For the phenol-chloroform and the Dynabeads DNA DIRECT purification strategies, 12.5% of the purified DNA was used in the subsequent 50-μl PCR mixtures. Relative to the number of cells used in each PCR, the detection limits were about 500 cells for both reference methods. This is considerably higher than the detection limit of less than 50 cells obtained for the strategy developed in this work. As for most other DNA purification strategies, the phenol-chloroform and the Dynabeads DNA DIRECT strategies are designed for DNA purification from cell cultures and tissues (where the amount of material is not limited). At low cell concentrations, the DNA was probably lost during the many steps required for the phenol-chloroform method of DNA purification (see Materials and Methods). The Dynabeads DNA DIRECT strategy is based on coaggregation of beads and DNA (13). This aggregate was not formed at low cell concentrations, which might have resulted in the poor DNA recovery. However, with ethanol precipitation, the association of the beads and the DNA is tighter, probably leading to the high recovery at low DNA concentrations for our method.
Finally, the combined cell concentration and DNA purification protocol was compared to direct lysis of the cells without subsequent DNA purification (9). Direct lysis of cultures of known density and subsequent PCR directly on the lysate (5 μl of lysed culture in a 50-μl PCR mixture) gave a detection limit of 1,000 cells/ml, which is 10 and 100 times higher than for the small-scale and large-scale protocols, respectively. The number of directly lysed cells tolerated in a 50-μl PCR volume varied considerably, i.e. from more than 105 to less than 104 cells, for 19 different cyanobacterial strains tested. Thus, direct lysis of the cells can be used only in some cases and is not suitable for methods intended for several types of environmental samples. Our DNA purification strategy, however, gave reproducible amplification for more than 105 cells per sample test for all these strains, indicating removal of the PCR-inhibitory substances (results not shown).
Most of the previously developed methods for processing environmental water samples have been designed with the aim of detecting specific pathogenic microorganisms (e.g., Legionella, Salmonella, and Shigella) (1). For research purposes, methods have been developed for qualitative DNA purification from large volumes of water processed by filtration through a plankton net (23) or by tangential-flow filtration of 1,000 to 8,000 liters of water (7). None of these methods can be universally applied to different types of environmental samples (2). In most cases, detection and quantitation of naturally occurring bacteria (e.g., cyanobacteria) in water require higher detection limits than for pathogenic bacteria. Thus, simpler protocols can be used for detection and quantitation of naturally occurring bacteria than of pathogenic bacteria. We conclude that our combined cell concentration and DNA purification protocol is favorable for the detection and quantitation of samples containing more than 10 cells of target organisms per ml. For lower detection limits, however, a separate cell concentration step may be required.
Complete assay for routine monitoring.
The simplicity of the method—without filtration or centrifugation steps—allows for high throughput of samples and makes it adaptable for automation. Furthermore, the detection limits obtained suggest that the method is suitable for early detection of water blooms formed by potential toxic cyanobacteria (4). However, the versatility of the method has to be further tested empirically by large-scale screenings, since both the biological and chemical compositions of environmental water can be diverse and difficult to define. In this regard, we are currently developing competitive PCR strategies and colorimetric detection assays for complete high-throughput systems suitable for such screenings (14).
ACKNOWLEDGMENTS
This work has been supported by the Norwegian Research Council (NFR) to K.S.J. (grant 107622/420).
We are grateful to Olav M. and Randi Skulberg for preparing and cultivating the cyanobacterial species used in this work, to Anne-Brit Kolstø for the Bacillus cereus strain, and to Heidi Rudi and John Stacy for critically reading the manuscript.
REFERENCES
- 1.Bei A K. PCR amplification of DNA recovered from the aquatic environment. In: Trevors J T, van Elsas J D, editors. Nucleic acids in the environment. Heidelberg, Germany: Springer-Verlag KG; 1995. pp. 179–218. [Google Scholar]
- 2.Bei A K, Mahbubani M H. Applications of the polymerase chain reaction (PCR) in vitro DNA-amplification method in environmental microbiology. In: Griffin H G, Griffin A M, editors. PCR technology: current innovations. Boca Raton, Fla: CRC Press, Inc.; 1994. pp. 327–339. [Google Scholar]
- 3.Bowman J P, Sayler G S. Nucleic acid techniques in the environmental detection of microorganisms and their activities. In: Pickup R W, Saunders J R, editors. Molecular approaches to environmental microbiology. Chichester, United Kingdom: Ellis Horwood; 1996. pp. 63–97. [Google Scholar]
- 4.Carmichael W W. Cyanobacterial toxins. In: Hallegraeff G M, Anderson D M, Cembella A D, editors. Manual on harmful marine microalgae. Paris, France: UNESCO; 1995. pp. 163–175. [Google Scholar]
- 5.Enroth H, Engstrand L. Immunomagnetic separation and PCR detection of Helicobacter pylori in water and stool specimens. J Clin Microbiol. 1995;33:2162–2165. doi: 10.1128/jcm.33.8.2162-2165.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falconer, I. R. 1996. Potential impact on human health of toxic cyanobacteria. Phycologia 35(Suppl. 6):6–11.
- 7.Giovannoni S J, DeLong E F, Schmidt T M, Pace N R. Tangential flow filtration and preliminary phylogenetic analysis of marine picoplankton. Appl Environ Microbiol. 1990;56:2572–2575. doi: 10.1128/aem.56.8.2572-2575.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmberg A, Deggerdal A, Larsen F. AMS ’95. Third International Conference on Automation in Mapping and DNA Sequencing. 1995. Automated DNA extraction from whole blood, abstr. A10. [Google Scholar]
- 9.Howitt C A. Amplification of DNA from whole cells of cyanobacteria using PCR. BioTechniques. 1996;21:32–34. doi: 10.2144/96211bm05. [DOI] [PubMed] [Google Scholar]
- 10.Matsiota-Bernard P, Pitsouni E, Legakis N, Nauciel C. Evaluation of commercial amplification kit for detection of Legionella pneumophila in clinical specimens. J Clin Microbiol. 1994;32:1503–1505. doi: 10.1128/jcm.32.6.1503-1505.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer C J, Bonilla G F, Roll B, Paszko-Kolva C, Sangermano L R, Fujioka R S. Detection of Legionella species in reclaimed water and air with the EnviroAmp Legionella PCR kit and direct fluorescent-antibody staining. Appl Environ Microbiol. 1995;61:407–412. doi: 10.1128/aem.61.2.407-412.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul J H, Jeffrey W H, David A W, DeFlaun M F, Cazares L H. Turnover of extracellular DNA in eutrophic and oligotrophic freshwater environments of southwest Florida. Appl Environ Microbiol. 1989;55:1823–1828. doi: 10.1128/aem.55.7.1823-1828.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudi K, Kroken M, Dahlberg O J, Deggerdal A, Jakobsen K S, Larsen F. Rapid, universal method to isolate PCR-ready DNA using magnetic beads. BioTechniques. 1997;22:506–511. doi: 10.2144/97223rr01. [DOI] [PubMed] [Google Scholar]
- 14.Rudi, K., O. M. Skulberg, F. Larsen, and K. S. Jakobsen. Unpublished data.
- 15.Rudi K, Skulberg O M, Larsen F, Jakobsen K S. Strain characterization and classification of oxyphotobacteria in clone cultures on the basis of 16S rRNA sequences from the variable regions V6, V7, and V8. Appl Environ Microbiol. 1997;63:2593–2599. doi: 10.1128/aem.63.7.2593-2599.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 17.Skulberg, O. M., and R. Skulberg. 1997. Personal communication.
- 18.Skulberg R, Skulberg O M. Research with algal cultures. NIVA Culture Collection of Algae. Oslo, Norway: Norsk Institutt for Vannforskning; 1990. . (In Norwegian.) [Google Scholar]
- 19.Steffan R J, Atlas R M. Polymerase chain reaction: applications in environmental microbiology. Annu Rev Microbiol. 1991;45:137–161. doi: 10.1146/annurev.mi.45.100191.001033. [DOI] [PubMed] [Google Scholar]
- 20.Walsby A E. Gas vesicles. Microbiol Rev. 1994;58:94–144. doi: 10.1128/mr.58.1.94-144.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Way J S, Josephson K L, Pillai S D, Abbaszadegan M, Gerba C P, Pepper I L. Specific detection of Salmonella spp. by multiplex polymerase chain reaction. Appl Environ Microbiol. 1993;59:1473–1479. doi: 10.1128/aem.59.5.1473-1479.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winters D K, Slavik M F. Evaluation of a PCR based assay for specific detection of Campylobacter jejuni in chicken washes. Mol Cell Probes. 1995;9:307–310. doi: 10.1016/s0890-8508(95)91556-7. [DOI] [PubMed] [Google Scholar]
- 23.Zehr J P, McReynolds L A. Use of degenerate oligonucleotides for amplification of the nifH gene from the marine cyanobacterium Trichodesmium thiebautii. Appl Environ Microbiol. 1989;55:2522–2526. doi: 10.1128/aem.55.10.2522-2526.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]