

Abstract
Background:
Microvascular rarefaction is a feature of HFpEF which may underlie associated rhythm disturbances. Angiotensin II (AngII) signaling has been implicated, but its role in SA node dysfunction remains unclear.
Objectives:
We tested whether changes in SA node microvascular architecture contribute to pacemaker dysfunction in early HFpEF.
Methods:
Mice received a 28-day subcutaneous infusion of a sub-pressor dose of AngII. ECG, echocardiography, confocal imaging, spatial RNA detection, and optical mapping were used to assess SA node structure and function.
Results:
Heart rate declined progressively during AngII infusion, with males falling from 605 ±6 to 490 ±6 bpm and females from 646 ±23 to 511 ±10 bpm by day 28. Bradycardia was accompanied by increased beat-to-beat variability: the percentage of consecutive heartbeats that differed in duration by >6 milliseconds increased from 3.5 ±1.3% to 32.1 ±4.5% in males and from 3.8 ±1.1% to 27.7 ±2.5% in females. These changes coincided with reduced microvessel density in the superior SA node (males: 6.1 ±0.5 to 3.9 ± 0.2 nm/μm3; females: 5.6 ±0.4 to 2.8 ±0.5 nm/μm3), while vessels in the inferior SA node remained unchanged. Despite preserved myocyte density, these changes were accompanied by upregulation of oxidative stress and the hypoxia-inducible factor 1α and vascular endothelial growth factor signaling pathways.
Conclusions:
These findings highlight microvascular rarefaction in the superior SA node as a key early event in HFpEF pathology. The loss of redundant vascular loops compromises metabolic support for pacemaking, illustrating a broader principle: rarefaction can impair excitability in metabolically demanding excitable tissues.
Condensed Abstract
SA node dysfunction is common in HFpEF, but its underlying mechanisms are not fully understood. Using a sub-pressor dose angiotensin II infusion model of early HFpEF, we show that microvascular rarefaction develops in the superior SA node alongside bradycardia and heart rate variability changes. This rarefaction, marked by loss of redundant vascular loops, coincided with increased oxidative stress and HIF1α/VEGF signaling despite preserved myocyte density. Intrinsic pacemaker activity declined, suggesting that compromised vascular support reduces excitability. These findings support the general concept that microvascular rarefaction can impair excitability of surrounding cells across cardiovascular contexts.
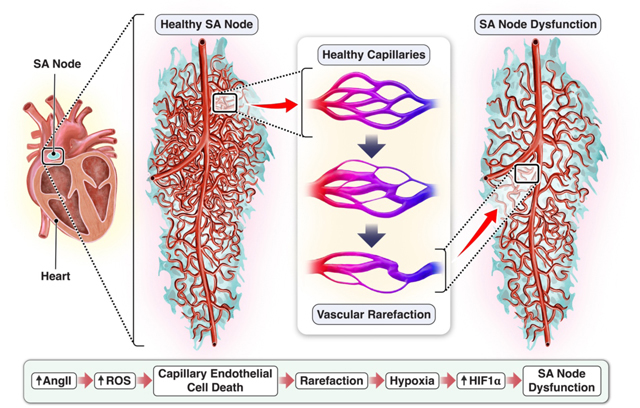
Central Illustration.
Vascular rarefaction in the sinoatrial node during low-dose angiotensin-II infusion. This study reveals the extent of microvascular rarefaction in the mouse sinoatrial node following 28-day infusion of low-dose angiotensin-II. This model of correlates microvascular rarefaction with the bradycardia and rhythm abnormalities caused by abnormal sinoatrial pacemaker function in the early stages of HFpEF and thus may represent an important determining factor in heart failure progression.
Introduction
The heart pumps blood into the pulmonary and systemic circulations to supply the body with oxygen and nutrients. Each beat begins with an action potential generated by pacemaker cells in the sinoatrial (SA) node, which then spreads through the atria, atrioventricular (AV) node, and ventricles, ensuring coordinated contractions and effective blood flow. This rhythmic activity depends on two main pacemaker mechanisms. The “membrane clock” involves hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels that depolarize cells from their maximum diastolic potential (1). In parallel, the “Ca2+ clock” features spontaneous Ca2+ release via ryanodine receptors located in the sarcoplasmic reticulum (SR) that activates the Na+/Ca2+ exchanger, driving further depolarization and helping the SA node reach action potential threshold (2,3).
The cardiac cycle recurs approximately 100,000 times per day in humans and nearly 1 million times per day in mice, requiring substantial energy, largely in the form of adenosine triphosphate (ATP) (4,5). ATP is consumed to maintain ionic gradients via the Na+/K+ ATPase and to power Ca2+ reuptake by the SR Ca2+ ATPase (SERCA) (6). Although SA node myocytes contain fewer sarcomeres than working cardiomyocytes, they still expend ATP during cross-bridge cycling. Moreover, continuous production of cyclic AMP (cAMP) by adenylate cyclases is energetically demanding, as it supports phosphorylation cascades regulating components of both the Ca2+ and the membrane clocks (7).
To meet these metabolic demands, SA node myocytes depend on a dense vascular network that delivers oxygen and nutrients. The SA node artery, typically branching from the right coronary or left circumflex artery (8), supplies blood to the node, with intranodal capillaries facilitating local diffusion (9–11). In both mice and humans, this artery spans from the superior to the inferior region of the node, giving rise to arterioles and capillary networks. Deoxygenated blood exits via venules and Thebesian veins draining into the right atrium (12).
Several studies highlight marked regional heterogeneity in SA node structure and function (13,14). In particular, Grainger, et al. (14) demonstrated that the superior SA node contains a higher density of microvessels and faster-firing myocytes than the inferior region. This spatial correlation between vascular density and electrical activity is consistent with a model in which dense vascularization in the superior node supports energetically demanding, high-frequency pacemaking. In contrast, the more sparsely vascularized inferior node region is populated by slower-firing myocytes, which contribute stochastic voltage fluctuations to pacemaking via stochastic resonance mechanisms (14–16).
Elevated angiotensin II (AngII) signaling is implicated in a range of cardiovascular diseases, including hypertension and heart failure (17,18). One well-established consequence of chronic AngII activation is microvascular rarefaction — the loss or pruning of capillaries — which has been observed in the ventricular myocardium during heart failure with preserved ejection fraction (HFpEF) (19,20). AngII-induced capillary loss not only contributes to ventricular remodeling but may also undermine normal perfusion in critical conduction system structures, setting the stage for rhythm abnormalities.
HFpEF is a complex syndrome defined by preserved ejection fraction but compromised diastolic filling. Rhythm disturbances are common in HFpEF (21–23), and a recent study (21) identified altered HCN channel expression and disrupted SR Ca2+ handling in SA node myocytes in a model of HFpEF. However, whether microvascular changes contribute to SA node pacemaking deficits remains largely unknown, despite evidence that arterial damage in the SA node can increase the propensity for rhythm disorders (24). Interestingly, although HFpEF is more common in both pre- and post-menopausal women (25), male patients tend to exhibit a higher incidence of arrhythmias, whereas female patients are more vulnerable to contractile dysfunction (26,27).
Here, we tested the hypothesis that HFpEF-induced changes in SA node vascular architecture occur during the development of pacemaking dysfunction. The findings of this study support a new model in which chronic AngII signaling drives microvascular rarefaction in the SA node, contributing to rhythm disturbances in early HFpEF that arise sooner and more prominently in males, yet ultimately also manifest in females. We posit that sustained AngII exposure increases oxidative stress, triggering selective endothelial cell death and capillary pruning—especially in the superior region of the node—while sparing the inferior region. As vessels disappear, local hypoxia ensues, activating HIF1α and angiogenic pathways. Despite this compensatory response, the vascular network remains topologically disrupted, depriving pacemaker myocytes of adequate metabolic support. Functionally, these changes manifest as bradycardia, and heightened heart rate variability, all in the absence of conduction block or extensive fibrosis. Collectively, these observations illustrate how AngII-driven oxidative stress and endothelial cell death converge to undermine pacemaker function, implicating superior SA node rarefaction as an early driver of HFpEF-related rhythm abnormalities. These findings support the principle that microvascular rarefaction impairs excitability in metabolically demanding excitable tissues—a generalizable mechanism with relevance across cardiovascular and other organ systems.
Methods
Briefly, wild-type C57BL/6J mice (8–12 weeks old) were used in this study. All animal experiments were conducted in accordance with institutional guidelines and national regulations for the care and use of laboratory animals. The study protocol was approved by the University of California Institutional Animal Care and Use Committee (IACUC). All procedures followed the approved animal-handling protocol to ensure the minimization of pain and distress. Mice were housed in a temperature- and humidity-controlled facility under a 12-hour light/dark cycle with ad libitum access to food and water.
Results
AngII infusion induces bradycardia and increases heart rate variability in male and female mice
To determine how a chronic sub-pressor dose of Ang II (0.1 mg/kg/day) alters heart rate and rhythm, we implanted male and female mice with ECG telemeters and recorded continuously for 28 days (Figure 1). Signals were processed as 24-hour averages so that each data point integrates activity across both the light and dark phases, thereby avoiding artefacts caused by time-of-day sampling.
Figure 1. Cardiac telemetry and platform ECG reveal bradycardia, increased heart rate variability, and reduced β-adrenergic responsiveness in male mice following 28-day AngII infusion.
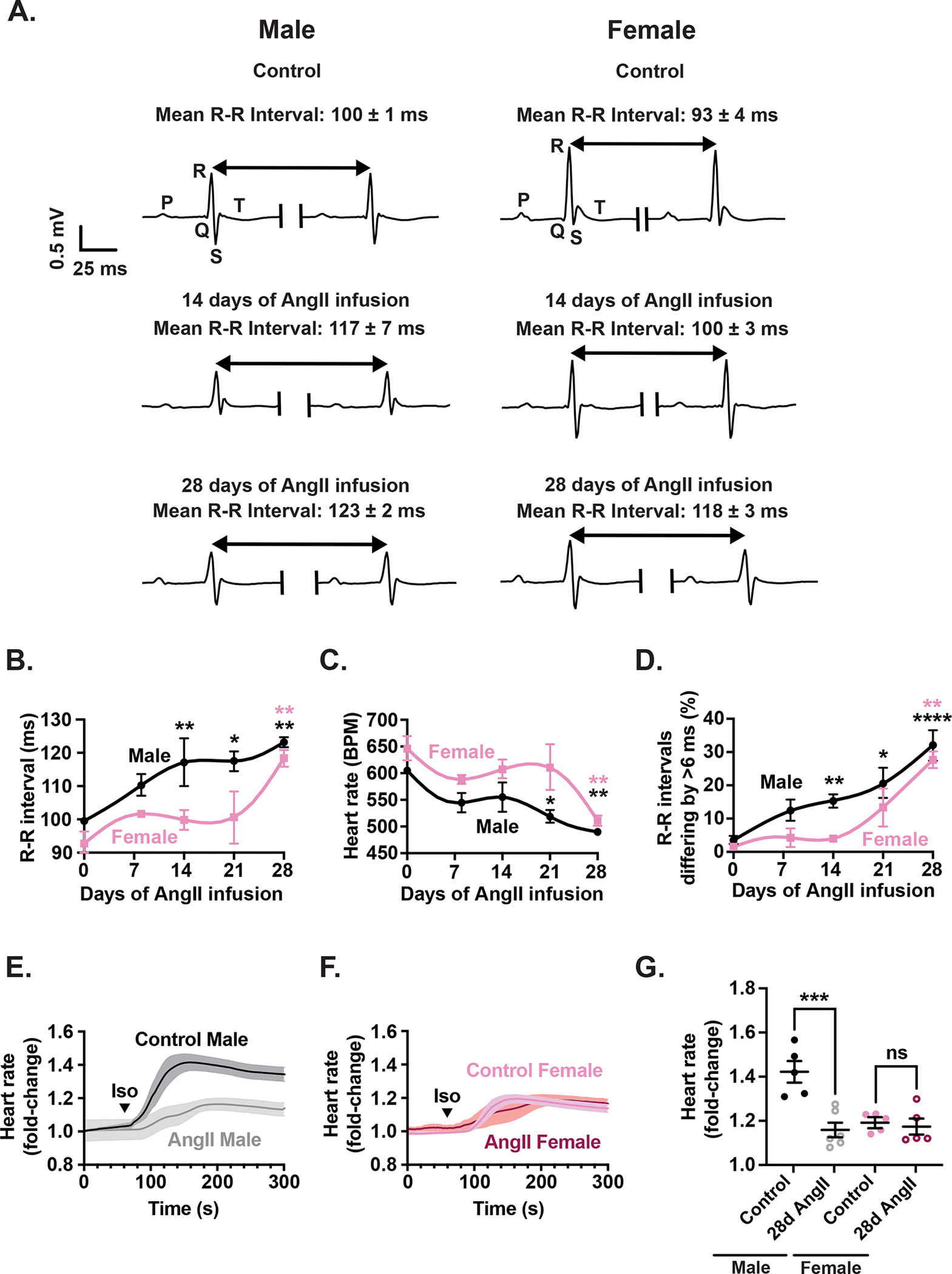
(A) Representative ECG telemetry traces recorded from male (right) and female (left) mice under control conditions as well as 14 and 28 days after AngII infusion. Time course of changes in R-R interval (B), heart rate (C), and (D) percentage of R-R intervals differing by greater than 6 ms increases during the 28-day infusion. Average time course of the effect of ISO injection on the HR of male (E) and female (F) control and AngII-infused (28 days) mice. (G) Scatter plot of the fold change in HR in upon ISO injection in control and AngII-infused mice.
Representative ECG traces (Figure 1A) show a progressive increase in R-R interval in both sexes during AngII infusion, indicative of bradycardia. Quantitative time-course data analysis revealed that bradycardia emerged earlier in males (Figure 1B), with R-R intervals significantly prolonged by day 14 (117 ± 7 ms; p = 0.0098), continuing through day 21 (117 ± 3 ms; p = 0.0250), and further lengthened to 123 ± 2 ms by day 28 (p = 0.0030). In females, it took longer for bradycardia to develop, with R-R intervals increasing from 93 ± 4 ms (day 0) to 118 ± 2 ms on day 28 (p = 0.0066). Correspondingly, male heart rates declined earlier than females (Figure 1C), falling from 605 ± 6 bpm at baseline to 519 ± 12 bpm on day 21 (p = 0.0295) and 490 ± 6 bpm on day 28 (p = 0.0033), whilst female HR fell from 646 ± 23 bpm on day 0 to 511 ± 10 bpm on day 28 (p = 0.0038). This was accompanied by an increase in the percentage of R-R intervals differing by more than 6 ms, which increased from 3.5 ± 1.3% at day 0 to 15.3 ± 2.0% at day 14 (p = 0.0019), 20.6 ± 4.7% at day 21 (p = 0.0114), and then peaked at 32.1 ± 4.5% by day 28 (p < 0.0001). In female mice, the onset of this index of HR variability was delayed, with a significant increase emerging by day 28 (27.7 ± 2.5%; p = 0.0022).
In addition to the ECG recordings described above, several ECG parameters from healthy and 28-day AngII-infused mice are summarized in Table 1. While P and R wave amplitudes, as well as QRS, corrected QT, and P wave durations, remained unchanged with AngII infusion, P-R intervals were significantly prolonged in both males (25.81 ± 1.07 ms in controls vs. 33.87 ± 1.27 ms in AngII-infused mice; p = 0.0014) and females (32.07 ± 1.34 ms in controls vs. 37.31 ± 1.71 ms in AngII-infused mice; p = 0.0208). This P-R interval prolongation may reflect impaired atrioventricular conduction in response to AngII treatment.
Table 1.
Detailed analysis of key ECG parameters.
| ECG Parameters | Control Male | 28d AngII Male | p Value | Control Female | 28d AngII Female | p Value |
|---|---|---|---|---|---|---|
|
| ||||||
| QRS Duration (ms) | 7.07 ± 0.09 ms | 7.46 ± 0.56 ms | 0.09116 | 10.04 ± 0.70 ms | 11.16 ± 0.87 ms | 0.4979 |
| QTc Interval (ms) | 54.29 ± 2.99 ms | 59.44 ± 2.80 ms | 0.0983 | 48.50 ± 1.48 ms | 52.33 ± 0.88 ms | 0.2409 |
| PR Interval (ms) | 25.81 ± 1.07 ms | 33.87 ± 1.27 ms | ** 0.0014 | 32.07 ± 1.34 ms | 37.31 ± 1.71 ms | * 0.0208 |
| P Duration (ms) | 6.52 ± 0.37 ms | 7.24 ± 0.28 ms | 0.641 | 8.41 ± 1.04 ms | 7.20 ± 0.62 ms | 0.3183 |
| P Amplitude (mV) | 0.10 ± 0.02 mV | 0.08 ± 0.01 mV | 0.3798 | 0.12 ± 0.01 mV | 0.09 ± 0.01 mV | 0.4006 |
| R Amplitude (mV) | 0.59 ± 0.06 mV | 0.56 ± 0.08 mV | 0.09591 | 0.71 ± 0.05 mV | 0.56 ± 0.10 mV | 0.3148 |
To evaluate whether these electrophysiological changes reflected impaired autonomic responsiveness, we next assessed β-adrenergic sensitivity using isoproterenol. To do this, we recorded using ECGs before and after an intraperitoneal bolus of isoproterenol (5 mg/kg) (Figure 1E–G). These experiments were performed at day 28 of AngII infusion, when bradycardia and HRV phenotypes were fully established, allowing us to determine whether sympathetic responsiveness remained intact or was diminished at this later stage of AngII infusion.
Healthy control male mice exhibited a robust increase in HR, reaching 1.42 ± 0.03-fold above baseline within minutes of drug administration (Figure 1E). In contrast, males subjected to 28-day AngII infusion showed a blunted response, with HR rising only to 1.16 ± 0.04-fold (p = 0.0001). In females, isoproterenol increased HR to 1.19 ± 0.04-fold in controls and 1.17 ± 0.08-fold in AngII-infused mice (p = 0.9102) (Figure 1F). These data indicate a sex-specific deficit in β-adrenergic responsiveness even under basal conditions, with impaired chronotropic reserve observed only in males (Figure 1G). Indeed, mixed effects analysis revealed a significant interaction between AngII treatment and sex (p = 0.0035), indicating that responses to β-adrenergic stimulation before and after AngII infusion differ markedly between males and females.
Together, these results demonstrate that AngII infusion is associated with progressive bradycardia, increased beat-to-beat variability, diminished atrioventricular conduction and reduced β-adrenergic sensitivity, particularly in males. Females show delayed onset of these changes and retain sympathetic responsiveness, highlighting important sex differences in the trajectory of pacemaker dysfunction in this model.
Amplification of heart rate variability across multiple frequency domains during AngII infusion
We next quantified heart-rate variability in the frequency domain during AngII infusion (Figure 2). As in Figure 1, telemetry data were averaged over 24 h, integrating light- and dark-phase activity and thereby eliminating time-of-day sampling artefacts. In male mice, very low frequency (VLF) power increased from 1.1 ± 0.1 ms2 at day 0 to 4.0 ± 0.5 ms2 at day 28 (p = 0.0002) (Figure 2A). Low frequency (LF) power rose from 4.2 ± 0.7 ms2 at day 0 to 14.9 ± 3.6 ms2 at day 14 (p = 0.0005), 13.1 ± 2.5 ms2 at day 21 (p = 0.0045), and 16.8 ± 1.2 ms2 at day 28 (p < 0.0001) (Figure 2B). High frequency (HF) power similarly increased significantly at all measured time points from baseline (Figure 2C). Female mice showed delayed HRV changes, with significant rises in VLF, LF, and HF power only fully manifesting by day 28. Mixed effects analysis of LF power indicated a significant interaction between AngII infusion and sex (p = 0.0070), suggesting that HRV responses differ markedly between sexes, particularly in the LF domain.
Figure 2. Frequency-domain telemetry ECG analysis reveals amplified HRV and altered autonomic balance during AngII infusion.
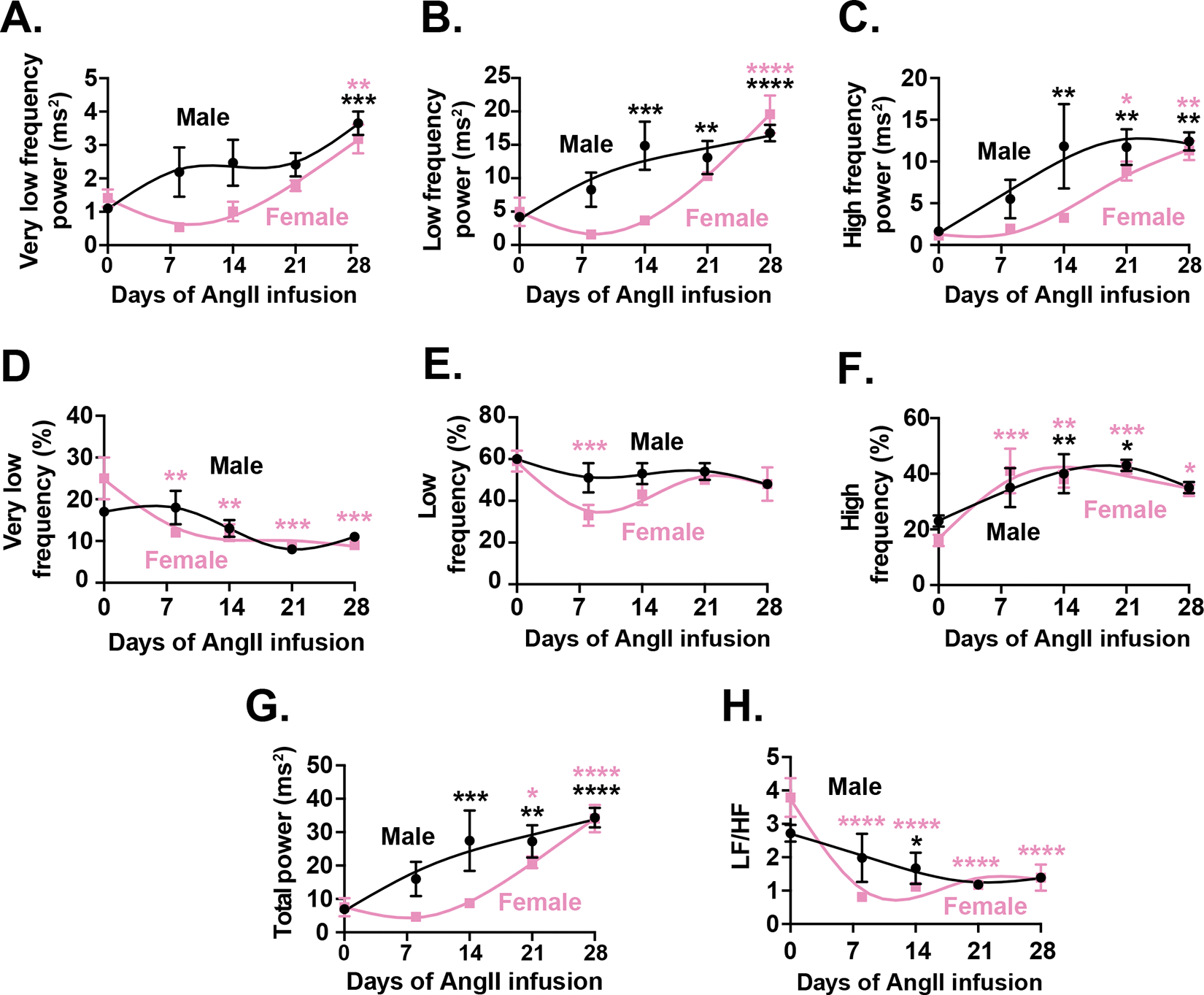
Time course of changes in very low (A), low (B), and high frequency (C), and (D) total power. (E) Time course of the LF/HF ratio. (F-G) Percentage contributions of very low (F), low (G), and high (H) frequency components to total HRV power shift over the 28-day infusion period. Data are presented as mean ± SEM for N = 6 male (black) and N = 5 female (pink) mice.
Next, we analyzed frequency component as a percentage of total HRV power, which allows us to discern the relative contributions of different autonomic mechanisms to heart rate regulation, independent of changes in overall variability under our experimental conditions. Furthermore, this normalization approach helps to clarify shifts in autonomic balance by accounting for inter-subject variability and ensuring that observed changes reflect true physiological adjustments rather than fluctuations in total variability alone. This analysis suggested that female mice exhibited an early and sustained reduction in VLF percentage, dropping significantly from 24.8 ± 5.1% at baseline to 9.3 ± 1.0% by day 28 (p = 0.0008) (Figure 2D). Conversely, the LF percentage initially declined but later returned toward baseline, indicating transient midrange autonomic shifts (Figure 2E). In contrast, males showed no significant percentage changes in VLF or LF components. Notably, HF power increased markedly as a percentage of total HRV power in both sexes, with females showing a pronounced rise as early as day 7, peaking at 41.8 ± 2.3% by day 21 (p = 0.0004), while males reached a similar peak more gradually by day 21 (p = 0.0107) (Figure 2F). Total HRV power increased significantly in both sexes but earlier in males, indicating a rapid onset of enhanced variability (Figure 2G). The LF/HF ratio, reflecting sympathovagal balance, declined prominently in females from 3.8 ± 0.6 at baseline to 0.8 ± 0.1 by day 7 (p < 0.0001) and remained suppressed, whereas males exhibited only modest, transient changes (Figure 2H). Taken together, these observations suggest that AngII infusion leads to heightened HRV primarily through increased short-term parasympathetic modulation, particularly in females, signaling a shift toward parasympathetic dominance alongside slower average heart rates.
AngII infusion induces diastolic dysfunction in male and female mice
Next, we focused our study on characterizing cardiac function in our AngII infusion model (Figure 3). Figures 3A and 3B present representative M- and Doppler-mode echocardiographic images, demonstrating the changes in left ventricular wall thickness and movement over the course of the experiment. Analyses of these images indicate an increase in left ventricular anterior wall (LVAW) thickness (Figure 3C). Males exhibited a progressive increase in LVAW thickness from 0.82 ± 0.01 mm at day 0 to 0.93 ± 0.16 mm at day 21 (p = 0.0057) and 0.99 ± 0.04 mm at day 28 (p = 0.0259). Similarly, LVAW increased in female mice from 0.73 ± 0.04 mm at day 0 to 0.86 ± 0.02 mm at day 21 (p = 0.0233) and 0.90 ± 0.02 mm at day 28 (p = 0.0083). Hence, both male and female mice showed a progressive increase in LVAW thickness over the 28-day period.
Figure 3. 28-day AngII infusion induces diastolic dysfunction in male and female mice.

Representative M-mode (A) and pulsed-wave Doppler mode echocardiographic images (B) from control as well as mice infused for 14, 21, or 28 days with AngII. Time courses for left ventricular anterior wall (LVAW) thickness (C), ejection fraction (D), fractional shortening decreases (E), myocardial performance index values (F), isovolumetric relaxation time (G), and E/A wave ratio (H). Data are presented for N = 6 female (pink) and N = 6 male mice (black).
Importantly, while the ejection fraction decreased compared to controls (male 0-day 86.5 ± 1.5 vs. 28-day 80.4 ± 0.6, p = 0.0038; female 0-day 87.5 ± 0.6 vs. 21-day 84.8 ± 0.9, p = 0.0037), it remained within the normal range (i.e., > 50%) throughout the study period for both sexes (one sample t-test, male and female p < 0.0001) confirming the preservation of systolic function characteristic of HFpEF (Figure 3D). However, we observed a decline in fractional shortening (Figure 3E), indicating changes in left ventricular contractility. This decline occurred faster in males, falling from 54.8 ± 0.7% at day 0 to 52.1 ± 0.3% at day 14 (p = 0.0103), 47.7 ± 1.1% at day 21 (p < 0.0001), and 47.4 ± 0.6% at day 28 (p < 0.0001) whereas females fell from 55.5 ± 0.8% at day 0 to 52.0 ± 1.1% at day 21 (p = 0.0074), and 50.8 ± 1.4% at day 28 (p = 0.0480).
To assess diastolic function, we examined several parameters. The myocardial performance index (Figure 3F) showed a gradual increase in male mice over the 28-day AngII infusion period, suggesting a sex-specific decline in overall cardiac performance. In males, myocardial performance index increased from 0.55 ± 0.02 at day 0 to 0.69 ± 0.02 at day 21 (p = 0.0169) and 0.71 ± 0.02 at day 28 (p = 0.02). Females exhibited a slower increase in myocardial performance index from 0.47 ± 0.04 at day 0 to 0.66 ± 0.03 at day 28 (p = 0.0138).
In males, the isovolumetric relaxation time (Figure 3G) also increased over the 28-day period, from 17.9 ± 0.7 ms at day 0 to 21.5 ± 0.8 ms at day 28 (p = 0.0243). On the other hand, a modest increase in females, from 16.1 ± 0.9 ms at day 0 to 18.9 ± 0.5 ms at day 28, did not reach statistical significance (p = 0.1213). These data indicate sex-specific impairment of left ventricular relaxation after 28-day AngII infusion.
Lastly, the E/A wave ratio (Figure 3H), another measure of diastolic function, showed a progressive decrease in both male and female mice. In males, the E/A wave ratio declined from 1.77 ± 0.05 at day 0 to 1.48 ± 0.03 at day 28 (p = 0.0026), whilst in females, the E/A wave ratio fell from 2.00 ± 0.17 at day 0 to 1.50 ± 0.10 at day 21 (p = 0.03) and 1.36 ± 0.07 at day 28 (p = 0.0455).
Beyond the conventional readouts of cardiac function outlined above, we analyzed several other parameters to further gauge whether the sub-pressor AngII model successfully induces diastolic dysfunction. Tissue Doppler recordings were measured in the mitral annulus (Figure S1A), measuring peak tissue velocity during early (e’ wave, Figure S1B) and late diastole (a’ wave, Figure S1C). No changes were observed in these tissue velocities over the course of AngII infusion in males or females, nor in the derivative ratios e’/a’ (Figure S1D) or E/e’ (Figure S1E). Neither male or female mice showed changes in peak blood velocity during early diastole (E wave, Figure S1F) and only females demonstrated an increased flow velocity during late diastole (A wave, Figure S1G). Estimations of left ventricular mass demonstrated significant increases in both males and females (Figure S1H). Meanwhile, only males displayed a transient increase in left ventricular end systolic volume (Figure S1I), and neither male nor female mice demonstrated any change in left ventricular end-diastolic volume (Figure S1J).
These results collectively demonstrate the induction of early diastolic dysfunction in the AngII mouse model of HFpEF. The preservation of ejection fraction, coupled with the development of many features of systolic and diastolic dysfunction, resemble key elements of the clinical presentation of HFpEF in humans (17,26,27).
Body and heart weights increase following AngII treatment
To further characterize the AngII infusion model of HFpEF, we next examined whether total body weight increased during AngII infusion (Figure S2). AngII-infused males were significantly heavier than age-matched controls (24.3 ± 0.3 g vs. 27.2 ± 0.6 g; p < 0.0001), as were females (19.0 ± 0.3 g vs. 21.9 ± 0.4 g; p < 0.0001) (Figure S2A). Longitudinal measurements before and after infusion confirmed weight gain in both sexes (Figure S2B), with males gaining 12.7 ± 1.3% and females gaining 18.1 ± 1.9% of their initial body weight (p = 0.0224) (Figure S2C).
We also assessed the weight of whole hearts excised from healthy and AngII-infused mice (Figure S2D). In males, heart weight increased from 161 ± 6 mg in controls to 197 ± 7 mg following AngII infusion (p = 0.0004), and in females from 145 ± 5 mg to 174 ± 7 mg (p = 0.0071). Interestingly, despite increases in both body and heart weights, body-to-heart weight ratios remained similar between AngII-treated and control groups (Figure S2E), suggesting proportional growth. Together, these findings indicate that chronic AngII infusion leads to systemic weight gain and increased heart mass consistent with cardiac hypertrophy.
AngII infusion induces vascular rarefaction in the superior but not in the inferior SA node in male and female mice
To assess whether AngII-induced HFpEF affects the vascular architecture of the sinoatrial (SA) node, we performed whole-mount immunohistochemistry on SA node preparations from male and female mice following 28 days of AngII infusion (Figure 4). Preparations were co-labeled with anti-HCN4 antibodies to identify pacemaker myocytes (Figure 4Ai) and anti-CD31 antibodies to visualize endothelial cells and delineate the vascular network (Figure 4Aii). Figure 4Aiii highlights the difference in vascular density between the superior and inferior SA node regions under baseline conditions. High-resolution confocal imaging and segmentation of CD31+ vessels were used to quantify regional vessel density in control and AngII-infused animals (Figure 4B). These 3D image stacks are represented in “maximum-Z” (above) and “maximum-Y” orientations (below).
Figure 4. AngII infusion induces microvascular rarefaction in the superior SA node.

Immunohistochemistry of the whole-mount sinoatrial preparation (A) delineates HCN4+ sinoatrial myocytes (green) and CD31+ microvasculature (red) (Ai). The microvascular network (Aii) was studied across multiple scales to assess evidence of regionalized rarefaction in the superior (sSAN) or inferior (iSAN) SA node regions (Aiii). High-resolution images were segmented (B) to analyze vessel density. Panels C and D show vessel tracing of CD31+ blood vessels from representative male (C) and female (D) mice. Panels E-F show scatter plots of vascular densities in superior and inferior regions of the node. Data are presented for n = 6–14 superior/inferior regional images from N = 3–5 male mice per group, and n = 5–6 superior/inferior regional images from N = 3 female mice per group.
Representative images indicate substantial remodeling in the vascular network following AngII infusion, particularly in the superior node region (Figures 4C–D). In male mice (Figure 4E), vessel density in the superior region of the SA node declined from 6.1 ± 0.5 nm/μm3 in controls to 3.9 ± 0.2 nm/μm3 following AngII infusion (p < 0.0001). Female mice showed a similar reduction, from 5.6 ± 0.4 nm/μm3 to 2.8 ± 0.5 nm/μm3 (p < 0.0001) (Figure 4F).
In contrast, vessel density in the inferior SA node region remained unaffected by AngII infusion. In males, vessel density was 1.8 ± 0.2 nm/μm3 in controls and 1.8 ± 0.3 nm/μm3 in AngII-treated mice (p > 0.9999). Similarly, in females, the vessel density was 1.7 ± 0.2 nm/μm3 in controls and 1.5 ± 0.3 nm/ μm3 after AngII infusion (p = 0.9971). A three-way ANOVA revealed a significant interaction between SA node region and AngII treatment (p < 0.0001), supporting the regional specificity of the rarefaction.
To further validate these findings, we applied two complementary quantification strategies. First, we assessed the percentage volume of each image stack occupied by vasculature (Figure S3A–B). This approach also revealed a significant interaction between SA node region and AngII infusion (p = 0.002; three-way ANOVA). Second, we quantified the number of branch-to-branch vessel segments within each stack (Figure S3C–D), which identified interactions between region and AngII treatment (p = 0.0043), and between region and sex (p = 0.0071), though no three-way interaction was observed (p = 0.9946). Interestingly, this microvessel loss was not accompanied by changes in average branch-to-branch vessel segment length, in either the superior or inferior SA node region of male or female mice (Figure S3E–F).
Together, these data show that chronic AngII infusion selectively induces microvascular rarefaction in the superior SA node, a region normally characterized by dense vascularization, while sparing the more sparsely vascularized inferior region. This pattern is consistent across both sexes and validated by multiple analytic methods, reinforcing the conclusion that early vascular remodeling in HFpEF is both regional and robust.
SA node myocyte density is preserved during chronic AngII infusion
To determine whether the rhythm disturbances observed in our AngII-induced HFpEF model are attributable to the loss of SA node myocytes, we assessed myocyte density in the superior and inferior regions of the node in male and female mice after 28 days of AngII infusion (Figure 5). Whole-mount SA node preparations were co-stained with antibodies against HCN4 to label pacemaker myocytes and CD31 to visualize vasculature (Figure 5Ai). The HCN4+ signal alone is shown in Figure 5Aii, with a focused view of the pacemaker region presented in Figure 5Aiii. High-resolution confocal images were obtained from the superior and inferior regions, and HCN4+ signal was segmented to quantify the percentage of tissue volume occupied by SA node myocytes (Figure 5B). Image stacks are represented in maximum-Z maximum-Y orientations.
Figure 5. SA node myocyte density in unaffected by 28-day AngII treatment.

The whole-mount sinoatrial preparation (A) showing HCN4+ sinoatrial myocytes (green) and CD31+ microvasculature (red) (Ai). Sinoatrial myocytes (Aii) were assessed in both the superior (sSAN) or inferior (iSAN) SA node regions (Aiii). High-resolution images were segmented (B) to quantify HCN4+ myocyte density. Representative myocyte density maps from a male (C) and female (D) mice. Panels E and F show scatter plots of myocyte densities in SA nodes from male and female hearts. Data are presented for n = 8–14 superior/inferior regional images from N = 4–5 male mice per group, and n = 6 superior/inferior regional images from N = 3 female mice per group.
Representative images of the myocyte layer in each region (Figures 5C and 5D) revealed no gross changes in cellular density following AngII infusion. In male mice (Figure 5E), there was no significant difference in myocyte density between control and AngII-infused groups in the superior region (63.8 ± 2.6% vs. 61.9 ± 4.9%; p = 0.9983) or the inferior region (53.7 ± 2.8% vs. 57.0 ± 3.9%; p = 0.9837). Female mice displayed a similar pattern (Figure 5F), with no significant changes in myocyte density in the superior (55.3 ± 5.7% in controls vs. 51.8 ± 6.9% in AngII-infused mice; p = 0.9924) or inferior region of the SA node (50.5 ± 10.2% vs. 60.7 ± 8.5%; p = 0.7206).
These findings indicate that chronic AngII signaling does not result in overt myocyte loss within the SA node, in either region or sex. Thus, the observed bradycardia and pacemaking dysfunction likely arise from non-lethal mechanisms — such as impaired vascular support, disrupted metabolic signaling, or altered ion channel function — rather than from myocyte death.
Vascular loops are reduced in the superior, but not in the inferior SA node, during AngII infusion
To investigate how chronic AngII infusion affects the topological organization of the SA node vasculature, we assessed vascular network complexity in male and female mice by quantifying the number of vascular loops — closed circular paths within the capillary network — across the superior and inferior regions of the node (Figure 6). CD31+ vessel networks were segmented and skeletonized to trace vessel paths and looped structures were automatically identified in white (Figure 6A). Image stacks are represented in maximum-Z maximum-Y orientations.
Figure 6. Microvascular rarefaction disrupts redundant vascular loops in the superior SA node.

Vascular loops were measured in the superior and inferior SA node regions using binary surface and filament tracing methods in Imaris (A). Loops are highlighted (white) against the terminal vessel segments (red). Representative vessel tracing images from male (B) and female (C) SA node immunohistochemistry experiments. Scatter plots of the number of vascular loops in males (D) and females (E). Data are presented for n = 7–14 superior/inferior regional images from N = 4–5 male mice per group, and n = 6 superior/inferior regional images from N = 3 female mice per group.
Representative images from the superior and inferior SA node regions in control and AngII-infused mice (Figures 6B and 6C) suggest a reduction in vascular loop complexity in the superior SA node region following AngII infusion, particularly in male mice.
Quantitative analysis confirmed this impression. In male mice (Figure 6D), the number of vascular loops in the superior SA node region declined significantly from 286.7 ± 34.5 loops per image stack in controls to 186.2 ± 16.7 following AngII infusion (p = 0.0077). No significant difference was observed in the inferior SA node region (54.2 ± 13.0 loops in controls vs. 42.9 ± 4.8 loops in AngII-treated mice; p = 0.8668).
Female mice showed a similar regional pattern (Figure 6E). In the superior region, vascular loops decreased from 128.7 ± 14.9 in controls to 57.7 ± 14.0 in AngII-infused mice (p = 0.0368). In the inferior node region, loop counts remained unchanged (44.3 ± 7.1 in controls vs. 35.8 ± 8.1 in AngII-treated mice; p = 0.8668). Notably, both regions exhibited lower baseline loop counts in females than in males, suggesting a potential sex-based difference in vascular network redundancy. A three-way ANOVA revealed interactions between SA node region and sex (p < 0.0001), and between region and AngII infusion (p = 0.015), but no three-way interaction.
These findings expand upon our earlier observations of selective rarefaction in the superior SA node by demonstrating that vascular loop structures are preferentially lost in this region during AngII-induced HFpEF. This loss of network complexity likely reflects a decline in vascular redundancy — the presence of multiple pathways for blood delivery. Such topological simplification may reduce the vascular system’s ability to buffer against increased metabolic demand or hemodynamic fluctuations, contributing to compromised pacemaker function despite preserved myocyte density and proximity to surviving vessels.
Myocyte-vessel proximity and contact are maintained in both the superior and inferior SA node during AngII infusion
To investigate whether the spatial relationship between pacemaker myocytes and the vasculature is disrupted during AngII-induced vascular rarefaction, we analyzed myocyte-vessel proximity in male and female mice following 28 days of AngII infusion (Figure 7). Whole-mount preparations were co-labeled with HCN4 and CD31 to visualize pacemaker cells and blood vessels, respectively. Figure 7A shows the merged signal across the SA node, while high-resolution images were segmented and processed using a watershed algorithm to approximate individual myocyte boundaries (Figure 7B). Image stacks are represented in maximum-Z maximum-Y orientations.
Figure 7. Sinoatrial myocytes maintain close contact with blood vessels during AngII-induced rarefaction.

Whole-mount SA node immunohistochemistry showing HCN4+ sinoatrial myocytes (green) and CD31+ blood vessels (red) (A) in the superior (sSAN) and inferior (iSAN) SA node regions. Respective signals were segmented and analyzed for proximity between myocytes and their nearest blood vessel, presented as heat maps (B). Heat maps of the distance from HCN4+ SA node myocytes to the nearest CD31+ blood vessel remains in males (C) and females (D) following 28-day. Scatter plots of the mean distance between myocytes and vessels in male and female SA node are shown in panels E and F, respectively. Data are presented for n = 6–14 superior/inferior regional images from N = 4–5 male mice per group, and n = 6 superior/inferior regional images from N = 3 female mice per group.
Color-coded maps of myocyte-to-vessel distance (Figures 7C and 7D) highlight regional differences in spatial organization between the superior and inferior regions of the SA node. However, no overt changes were observed in myocyte-vessel proximity between control and AngII-treated groups in either region or sex (Figures 7E and 7F). In the superior SA node, mean nearest-vessel distances remained consistently low across conditions — 0.38 ± 0.14 μm (male control), 0.18 ± 0.04 μm (male AngII; p = 0.9901), and 0.48 ± 0.17 μm (female control) vs. 0.26 ± 0.10 μm (female AngII; p = 0.9945). In the inferior region, average distances were greater but similarly unaffected by treatment: 1.65 ± 0.38 μm (male control) vs. 2.11 ± 0.55 μm (male AngII; p = 0.8276) and 2.61 ± 0.50 μm (female control) vs. 3.02 ± 0.74 μm (female AngII; p = 0.9465).
To complement these measurements, we assessed the percentage of SA node myocytes in direct contact with CD31+ vessels using a binary overlap analysis (Figure S4). In the superior SA node region of male mice (Figure S4A), contact was nearly universal in both control (96.5 ± 1.7%) and AngII-infused animals (96.4 ± 0.7%; p = 0.6911). In the inferior node region, contact rates were lower but unchanged: 71.7 ± 5.8% (control) vs. 70.4 ± 5.4% (AngII; p = 0.9988). Female mice displayed the same pattern (Figure S4B), with no significant differences in contact rates between control and AngII-infused groups in either the superior (94.3 ± 1.6% vs. 95.2 ± 1.4%; p = 0.9998) or inferior SA node regions (65.5 ± 4.1% vs. 63.1 ± 5.4%; p = 0.9941).
Together, these findings suggest that AngII-induced microvascular rarefaction reduces the overall number of vessels near SA node myocytes, particularly in the superior region. However, the distance between each myocyte and its nearest surviving vessel remains unchanged. As a result, while the microanatomy of existing myocyte-vessel connections is preserved, as noted above, the redundancy of vascular supply is diminished.
Vessel topography remains unaltered as microvessels are lost during AngII infusion
To further characterize vascular remodeling in the SA node during HFpEF progression, we analyzed the geometric properties of individual vessel segments, focusing on branching angle and straightness (Figure S5). In both male and female mice, vessel branching angles did not differ between control and AngII-infused groups in either the superior or inferior SA node regions (Figure S5A–C). Similarly, segment straightness — a measure of how directly a vessel connects two points — was unaffected by AngII treatment in either region or sex (Figure S5D–F). These findings indicate that although vascular loops are selectively lost in the superior SA node, the geometry of surviving vessel segments remains intact. Thus, AngII-induced rarefaction reduces network complexity without altering the fundamental topography of the remaining vasculature.
AngII exposure increases oxidative stress in the SA node
Having established that vascular rarefaction selectively impacts the superior SA node during AngII-induced HFpEF, we next sought to explore upstream mechanisms driving this regional microvascular remodeling. Given the well-established role of oxidative stress in vascular dysfunction (28–30), we asked whether acute AngII exposure is sufficient to trigger reactive oxygen species (ROS) production in the SA node.
To address this, we performed ex vivo ROS imaging in SA node preparations from healthy male and female mice before and during a 30-minute exposure to 100 nM AngII. Low-magnification imaging captured the entire node (Figure 8A), and high-resolution images were collected every five minutes in both the superior and inferior regions (Figure 8B). In male SA nodes, AngII increased ROS levels in both regions: to 1.31 ± 0.06 F/F0 over baseline in the superior (p = 0.0009) and 1.23 ± 0.08 F/F0 in the inferior (p = 0.0113) (Figure 8C). A similar response was observed in female mice, with ROS rising to 1.20 ± 0.05 F/F0 in the superior (p = 0.0091) and 1.21 ± 0.05 F/F0 in the inferior region (p = 0.0051) (Figure 8D). These data demonstrate that AngII rapidly induces ROS production across both major SA node regions and sexes.
Figure 8. AngII increases reactive oxygen species generation in male and female SA nodes.

(A) Representative image of the whole SA node preparation incubated with 5 μM CellRox Green, delineating the superior region in red and inferior region in blue. (B) Representative zoomed in images of superior (red) and inferior (blue) regions before and after application of 100 nM AngII for 30 minutes. Time course of the normalized CellROX Green fluorescence intensity over 30 minutes in response to 100 nM AngII from male SAN (C) and female SAN (D). Each solid line of the time course is presented as an average fit of all traces with the SEM plotted in the shaded regions. Data are presented for N = 4 males and N = 6 females.
Chronic AngII infusion increases regional HIF1α and VEGF expression in the SA node
Having established that chronic AngII exposure initiates oxidative stress and leads to microvascular rarefaction, particularly affecting vascular supply in the superior region of the SA node, we next investigated whether this triggers a sustained hypoxic response. We hypothesized that regional microvascular rarefaction would create localized tissue hypoxia, consequently activating canonical oxygen-sensing pathways mediated by hypoxia-inducible factor 1α (HIF1α) and VEGF (31–33). To test this hypothesis, we performed RNAscope® in situ hybridization (Figure 9) to quantify HIF1α and VEGF mRNA expression in both superior and inferior SA node regions from male and female mice following 28 days of AngII infusion, compared to age-matched controls (Figure 9A).
Figure 9. Chronic angiotensin II infusion increases Hif1α and Vegf mRNA expression in the SA node.

Representative image of a healthy male SA node slice preparation (A) subjected to RNAscope in situ hybridization, revealing Hif1α+ (magenta) and Vegf+ (yellow) mRNA puncta, as well as DAPI nuclear staining (blue). Super and inferior SA node regions of interest are presented, inset. Representative regions of interest are presented from AngII-infused and healthy control male and female mice (B). HIF1α mRNA puncta per mm2 (ln-transformed) are presented in the superior and inferior regions of the SA node from male (C) and female mice (D). Quantification of VEGF mRNA puncta per mm2 (ln-transformed) in the same regions from male (E) and female mice (F). Data are presented for n = 30–42 superior/inferior regional images from N = 3–4 male mice (black) and n = 19–29 superior/inferior regional images from N = 3 female mice (pink).
Quantitative analysis revealed significant AngII-induced upregulation of both HIF1α+ and VEGF+ across all regions and in both sexes (Figure 9B). Normalized gene expression levels were quantified as Ln-transformed densities of HIF1α+ and VEGF+ puncta per mm2 tissue area. In males, HIF1α expression rose from 9.72 ± 0.20 to 11.00 ± 0.28 in the superior SA node region (p < 0.0001), and from 9.75 ± 0.24 to 10.60 ± 0.17 in the inferior region (p < 0.0001) (Figure 9C). In females, HIF1α expression increased from 10.30 ± 0.03 to 10.70 ± 0.10 in the superior SA node (p < 0.0001) and from 9.97 ± 0.18 to 10.60 ± 0.06 in the inferior region (p < 0.0001) (Figure 9D).
Consistent with HIF1α-mediated transcriptional activation (34), VEGF transcript density was also significantly elevated in AngII-treated mice. In males, VEGF expression increased from 9.95 ± 0.04 to 10.50 ± 0.04 in the superior region (p < 0.0001), and from 9.37 ± 0.15 to 9.95 ± 0.11 in the inferior region (p < 0.0001) (Figure 9E). Female VEGF expression also rose from 9.80 ± 0.06 to 10.30 ± 0.04 in the superior region (p = 0.0002) and from 9.65 ± 0.09 to 10.20 ± 0.07 in the inferior region (p < 0.0001) (Figure 9F).
These findings support a model in which localized vascular rarefaction drives a regional hypoxic transcriptional program aimed at restoring perfusion. However, as rarefaction persists despite VEGF upregulation, this response may be insufficient to preserve microvascular integrity or function during chronic AngII exposure.
AngII infusion causes fibrosis in the ventricles but not in the SA node
We investigated whether the robust hypoxic transcriptional response observed in the SA node during chronic AngII infusion was accompanied by structural remodeling, specifically fibrosis. Increased fibrosis is characteristic of advanced or late-stage HFpEF and is well-established in the ventricular myocardium (20). To assess fibrosis, we performed Masson’s trichrome staining on ventricular and SA node sections from male and female mice following 28 days of AngII infusion (Figure 10).
Figure 10. AngII-infusion increases fibrosis in the ventricle but not in the SA node.

(A) Representative Masson’s trichrome-stained images of ventricular tissue from control and AngII-infused mice. (B) Quantification of collagen-positive area in ventricular sections. (C–D) Representative Masson’s trichrome-stained images of sinoatrial node tissue from control and AngII-infused mice. (E–F) Quantification of collagen-positive area in sinoatrial node sections from male and female mice. Ventricular collagen was measured in n = 26–38 samples from N = 4–7 mice per group. Sinoatrial node collagen was measured in n = 15–20 samples from N = 3–4 mice per group.
In both male and female mice, AngII infusion significantly increased ventricular collagen deposition, with collagen-positive areas rising from 1.8 ± 0.2% to 3.9 ± 0.4% in males (p < 0.0001) and from 1.9 ± 0.2% to 4.1 ± 0.3% in females (p < 0.0001) (Figure 10A–B). In contrast, despite inherently greater baseline fibrosis compared to surrounding myocardium, the SA node did not exhibit further increases in fibrosis following AngII treatment (Figure 10C–F). In male mice, collagen abundance remained unchanged in both superior (29.7 ± 2.7% vs. 26.5 ± 2.1%; p = 0.8019) and inferior regions (37.2 ± 2.6% vs. 30.3 ± 2.5%; p = 0.3092). Similarly, female mice showed no significant changes (superior: 33.6 ± 2.4% vs. 31.1 ± 1.7%; p = 0.8943; inferior: 33.4 ± 2.1% vs. 30.4 ± 2.1%; p = 0.8063).
These findings indicate that the SA node remains structurally sf while fibrotic remodeling has begun in the ventricular myocardium by day 28 of sub-pressor AngII infusion. The absence of increased fibrosis despite significant microvascular rarefaction suggests that SA node dysfunction in early HFpEF arises primarily through non-fibrotic mechanisms, potentially involving impaired metabolic support and altered excitability.
SA node firing decreases and becomes less periodic during HFpEF progression
Given the observed vascular rarefaction and oxidative stress in the SA node, but without concurrent fibrosis, we assessed whether these changes affect the intrinsic pacemaking activity of SA node myocytes during HFpEF progression. Using high-resolution optical mapping, we recorded spontaneous action potentials (APs) from isolated SA node preparations of male and female mice after 28 days of AngII infusion (Figure 11).
Figure 11. Optical recordings of intrinsic SA node activity following 28-day AngII infusion.

(A) Representative optical action potential (AP) traces from excised SA node preparations from control and AngII-infused (28-day) mice. (B) Scatter plots of spontaneous beating rates recorded from individual SA node preparations from male mice. (C) Representative optical recordings of APs from the SA node and adjacent right atrium. (D) Poincaré plots illustrating inter-AP interval variability in SA node preparations from male and female mice. Data are presented for N = 4 control male mice (black), N = 5 control females (pink), N = 5 AngII-infused males (grey), and N = 4 AngII-infused females (pink circles).
Consistent with our in vivo ECG findings, spontaneous AP frequency was significantly reduced in both sexes. Specifically, inter-AP intervals prolonged markedly from 205 ± 18 ms to 755 ± 61 ms in males (p = 0.0003) and from 217 ± 20 ms to 691 ± 148 ms in females (p = 0.001), corresponding to reduced spontaneous firing rates from 315 ± 29 bpm to 144 ± 15 bpm (males, p < 0.0001) and from 288 ± 22 bpm to 165 ± 15 bpm (females, p = 0.0019) (Figure 11A–B).
To determine whether conduction abnormalities accompanied this reduction in firing frequency, we simultaneously monitored activation in the SA node and the adjacent right atrium (Figure 11C). Every recorded SA node AP triggered subsequent activation of the right atrium, confirming intact conduction. Thus, the observed abnormalities reflect impaired pacemaker initiation rather than propagation block.
We further examined the periodicity of SA node firing by plotting joint interval distributions (Figure 11D). AngII-infused mice exhibited increased dispersion of inter-beat intervals compared to controls, indicating enhanced cycle length variability. Coefficient of variation (CV) analysis confirmed significant increases in variability in both males (0.06 ± 0.03 to 0.29 ± 0.06, p = 0.0013) and females (0.04 ± 0.01 to 0.27 ± 0.1, p = 0.0013).
Collectively, these findings demonstrate that chronic AngII exposure impairs intrinsic SA node pacemaker function by reducing spontaneous firing frequency and disrupting rhythm regularity. Importantly, these deficits occur in the absence of structural fibrosis or conduction defects, implicating alternative mechanisms such as impaired metabolic support or altered ion channel function secondary to microvascular rarefaction.
Discussion
The findings of this study support a model in which sustained AngII signaling triggers a cascade of events that culminate in selective microvascular rarefaction within the SA node, contributing to diastolic dysfunction in both male and female hearts. Specifically, prolonged AngII exposure elevates oxidative stress in the SA node, instigating targeted endothelial cell death—particularly in the superior region where vascular loops are abundant. We propose that as these redundant vessels are pruned, local hypoxia ensues, triggering the upregulation of HIF1α and its downstream angiogenic pathways. Despite this attempted compensatory response, the overall vascular network remains topologically disrupted, diminishing metabolic support to pacemaker myocytes. Functionally, these changes manifest as bradycardia, elevated heart rate variability, and reduced β-adrenergic reserve, all occurring without overt conduction block or significant fibrosis in the node. Notably, these arrhythmic and structural changes emerge earlier in males, underscoring sex-specific differences in disease progression. Taken together, our data reveal how AngII-induced oxidative stress and endothelial cell injury converge to compromise SA node function at an early stage of HFpEF. The implications of these findings are discussed below.
Chronic AngII infusion causes sex-specific differences in ECG, HRV, and diastolic function
An important aspect of this study is the longitudinal tracking of electrical (i.e., ECG) and functional (e.g., echocardiography) changes during the development of diastolic dysfunction under chronic AngII infusion. We observed progressive prolongations of the R–R interval and frequent rhythm irregularities in both male and female mice, indicative of nascent SA node dysfunction; however, these occurred earlier in males. Correspondingly, HRV data revealed compromised autonomic regulation in both sexes—again, with a delayed onset in females. In particular, the mice often exhibited bradycardia coupled with an overall rise in total HRV power, a decrease in VLF, no significant change in LF, and an increase in HF components, leading to a lower LF/HF ratio. Functionally, this pattern suggests that, despite the reduced sinus rate, there is robust beat-to-beat variability at shorter timescales driven largely by parasympathetic or respiratory oscillations.
Moreover, the diminished β-adrenergic reserve seen in AngII-infused mice—particularly in males—further reinforces the idea that the sinus node lacks the capacity to mount normal sympathetic responses, thereby “defaulting” to a bradycardic baseline while magnifying rapid vagal fluctuations. Although baseline β-adrenergic responsiveness was lower in female mice, it declined more sharply in males during the emergence of AngII-driven diastolic dysfunction. While our study does not dissect the underlying molecular mechanisms behind these sex-specific differences, future work should explore potential factors such as hormonal regulation, ion channel expression, and autonomic feedback loops. Notably, our observation that electrical remodeling appears earlier in males aligns with clinical reports of a higher incidence of arrhythmias in men with HFpEF, suggesting a potential mechanistic link between earlier SA node remodeling and subsequent arrhythmic vulnerability (26,27). Notably, whilst several recent studies have identified fibrosis as a key driver of pacemaker dysfunction (35–37), we did not observe significant SA node fibrosis in our model. Instead, our findings suggest that AngII-induced microvascular rarefaction may represent an earlier pathological event—preceding fibrosis—that contributes to impaired pacemaker function and progressive conduction abnormalities in the sinus node.
Complementing our findings on pacemaker remodeling, parallel echocardiographic assessments showed that changes in fractional shortening, myocardial performance index, isovolumetric relaxation time, and E/A wave ratio emerged at comparable times in both sexes, with most indices showing significant shifts by day 21. In males, these echo-based hallmarks of diastolic dysfunction preceded overt bradycardia and HRV changes, suggesting that electrophysiological remodeling may develop prior to—or at least in tandem with—the onset of evident contractile dysfunction. Collectively, these data highlight the interconnected nature of electrical and structural remodeling in early HFpEF. Moreover, the accelerated decline of SA node function and HRV in males underscores the importance of recognizing sex-specific disease trajectories, which could inform more personalized therapeutic strategies and clinical management.
What are the mechanisms driving SA node vascular rarefaction in AngII-induced HFpEF?
Building on the sex-specific functional changes in our model and prior studies (17,25), several mechanisms emerge to explain the AngII-driven loss of the SA node microvasculature. First, AngII activates AT1 receptors on endothelial cells and pericytes, triggering Gq-coupled signaling that promotes ROS generation, inflammatory cytokine expression (e.g., IL-6), and leukocyte recruitment (29,30,38–40). These factors compromise endothelial integrity and can ultimately cause capillary endothelial cell death. In line with this sequence, we detected a rapid rise in ROS following acute AngII exposure. Although brief ROS elevations may transiently augment blood flow, sustained AngII signaling likely drives oxidative damage, endothelial apoptosis, and vascular pruning (28).
Our findings demonstrate that chronic AngII exposure induces a robust region-specific signaling cascade in the SA node, likely as a compensatory response to microvascular rarefaction. The upregulation of HIF1α and its downstream target VEGF suggests the activation of canonical angiogenic pathways aimed at restoring tissue perfusion (28). HIF1α is known to rise under low-oxygen or high-ROS conditions in the working myocardium, thereby inducing VEGF expression (31,34). Although both ROS and hypoxia can independently stabilize HIF-1α, the observed microvascular rarefaction in our model provides strong anatomical evidence for a local hypoxic milieu. Thus, while AngII-induced ROS may contribute to HIF-1α activation, the concomitant rarefaction suggests that tissue hypoxia is likely a principal driver in sustaining HIF-1α signaling under chronic AngII stimulation.
However, despite robust HIF1α and VEGF upregulation in our model, the lack of revascularization within 28 days suggests a potentially delayed or ineffective angiogenic response that is insufficient to maintain microvascular integrity during prolonged AngII exposure. Several mechanisms may contribute to these findings. Specifically, chronic AngII signaling may be blunting VEGF signaling via: 1) Persistent oxidative stress and ROS production via sustained NADPH oxidase activation and mitochondrial uncoupling, resulting in VEGF inactivation or impaired downstream targets of VEGF (28,41); 2) VEGF receptor desensitization from the consequent downregulation of VEGFR2/Flk-1 expression or function through AT1R-mediated pathways (42,43); and 3) An inflammatory microenvironment partly mediated by TLR4 activation that creates an unfavorable environment for appropriate angiogenesis (44,45).
The SA node’s unique electrophysiological demands, including a high metabolic rate for highly specialized functions, may render it particularly vulnerable to ineffective pro-angiogenic and effective vascular restoration. Since VEGF shapes SA node size, vascular density, and Connexin-43 expression during development (33), disruption of these normal adaptive processes may impair not only vascularization but also electrical coupling essential for pacemaker function, disrupting gap junction networks that impair SA node-atrial conduction (46,47). This may be further exacerbated by the localized and region-specific impact that may explain the bradycardia and sinus node dysfunction frequently observed in heart failure (17,26,48). Furthermore, our observation of preserved vessel segment morphology and myocyte-vessel contact despite extensive rarefaction implies selective vascular pruning of “redundant” loops rather than a complete collapse of the entire vascular network. These loops likely serve as critical reserves during increased metabolic demands for maintaining steady perfusion under stress, so their loss compromises the node’s functional reserve and local metabolic support for nearby pacemaker myocytes (33).
Overall, we propose that chronic AngII signaling triggers a pathological positive feedback loop which amplifies ROS production, disrupts blood flow regulation, and further exacerbates endothelial dysfunction, eventually causing anatomical rarefaction (28,41,44). This triggers a hypoxic cascade and upregulation of HIF1α and VEGF, which proves insufficient to rebuild the vasculature. As a result, local oxygen supply is compromised, potentially contributing to early rhythm disturbances in HFpEF.
Does metabolic insufficiency drive remodeling of SA node myocytes during AngII infusion?
Although HIF1α can shield cardiomyocytes from apoptosis (49), sustained overexpression leads to chronic shifts in intracellular Ca2+ signaling (32). In atrial fibrillation models, HIF1α downregulates SERCA2a and RyR2 while upregulating NCX expression (50), thereby reducing sarcoplasmic Ca2+ load and release (51–53). While similar experiments have not been performed in the SA node, such remodeling could weaken the intrinsic “Ca2+ clock” (2,3), contributing to the bradycardia observed here. Accordingly, Mesquita, et al. (21) reported decreased SR Ca2+ release in a multi-hit HFpEF model. Future studies should determine whether there is a causal between HIF1α activation and ion channel and Ca2+-handling protein expression in SA node myocytes during the development of HFpEF.
Our data show that AngII-induced rarefaction remains confined to the superior SA node, paralleling earlier findings that this region’s high-frequency pacemaker activity demands more robust vascularization (14). This aligns with broader cardiac patterns, where sites with higher contractile or metabolic load—such as the left ventricle—exhibit denser capillary networks (54) (55). Because cytosolic ATP levels in cardiac muscle remain low (~0.5 mM) and fluctuate on a beat-to-beat basis (4), even modest reductions in microvessels and hence local blood supply can quickly undermine myocyte excitability. Although such measurements have not yet been performed in SA node cells, future research should examine how ATP levels in these pacemaker myocytes evolve during HFpEF progression.
In our earlier work, we introduced a “stochastic resonance” model of SA node function in which electrical heterogeneity between the superior and inferior regions enhances pacemaking rather than hindering it (14–16). In this framework, the superior SA node generates strong, periodic action potentials, while inferior myocytes provide stochastic subthreshold oscillations that increase the probability of firing when integrated with the superior region’s signals. By effectively “boosting” the superior node’s rhythmic output, these noisier inputs from the inferior node help sustain consistent pacemaker activity at higher rates. Extending this model with our current findings, we now propose that microvascular remodeling in the superior node may lead to bradycardia. The superior region is the principal and most crucial site of periodic oscillation and relies on a robust blood supply to support its high-frequency firing.
Cardiac muscle has limited energetic reserves (4,5), making myocytes particularly vulnerable to ischemia once vascular loops are pruned. This can weaken their oscillatory strength, reducing the overall synergy with the subthreshold signals in the inferior region of the node. Consequently, both pacemaking rate and regularity decline, manifesting as the bradycardia and heightened beat-to-beat variability observed in our AngII model.
The SA node’s vulnerability to chronic AngII exposure underscores its potential role as an early marker of cardiovascular dysfunction in conditions such as HFpEF (21,56). Comorbidities such as obesity, diabetes, and hypertension can exacerbate oxidative stress and microvascular rarefaction (20,28,57–60), further impairing angiogenesis, aggravating endothelial dysfunction, and intensifying SA node hypoxia. Improved understanding of microvascular rarefaction-driven hypoxia and dysregulated HIF1α/VEGF-mediated repair mechanisms may provide a unifying framework for understanding SA node dysfunction across the spectrum of cardiovascular diseases while potentially explaining observed sex-specific differences in disease onset and progression. Whether this microvascular vulnerability is universal across HFpEF variants and whether rarefaction precedes or follows pacemaker remodeling remain open questions. Multi-hit animal models that incorporate both metabolic and hypertensive stressors—mirroring the clinical profile of HFpEF patients—will be instrumental in elucidating these temporal relationships and developing targeted therapeutic approaches.
Limitations of the study
This study employed a low-dose AngII infusion model (18) designed to isolate vascular changes without the confounding factors of hypertension, obesity, or diabetes. Although it induced diastolic dysfunction and sex-specific traits (e.g., male chronotropic incompetence), we did not observe classic HFpEF markers such as elevated E/e′ or e′/a′ ratios, and fractional shortening declined—an atypical feature of fully established HFpEF. These findings likely represent an earlier or transitional disease stage before overt hemodynamic compromise, offering insight into the incipient mechanisms of SA node dysfunction.
Conclusions
Our results identify region-specific vascular rarefaction in the superior SA node as an early driver of arrhythmic changes in AngII-induced HFpEF—occurring in the absence of fibrosis or widespread myocyte loss. AngII-triggered oxidative stress and endothelial cell injury diminish the vascular reserve around high-demand pacemaker myocytes, sparking a hypoxic response that upregulates HIF1α and VEGF but fails to restore capillary density. This rarefaction weakens the superior node’s contribution to sinus rhythm, reducing intrinsic firing and heightening heart rate variability. More broadly, our findings support the concept that microvascular rarefaction can impair excitability in metabolically demanding excitable tissues, providing a generalizable mechanism relevant across cardiovascular and possibly other organ systems.
Supplementary Material
Clinical Perspectives.
Competency in Medical Knowledge
This study enhances medical knowledge by identifying the SA node as an early and regionally vulnerable site of dysfunction in HFpEF, driven by AngII–induced microvascular rarefaction and oxidative stress. By demonstrating that these changes occur prior to fibrosis or conduction block, and differ by sex, the findings expand current understanding of nodal remodeling and arrhythmic risk in early-stage HFpEF. Clinician readers may use this mechanistic insight to better appreciate the role of microvascular support in pacemaker function and to inform emerging diagnostic or therapeutic approaches aimed at preserving conduction system integrity.
Translational Outlook
These findings position SA node microvascular rarefaction as a potential early driver of rhythm disturbances in HFpEF, occurring prior to the development of structural fibrosis within the SA node. Translating this insight into clinical practice will require non-invasive tools to detect regional vascular remodeling within the cardiac conduction system and validation of rarefaction as a predictive biomarker for arrhythmic risk. A key challenge is that existing preclinical models often fail to capture the complex, multi-hit pathophysiology of human HFpEF. Future research should integrate hypertensive, metabolic, and inflammatory stressors to better reflect the clinical disease landscape and investigate whether restoring vascular support through pro-angiogenic or anti-oxidative strategies can preserve nodal function. Progress in this area will depend on multidisciplinary collaboration among basic scientists, cardiovascular imaging specialists, and electrophysiologists to bridge the gap between mechanistic discovery and clinical application.
Acknowledgments
The project was supported by NIH grants HL168874 (LFS), TR001860 (TL1 TR001861) (EJR), HL163930 (JEC), UL1 TR001860 (GB), TR001859 (GB), GM130459 (NG), HL085727 (NC), HL085844 (NC), HL170520 (NC), HL160274 (NC), and OD010389. The work was further supported by AHA grants 25POST1378853 (PR), 24CDA1276831 (PNT), 23SFRNCCS1052478 (NC), 23SFRNPCS1060482 (NC), as well as a Harold S. Geneen Charitable Trust Awards for Coronary Heart Disease (PNT).
We would like to acknowledge Michele Persiani for his technical assistance with in situ hybridization experiments.
Abbreviations
- AngII
Angiotensin II
- HFpEF
Heart Failure with Preserved Ejection Fraction
- SA
Sinoatrial
- HCN
Hyperpolarization-activated, Cyclic Nucleotide-gated
- CD31
Cluster of Differentiation 31
- ATP
Adenosine Triphosphate
- ECG
Electrocardiogram
- HRV
Heart Rate Variability
- HIF1α
Hypoxia-Inducible Factor-1α
- VEGF
Vascular Endothelial Growth Factor
Footnotes
The authors have no conflicts of interest to declare. All co-authors have seen and agree with the contents of the manuscript and there is no financial interest to report. We certify that the submission is original work and is not under review at any other publication.
References
- 1.DiFrancesco D Characterization of single pacemaker channels in cardiac sino-atrial node cells. Nature 1986;324:470–3. [DOI] [PubMed] [Google Scholar]
- 2.Bogdanov KY, Maltsev VA, Vinogradova TM et al. Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ Res 2006;99:979–87. [DOI] [PubMed] [Google Scholar]
- 3.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res 2010;106:659–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhana P, Matsumoto C, Fong Z et al. Fueling the heartbeat: Dynamic regulation of intracellular ATP during excitation-contraction coupling in ventricular myocytes. Proc Natl Acad Sci U S A 2024;121:e2318535121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol 2018;15:457–470. [DOI] [PubMed] [Google Scholar]
- 6.Smith IC, Bombardier E, Vigna C, Tupling AR. ATP consumption by sarcoplasmic reticulum Ca2+ pumps accounts for 40–50% of resting metabolic rate in mouse fast and slow twitch skeletal muscle. PLoS One 2013;8:e68924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yaniv Y, Juhaszova M, Lyashkov AE, Spurgeon HA, Sollott SJ, Lakatta EG. Ca2+-regulated-cAMP/PKA signaling in cardiac pacemaker cells links ATP supply to demand. J Mol Cell Cardiol 2011;51:740–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodwill AG, Dick GM, Kiel AM, Tune JD. Regulation of Coronary Blood Flow. Compr Physiol 2017;7:321–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.James TN. Anatomy of the human sinus node. Anat Rec 1961;141:109–39. [DOI] [PubMed] [Google Scholar]
- 10.Titus JL. Normal anatomy of the human cardiac conduction system. Mayo Clin Proc 1973;48:24–30. [PubMed] [Google Scholar]
- 11.Opthof T The mammalian sinoatrial node. Cardiovasc Drugs Ther 1988;1:573–97. [DOI] [PubMed] [Google Scholar]
- 12.Wearn JT. The Role of the Thebesian Vessels in the Circulation of the Heart. J Exp Med 1928;47:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brennan JA, Chen Q, Gams A et al. Evidence of Superior and Inferior Sinoatrial Nodes in the Mammalian Heart. JACC Clin Electrophysiol 2020;6:1827–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grainger N, Guarina L, Cudmore RH, Santana LF. The Organization of the Sinoatrial Node Microvasculature Varies Regionally to Match Local Myocyte Excitability. Function (Oxf) 2021;2:zqab031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guarina L, Moghbel AN, Pourhosseinzadeh MS et al. Biological noise is a key determinant of the reproducibility and adaptability of cardiac pacemaking and EC coupling. J Gen Physiol 2022;154. [Google Scholar]
- 16.Clancy CE, Santana LF. Evolving Discovery of the Origin of the Heartbeat: A New Perspective on Sinus Rhythm. JACC Clin Electrophysiol 2020;6:932–934. [DOI] [PubMed] [Google Scholar]
- 17.Withaar C, Lam CSP, Schiattarella GG, de Boer RA, Meems LMG. Heart failure with preserved ejection fraction in humans and mice: embracing clinical complexity in mouse models. Eur Heart J 2021;42:4420–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Regan JA, Mauro AG, Carbone S et al. A mouse model of heart failure with preserved ejection fraction due to chronic infusion of a low subpressor dose of angiotensin II. Am J Physiol Heart Circ Physiol 2015;309:H771–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng H, Chen JX. Microvascular Rarefaction and Heart Failure With Preserved Ejection Fraction. Front Cardiovasc Med 2019;6:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015;131:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mesquita T, Zhang R, Cho JH et al. Mechanisms of Sinoatrial Node Dysfunction in Heart Failure With Preserved Ejection Fraction. Circulation 2022;145:45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borlaug BA, Melenovsky V, Russell SD et al. Impaired chronotropic and vasodilator reserves limit exercise capacity in patients with heart failure and a preserved ejection fraction. Circulation 2006;114:2138–47. [DOI] [PubMed] [Google Scholar]
- 23.Shah SJ, Borlaug BA, Kitzman DW et al. Research Priorities for Heart Failure With Preserved Ejection Fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation 2020;141:1001–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaudino M, Nasso G, Minati A et al. Early and late arrhythmias in patients in preoperative sinus rhythm submitted to mitral valve surgery through the superior septal approach. Ann Thorac Surg 2003;75:1181–4. [DOI] [PubMed] [Google Scholar]
- 25.Beale AL, Meyer P, Marwick TH, Lam CSP, Kaye DM. Sex Differences in Cardiovascular Pathophysiology: Why Women Are Overrepresented in Heart Failure With Preserved Ejection Fraction. Circulation 2018;138:198–205. [DOI] [PubMed] [Google Scholar]
- 26.Curtain JP, Adamson C, Kondo T et al. Investigator-reported ventricular arrhythmias and mortality in heart failure with mildly reduced or preserved ejection fraction. Eur Heart J 2023;44:668–677. [DOI] [PubMed] [Google Scholar]
- 27.Vaduganathan M, Claggett BL, Chatterjee NA et al. Sudden Death in Heart Failure With Preserved Ejection Fraction: A Competing Risks Analysis From the TOPCAT Trial. JACC Heart Fail 2018;6:653–661. [DOI] [PubMed] [Google Scholar]
- 28.Manning D, Rivera EJ, Santana LF. The life cycle of a capillary: Mechanisms of angiogenesis and rarefaction in microvascular physiology and pathologies. Vascul Pharmacol 2024;156:107393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan I, Schmidt MO, Kallakury B et al. Low Dose Chronic Angiotensin II Induces Selective Senescence of Kidney Endothelial Cells. Front Cell Dev Biol 2021;9:782841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimmeler S, Rippmann V, Weiland U, Haendeler J, Zeiher AM. Angiotensin II induces apoptosis of human endothelial cells. Protective effect of nitric oxide. Circ Res 1997;81:970–6. [DOI] [PubMed] [Google Scholar]
- 31.Datta Chaudhuri R, Banerjee D, Banik A, Sarkar S. Severity and duration of hypoxic stress differentially regulates HIF-1alpha-mediated cardiomyocyte apoptotic signaling milieu during myocardial infarction. Arch Biochem Biophys 2020;690:108430. [DOI] [PubMed] [Google Scholar]
- 32.Holscher M, Schafer K, Krull S et al. Unfavourable consequences of chronic cardiac HIF-1alpha stabilization. Cardiovasc Res 2012;94:77–86. [DOI] [PubMed] [Google Scholar]
- 33.Calkoen EE, Vicente-Steijn R, Hahurij ND et al. Abnormal sinoatrial node development resulting from disturbed vascular endothelial growth factor signaling. Int J Cardiol 2015;183:249–57. [DOI] [PubMed] [Google Scholar]
- 34.Forsythe JA, Jiang BH, Iyer NV et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 1996;16:4604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalyanasundaram A, Li N, Gardner ML et al. Fibroblast-Specific Proteotranscriptomes Reveal Distinct Fibrotic Signatures of Human Sinoatrial Node in Nonfailing and Failing Hearts. Circulation 2021;144:126–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li N, Artiga E, Kalyanasundaram A et al. Altered microRNA and mRNA profiles during heart failure in the human sinoatrial node. Sci Rep 2021;11:19328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li N, Kalyanasundaram A, Hansen BJ et al. Impaired neuronal sodium channels cause intranodal conduction failure and reentrant arrhythmias in human sinoatrial node. Nat Commun 2020;11:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuroda J, Ago T, Nishimura A et al. Nox4 is a major source of superoxide production in human brain pericytes. J Vasc Res 2014;51:429–38. [DOI] [PubMed] [Google Scholar]
- 39.Murphy TJ, Alexander RW, Griendling KK, Runge MS, Bernstein KE. Isolation of a cDNA encoding the vascular type-1 angiotensin II receptor. Nature 1991;351:233–6. [DOI] [PubMed] [Google Scholar]
- 40.Bhullar SK, Dhalla NS. Angiotensin II-Induced Signal Transduction Mechanisms for Cardiac Hypertrophy. Cells 2022;11. [Google Scholar]
- 41.Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler Thromb Vasc Biol 2006;26:826–32. [DOI] [PubMed] [Google Scholar]
- 42.Sano H, Hosokawa K, Kidoya H, Takakura N. Negative regulation of VEGF-induced vascular leakage by blockade of angiotensin II type 1 receptor. Arterioscler Thromb Vasc Biol 2006;26:2673–80. [DOI] [PubMed] [Google Scholar]
- 43.Buharalioglu CK, Song CY, Yaghini FA et al. Angiotensin II-induced process of angiogenesis is mediated by spleen tyrosine kinase via VEGF receptor-1 phosphorylation. Am J Physiol Heart Circ Physiol 2011;301:H1043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol 2005;25:2106–13. [DOI] [PubMed] [Google Scholar]
- 45.Nakashima T, Umemoto S, Yoshimura K et al. TLR4 is a critical regulator of angiotensin II-induced vascular remodeling: the roles of extracellular SOD and NADPH oxidase. Hypertens Res 2015;38:649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin YQ, Zhong Y, Zhu Y, Tian L. Changes in gap junction proteins Connexin30.2 and Connexin40 expression in the sinoatrial node of rats with dexmedetomidine-induced sinus bradycardia. Braz J Anesthesiol 2022;72:768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li N, Csepe TA, Hansen BJ et al. Molecular Mapping of Sinoatrial Node HCN Channel Expression in the Human Heart. Circ Arrhythm Electrophysiol 2015;8:1219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borlaug BA, Sharma K, Shah SJ, Ho JE. Heart Failure With Preserved Ejection Fraction: JACC Scientific Statement. J Am Coll Cardiol 2023;81:1810–1834. [DOI] [PubMed] [Google Scholar]
- 49.Datta Chaudhuri R, Banik A, Mandal B, Sarkar S. Cardiac-specific overexpression of HIF-1alpha during acute myocardial infarction ameliorates cardiomyocyte apoptosis via differential regulation of hypoxia-inducible pro-apoptotic and anti-oxidative genes. Biochem Biophys Res Commun 2021;537:100–108. [DOI] [PubMed] [Google Scholar]
- 50.Bekeredjian R, Walton CB, MacCannell KA et al. Conditional HIF-1alpha expression produces a reversible cardiomyopathy. PLoS One 2010;5:e11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol 2002;34:951–69. [DOI] [PubMed] [Google Scholar]
- 52.Schillinger W, Janssen PM, Emami S et al. Impaired contractile performance of cultured rabbit ventricular myocytes after adenoviral gene transfer of Na(+)-Ca(2+) exchanger. Circ Res 2000;87:581–7. [DOI] [PubMed] [Google Scholar]
- 53.Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol 2014;76:107–27. [DOI] [PubMed] [Google Scholar]
- 54.Goo S, Joshi P, Sands G et al. Trabeculae carneae as models of the ventricular walls: implications for the delivery of oxygen. J Gen Physiol 2009;134:339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicolas N, Roux E. 3D Imaging and Quantitative Characterization of Mouse Capillary Coronary Network Architecture. Biology (Basel) 2021;10. [Google Scholar]
- 56.Dorey TW, Moghtadaei M, Rose RA. Altered heart rate variability in angiotensin II-mediated hypertension is associated with impaired autonomic nervous system signaling and intrinsic sinoatrial node dysfunction. Heart Rhythm 2020;17:1360–1370. [DOI] [PubMed] [Google Scholar]
- 57.Tucsek Z, Toth P, Tarantini S et al. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J Gerontol A Biol Sci Med Sci 2014;69:1339–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Dinther M, Voorter PH, Jansen JF et al. Assessment of microvascular rarefaction in human brain disorders using physiological magnetic resonance imaging. J Cereb Blood Flow Metab 2022;42:718–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whelton PK, Carey RM, Aronow WS et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2018;71:e127–e248. [DOI] [PubMed] [Google Scholar]
- 60.Zeng H, Li L, Chen JX. Loss of Sirt3 limits bone marrow cell-mediated angiogenesis and cardiac repair in post-myocardial infarction. PLoS One 2014;9:e107011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.