

Abstract
Background
Opsoclonus-myoclonus ataxia syndrome (OMAS) is a rare neurological disorder, with involuntary rapid saccadic conjugate eye movements as one of characteristics, primarily affecting the cerebellum. While the exact pathogenesis remains unclear, genetic and autoimmune factors have been suggested to contribute to its development.
Methods
We enrolled patients diagnosed with OMAS before the age of 18 years at a pediatric neuroimmunology clinic in Boston, USA, using the 2004 Genoa Criteria. WGS was conducted for the patients and their biological parents in all cases, with one case including an unaffected sibling.
Results
De novo germline variants (DNVs) in probands were identified and validated, and analyses of structural variants (SVs), recessive variants in neuroimmune-associated genes, and high-resolution HLA typing were performed. Our study included 42 patients, 23 of whom had neuroblastoma. We found 12 confirmed DNVs in protein-coding regions in nine patients (29.0% of 31 from 30 trios and 1 quartet). Ten patients (23.8% of 42) had rare homozygous, or compound heterozygous variants known to alter protein function, affecting 11 genes. Notably, the HLA-DRB1*01 allele was observed in 27 out of 84 (32.1%) alleles in the patients, significantly higher than that in the general population (chi-square test, p < 0.0001). In one case, a potential genetic modifier of OMAS with severe cerebellar atrophy was identified, associated with a protein-truncating DNV in the CACNA2D2 gene.
Conclusions
This first genome sequencing study reveals potential genetic contributors to OMAS, implicating polygenic predisposition—with HLA-DRB1*01 as a possible factor—combined with non-genetic risk factors like neuroblastoma.
Keywords: opsoclonus-myoclonus ataxia syndrome, linked-reads genome sequencing, de novo germline variants, recessive, compound heterozygous
Introduction
Opsoclonus-myoclonus ataxia syndrome (OMAS) is a rare neurological disorder characterized by involuntary rapid saccadic conjugate eye movements, myoclonus, and ataxia which are often accompanied by sleep disturbance and behavioral symptoms. 1 Approximately 50% of children with OMAS have a neural crest tumor such as neuroblastoma (NB, MIM: 256700) and ganglioneuroblastoma.2 The association of pediatric-onset OMAS with neuroblastoma fits within a paraneoplastic paradigm in which there is a shared antigen between tumor cells and neurons leading to a cross-reactive immune response directed against both the tumor and the cerebellum.3, 4 Magnetic resonance imaging (MRI) of the brain is typically normal at onset. Over time, some patients develop cerebellar atrophy which is typically mild if it occurs.5 The proposed autoantigen in OMAS has yet to be identified and therefore OMAS remains a clinical diagnosis. In addition, only 2% to 4% of pediatric patients with neuroblastoma develop OMAS, and nearly half of OMAS cases occur sporadically without coexisting neural crest tumors, suggesting that other factors, which may include genetic background, contribute to the development of OMAS.6 In addition, OMAS has significant heterogeneity in terms of clinical course, treatment response and outcomes, which may be influenced by genetic background, particularly in genes involved in the immune or neuronal/glial cells of the central nervous systems.
A limited prior literature suggests that heritable genetic factors may contribute to OMAS. In one study, low resolution human leukocyte antigen (HLA) typing by PCR for loci A, B and DRB1 in 43 children with OMAS (21 with neuroblastoma) and 100 healthy controls showed a higher percentage of patients with HLA-DRB1*01 compared to healthy controls.7 Another recent study which inferred HLA types from tumor derived transcriptome data showed a trend towards a higher frequency of HLA-DRB1*01 in OMAS with NB compared to NB without OMAS, but did not include patients with OMAS without NB.8 This study also showed a showed a higher frequency of HLA-DOB*01:01 in OMAS with NB compared to NB without OMAS. Two studies have also shown that a higher percentage of parents of children with OMAS have an autoimmune disease compared to controls.9, 10
Alternatively, particularly in patients who do not have a NB, it is possible that monogenic neurological disorders could mimic OMAS. Such patients may have earlier than typical onset of OMAS and/or atypical features such as epileptic seizures.11 Published examples of this situation include KCTD7,12 SLC2A1,13 and SGCE 14 which likely do not involve immune mediated mechanisms. Finally, monogenic disorders involving immune function may lead to symptoms suggestive of OMAS through immune mediated mechanisms as has been described in Aicardi Goutieres syndrome with mutations in TREX1 and RNASEH2B.15
Within this complex background, we used whole genome sequencing (WGS) in a cohort of children with OMAS to discover de novo germline variants (DNVs) and inherited rare high-impact variants which could contribute to OMAS presentation, course and outcomes within the paraneoplastic/autoimmune paradigm, or genetic mimics of the syndrome. We also assessed HLA typing in the cohort. For an unbiased discovery of DNVs across the genome, third-generation long-read sequencing technique to minimize false negatives was used.16
Patients and Methods
Patient Recruitment and Samples
Potentially eligible patients were identified through prospective review of one of the author’s (MPG) specialized pediatric neuroimmunology clinic, supplemented by automated text search of a Boston Children’s Hospital (BCH) medical records database to identify potentially eligible patients who were no longer followed in clinic at the time of the study. Patients with a diagnosis of OMAS (as defined by the 2004 Genoa Criteria17) before the age of 18 years, along with their biological mother and father (and, in the case of the quartet, unaffected sibling), were eligible for inclusion. Of the 42 patients, one of the authors (MPG) evaluated 41 of them in clinic. The remaining patient’s diagnosis was confirmed by review of medical records. Year of OMAS onset ranged from 1992 to 2017. Study enrollment occurred between April 2018 and October 2019. Over this time frame, one of the authors (MPG) evaluated one patient for possible OMAS but suspected a monogenic cause which was confirmed clinically. This patient was not enrolled in the WGS study but is described below. Clinically obtained brain MRI reports were reviewed to assess for the presence of cerebellar atrophy.
Biological samples were obtained, processed and accessed through the Biorepository in the Department of Neurology and the PrecisionLink Health Discovery biorepository at BCH. To ensure biological relatedness, the ABI Prism linkage mapping set (panels 1–28) was used, and non-biological parents, such as step-mothers or step-fathers, were excluded from the WGS analysis. This included 30 complete trios and one quartet, which consisted of a family including an unaffected twin sibling, and 11 unrelated patients for a total of 105 subjects.
Whole-genome sequencing and Analysis
The genomic DNA (gDNA) samples were extracted from the whole blood of 89 individuals using Gentra Purgene and Chemagic B5k extraction kits, and from saliva of the remaining 16 individuals using Prep-iT kit. DNA quality was measured using Tapestation (Agilent, Santa Clara, CA, USA), a gel-based assay, for approval to proceed with sequencing. The Biobanks used a standard operating procedure to calculate and perform normalization on approved samples. Subsequently, 56 samples were sent to the Broad Institute, and 49 samples were sent to the New York Genome Center (NYGC) for WGS. The gDNA samples were prepared with a third generation linked-read (LR-WGS) protocol, followed by sequencing on the Illumina platform. The DNA libraries were constructed using the 10X Chromium Genome Library and Gel Bead Kit (10x Genomics, Pleasanton, CA, USA) according to the manufacturer’s protocols, starting with at least 1.2 ng of template gDNA. The Chromium Controller was utilized to combine a library of 10X Genome Gel Beads with high molecular weight template genomic DNA (1.25 ng), a master mix of enzymes and buffer, and partitioning oil to create droplets containing single gel beads and DNA. During the LR-WGS process, gDNA sample was partitioned into approximately 1 million 10X Gel Bead-In Emulsions (GEMs). The partitioned reactions were incubated isothermally for 3 hours at 30°C, followed by 10 minutes at 65° C, and held at 4°C. This generated barcoded fragments ranging from a few to several hundred base pairs. After amplification, the emulsion was collected, and GEMs were broken. The recovered barcoded DNA was then size-selected for library preparation. At the Broad Institute, the resulting library fragment sizes were determined using the DNA 1000 Kit and 2100 BioAnalyzer (Agilent Technologies). The barcoded fragments were then indexed with Illumina-specific samples according to the manufacturer’s instructions to generate libraries. Finally, the barcoded sequencing libraries were quantified using qPCR (KAPA Biosystems Library Quantification Kit).
In a similar manner, at NYGC, high Molecular Weight (HMW) DNA underwent qualitative checks for size distributions on the Fragment Analyzer using the High Sensitivity gDNA assay (Advanced Analytical™) before library preparation to ensure intactness of the input material. Subsequently, samples were quantified to ensure precisely 2.5ng of gDNA was used as input for library preparation. The samples were processed with the Chromium™ Genome Reagent Kits v2 Rev B following the manufacturer’s instructions. Resulting libraries were assessed for quality with the Picogreen assay (Invitrogen, Waltham, MA, USA) to determine concentrations and subsequently underwent qualitatively checks for library size distributions on the Fragment Analyzer using the High Sensitivity NGS library assay (Agilent, Santa Clara, CA, USA).
All DNA libraries were sequenced to meet the goal of 30x or higher mean coverage, using paired 151 base pair reads on Illumina HiSeqX at the Broad Institute, and paired 150 base pair reads on Illumina Novaseq 6000 at NYGC. Sequencing data were received in FASTQ format from NYGC (n=49) and BCL files from Broad (n=56). The BCL files were converted to FASTQ using mkfastq (v2.20.0.422), a wrapper for Illumina’s bcl2fastq. The resulting FASTQ files were processed using the 10x Genomics Long Ranger analysis pipeline (v2.2.2). Long Ranger is a collection of analysis pipelines that processes the 10x Genomics sequencing output to align reads and call and phase SNVs, indels, and structural variants. For the analysis, the Genome Reference Consortium Human build 37 (GRCh37), as in the reference datafile provided by 10x Genomics (refdata-b37–2.1.0), was used. The alignment in Long Ranger uses Lariat, an aligner for barcoded linked reads using a Random Field Aligner (RFA) method.18 Lariat can map all reads for a single barcode simultaneously by using the prior knowledge that all the reads for a single barcode are from a small number of 10kb to 200kb DNA molecules. For variant discovery, we configured Long Ranger to use Genome Analysis Toolkit (GATK, v4.0.8.1) with default parameters. The final variants were annotated based on Ensembl canonical transcript using Variant Effect Predictor (VEP, release 94.5).19 Variants with a minimum read depth of 20x with quality score > 30 were used for further analysis and annotation. The high-resolution haplotyping of class I and II HLA regions was conducted using HLA-HD (version 1.3.0).20
Variant prioritization workflow to identify de novo variant candidates
We developed a workflow for discovering DNVs in trios and applied to LR-WGS datasets from patients with OMAS and their family members.21 Briefly, we restricted our analysis to protein-coding exons that were covered by at least 20 sequencing reads in all family members (trios or a quartet), ensuring sufficient coverage across the entire family unit. Then, we searched for loci where the proband was heterozygous and both parents were homozygous. We visually inspected the aligned reads for selected DNV candidates and validated them using Sanger sequencing. We selected very rare (i.e., population minor allele frequency < 0.001) or novel homozygous or compound heterozygous variants with protein-altering impacts by checking the pattern of inheritance in trios and a quartet. For patient-only data, we focused on very rare or novel homozygous variants with protein-altering impacts and very rare or novel heterozygous variants in the candidate genes discovered from trio and quartet analysis. We confirmed germline DNV candidates using Sanger sequencing (Genewiz, Cambridge, MA, USA).
Statistical Analysis
To determine the cellular location of protein products of candidate genes, as well as the cell and tissue types expressing these genes, we used the DAVID/EASE functional annotation system,22 Allen Brain Atlas,23 and GTEx portal.24 Statistical significance with 95% confidence intervals (CIs) was calculated using a Chi-squared test for categorical data. We performed all statistical analysis using the R programming language, version 4.1.2 (R Foundation for Statistical Computing).
Results
We enrolled 42 patients with OMAS, with clinical and demographic characteristics detailed in Table 1. On average, the age of onset was 28.4 months (standard deviation (SD) 22.7 months). Twenty-three patients (54.8%) were female, and the majority were Caucasian (n=28, 66.7%). Neuroblastoma was diagnosed in 23 cases (54.8%), with the majority (n=32, 76.2%) exhibiting a multiphasic OMAS clinical course. Brain MRI data were available for most patients, and five patients (12.8%) had cerebellar atrophy. WGS was performed for 105 participants, including 42 patients and 63 family members). Of these, 11 patients had no available parental samples and were analyzed as patient-only cases. In concordance with our previous report,10 sixteen patients (38.1%) had a positive family history of autoimmune disorders.
Table 1. Demographic and Clinical Characteristics of the Patients.
| Entire Cohort (n = 42) |
|
|---|---|
| Age of onset (months) | 28.4 ± 22.7 |
| Age at diagnosis (months) | 35.7 ± 29.9 |
| Sex (n female, % female) | 23 (54.8%) |
| Race/Ethnicity | |
| Caucasian African American Asian Hispanic (Caucasian) Native Hawaiian/Islander Not Recorded |
28 (66.7%) 1 (2.4%) 2 (4.8%) 3 (7.1%) 1 (2.4%) 7 (16.6%) |
| Tumor Identified | 23 (54.8%) |
| Clinical Course | |
| Monophasic Multiphasic (relapsing) |
10 (23.8%) 32 (76.8%) |
| Brain MRI available Atrophy present |
39 (92.8%) 5 (12.8%) |
| Family History of Autoimmunity | 16 (38.1%) |
De novo germline variants identified in probands through family-based analysis
The variant discovery workflow is depicted in Figure 1. For the loci where both parents had a reference concordant homozygous genotype, novel heterozygous variants with nonsynonymous effects in each proband were selected as previously described.21 We manually reviewed the aligned short reads at candidate DNV loci in the 31 probands eligible for DNV analysis (from 30 trio and 1 quartet) to refine the list for Sanger sequencing validation. On average, two candidate DNVs were identified per proband (range 1–6). Sanger sequencing subsequently confirmed a total of 12 DNVs in 9 of the 31 patients (29.0%), three of whome had two confirmed DNVs. Confirmed variants are listed in Table 2 (denoted as ‘de novo’ under ‘Variant type’), with corresponding chromatograms shown in Supplemental Figure 1. No gene with recurrent DNVs across probands was found. Several DNVs were identified in genes highly expressed in the brain, such as ADRA2C, CACNA2D2, FAM81B, KAT6B, SIRT5, ZFP69, NRP1, and ZBTB16. However, the clinical phenotypes associated with these genes are either unreported or linked to autosomal recessive inheritance.
Figure 1. Overview of the Variant Prioritization Pipeline.
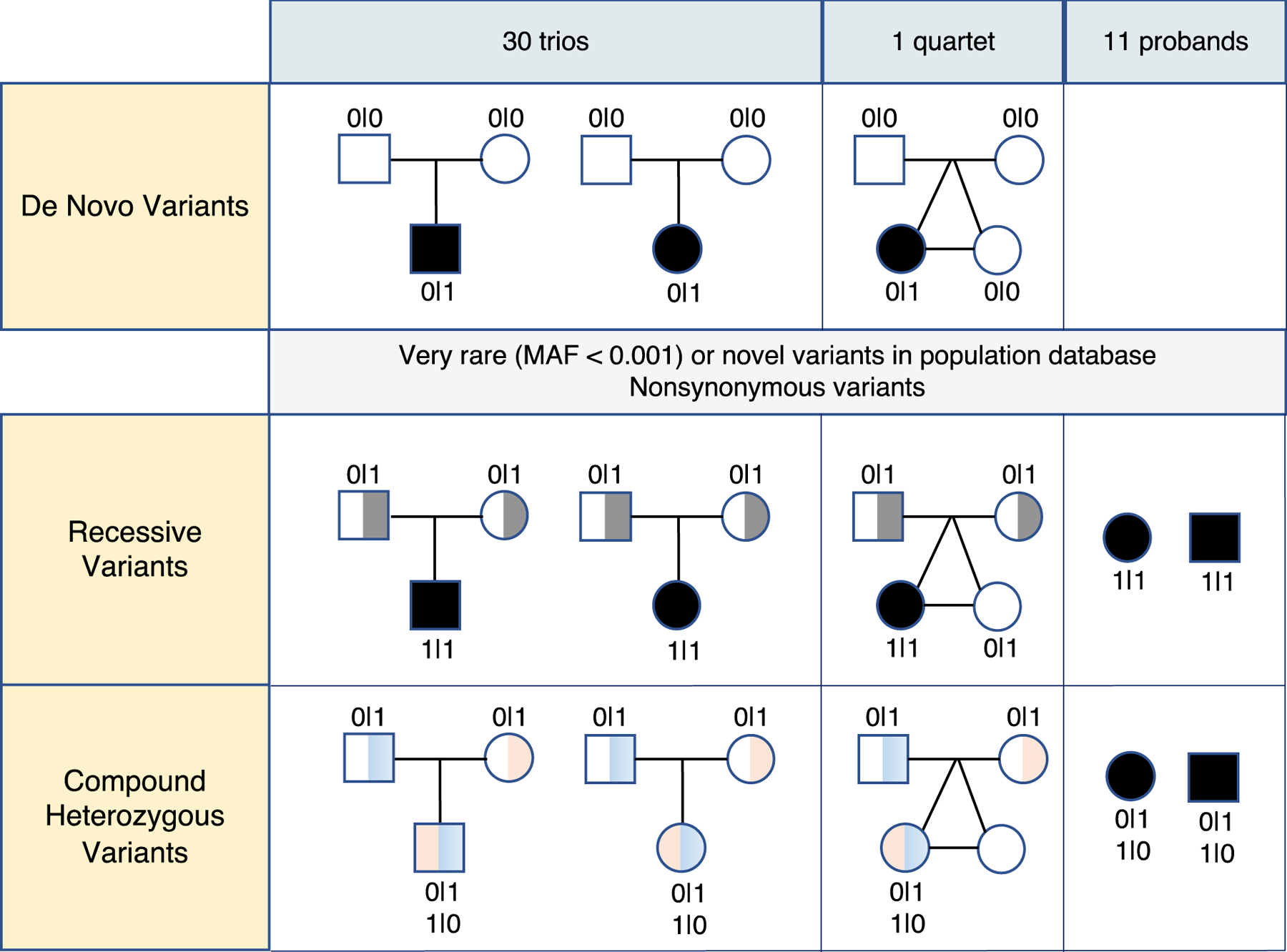
This figure depicts the workflows for identifying disease-associated variants in 30 trios, 1 quartet, and 11 unrelated patients. Pedigree charts represent the inheritance patterns, with squares for males, circles for females, and filled symbols indicating affected individuals. Genotypes are indicated as “0|0” for homozygous reference, “0|1” for heterozygous, and “1|1” for homozygous variant. De novo germline variants are identified by examining loci where both parents have “0|0”, and the proband has “0|1” in the trios. For the quartet with monozygotic twins, de novo variants are selected only if they are observed exclusively in the affected twin (top panel). Pedigree charts also show affected individuals with homozygous variants (“1|1”) inherited from carrier parents (“0|1”). These variants are very rare (MAF < 0.001) or novel in the population database and have nonsynonymous effects with reported disease-associated phenotypes in ClinVar (middle panel). Additionally, pedigree charts illustrate cases where the proband has two different heterozygous variants (“0|1”) inherited from each parent, resulting in a compound heterozygous genotype (“1|0”, “0|1”). These variants are also very rare or novel and have nonsynonymous effects with reported disease associations in ClinVar. Inheritance from each parent is confirmed with phased genome sequencing intervals (bottom panel). MAF: Minor Allele Frequency.
Table 2. Putative Disease-associated Variants in the Patients with OMAS.
| Patient ID | Gene (HGNC ID) | Variant Description (HGVS) | Variant type | Variant impact | PAF | Sex | Neuroblastoma | Family history of autoimmune disorders |
|---|---|---|---|---|---|---|---|---|
| OMAS-001 | R3HDM4 (28270) | c.163G>A (p.Val55Met) | de novo | missense | N.R. | M | Yes | No |
|
| ||||||||
| OMAS-002 | CACNA2D2 (1400) | c.2847del (p.Gly950AlafsTer66) | de novo | frameshift | N.R. | F | Yes | No |
|
| ||||||||
| OMAS-003 | USP28 (12625) | c.1683T>C (p.Thr561=) | de novo | synonymous | N.R. |
M | No | No |
| CTAGE9 (37275) | c.58G>C (p.Gly20Arg) | homozygous | missense | 0.0002037 | ||||
|
| ||||||||
| OMAS-004 | SSPOP (21998) | c.7511C>T (p.Ala2504Val) | compound heterozygous: paternal | missense | 0.00003225 | M | No | Yes |
| c.1210C>T (p.Gln404Ter) | compound heterozygous: maternal | stop gained | 0.000006301 | |||||
|
| ||||||||
| OMAS-005 | TUBA3E (20765) | c.132C>T (p.Gly44=) | de novo | synonymous | 0.00003539 | F | Yes | Yes |
| ZFP69 (24708) | c.1222G>C (p.Glu408Gln) | de novo | missense | N.R. | ||||
|
| ||||||||
| OMAS-008 | NRP1 (8004) | c.1084G>A (p.Val362Ile) | de novo | missense | N.R. |
F | No | No |
| ZBTB16 (12930) | c.1772C>T (p.Thr591Met) | de novo | missense | N.R. | ||||
|
| ||||||||
| OMAS-012 | ASAH2B (23456) | c.1592A>C (p.Asn531Thr) | homozygous | missense | 0 | F | No | No |
| LGALS9B (24842) | c.454A>G | homozygous | missense | 0 | ||||
|
| ||||||||
| OMAS-014 | DNAH1 (2940) | c.5794C>T (p.Arg1932Trp) | compound heterozygous: paternal | missense | 0.00001605 | M | No | No |
| c.12131A>T (p.Asp4044Val) | compound heterozygous: maternal | missense | 0.00001274 | |||||
|
| ||||||||
| OMAS-016 | CLDN34 (51259) | c.17A>G (p.Asn6Ser) | homozygous | missense | N.R. | M | Yes | No |
|
| ||||||||
| OMAS-021 | KAT6B (17582) | c.4723T>A (p.Tyr1575Asn) | de novo | missense | N.R. |
M | Yes | No |
| SIRT5 (14933) | c.637G>A (p.Gly213Arg) | de novo | missense | 0.00006014 | ||||
|
| ||||||||
| OMAS-023 | MAPKBP1 (29536) | c.3841C>T (p.Arg1281Ter) | de novo | stop gained | 0.000007073 | M | No | Yes |
|
| ||||||||
| OMAS-024 | ATP10A (13542) | c.1996G>A (p.Ala666Thr) | compound heterozygous: paternal | missense | 0.00005767 | M | Yes | Yes |
| c.3064C>T (p.Leu1022Phe) | compound heterozygous: maternal | missense | N.R. | |||||
|
| ||||||||
| OMAS-026 | IRS4 (6128) | c.2422T>C (p.Tyr808His) | homozygous | missense | N.R. | M | Yes | No |
|
| ||||||||
| OMAS-030 | ADRA2C (283) | c.1190C>A (p.Pro397His) | de novo | missense | N.R. | F | Yes | No |
|
|
|
|||||||
| OMAS-031 | PALD1 (23530) | c.89G>C (p.Arg30Pro) | compound heterozygous: paternal | missense | N.R. |
F | No | No |
| c.2241C>G (p.Ile747Met) | compound heterozygous: maternal | missense | 0.000004025 | |||||
|
| ||||||||
| NEU-0347 | RHOXF2B (33519) | c.277A>G (p.Asn93Asp) | homozygous | missense | N.R. | F | No | Yes |
|
| ||||||||
| NEU-0694 | FAM81B (26335) | c.755T>C (p.Leu252Pro) | de novo | missense | N.R. |
F | Yes | Yes |
| XIRP2 (14303) | c.7188T>G (p.Tyr2396Ter) | compound heterozygous: paternal | stop gained | 0.00004635 | ||||
| c.263G>A (p.Arg88Lys) | compound heterozygous: maternal | missense | N.R. | |||||
HGNC: HUGO (Human Genome Organization) Gene Nomenclature Committee
PAF: Population Allele Frequency
N.R.: Not Reported
Among the NB positive and NB negative groups, which included 16 and 15 patients respectively, confirmed DNVs were identified in 6 NB-positive and 3 NB-negative patients. No significant difference in DNV detection rates was observed between the two groups (Fisher’s exact test, p = 0.43).
Rare homozygous and compound heterozygous variants
We next examined autosomal recessive loci containing very rare or novel nonsynonymous variants. To confirm that the variants were truly homozygous, we manually inspected the sequence read alignments at each candidate site. We excluded any locus where even a few reads suggested the presence of a heterozygous allele in the proband. After this filtering, homozygous variants were identified in six genes—ASAH2B, CLDN34, CTAGE9, RHOXF2B, IRS4, and LGALS9B —each found in a different patient (Table 2, labeled as ‘homozygous’ under ‘Variant type’).
We further investigated genes harboring compound heterozygous variants using family-based data, taking the advantage of the long-range phasing information enabled by LR-WGS, which is not readily available in short-read sequencing. Five genes—ATP10A, DNAH1, PALD1, SSPOP, and XIRP2—were identified as carrying very rare or novel compound heterozygous variants predicted to alter protein function (Table 2, annotated as ‘compound heterozygous’ in the ‘Variant type’ column; Supplemental Figure 2). Notably, SSPOP and XIRP2 each harbored a very rare nonsense variant, suggesting the potential loss-of-protein-function mechanism in these genes.
Rare heterozygous variants in dominant genes
In our prior research, an increased prevalence of autoimmune diseases was observed among first-degree relatives of OMAS patients. Given the pleiotropic nature of many disease-associated genes, we analyzed rare heterozygous variants in genes reported to follow an autosomal dominant inheritance pattern for any phenotype in the ClinGen and OMIM databases. In seven patients, without confirmed de novo or compound heterozygous variants, we identified rare nonsynonymous variants in seven dominant or X-linked genes: ARX, GDF3, HRAS, KCNH2, PCDH19, PRRT2, and TTR. The associated clinical phenotypes are summarized in Table 3.
Table 3. Rare nonsynonymous variants in dominant genes.
| Patient ID | Gene | Variant Description (HGVS) | Variant type | Variant impact | PAF | Sex | Neuroblastoma | Family history of autoimmune disorders | Reported phenotype | MIM ID | Mode of inheritance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NEU-0233 | HRAS | c.179G>T (p.Gly60Val) | heterozygous | missense | N.R. | Female | Yes | Yes | Costello syndrome | # 218040 | AD |
| NEU-0295 | PRRT2 | c.649dup (p.Arg217fs) | homozygous | frameshift | 0.003741 | Female | Yes | Unknown | Paroxysmal nonkinesigenic dyskinesia | # 128200 | AD |
| NEU-0667 | ARX | c.1471dup (p.Leu491fs) | hemizygous | frameshift | N.R. | Male | No | Unknown | lissencephaly, X-linked 2 | # 300215 | XL |
| OMAS-006 | PCDH19 | c.1091dup (p.Tyr366fs) | hemizygous | frameshift | N.R. | Male | No | No | Developmental and epileptic encephalopathy, 9 | # 300088 | XL |
| OMAS-013 | KCNH2 | c.2775dup (p.Pro926fs) | heterozygous | frameshift | 0.000001958 | Female | No | Yes | Long QT syndrome 2 | # 613688 | AD |
| OMAS-022 | TTR | c.148G>A (p.Val50Met) | heterozygous | missense | 0.000057 | Female | Yes | No | Familial amyloid neuropathy | # 105210 | AD |
| OMAS-028 | GDF3 | c.796C>T (p.Arg266Cys) | heterozygous | missense | 0.002507 | Female | No | Yes | Klippel-Feil syndrome 3 | # 613702 | AD |
AD: Autosomal Dominant
XL: X-Linked
Genetic modifiers and mimics of OMAS
The protein-truncating DNV in the CACNA2D2 gene (p.Gly950AlafsTer66, Figure 2A) was notable due to its high expression level in the cerebellum and the reported association of ataxia with recessive mutations in this gene. Clinically, the patient harboring the DNV in CACNA2D2 exhibited an atypical course of OMAS, including very severe cerebellar atrophy, evident on brain MRI conducted 14.3 years after OMAS onset (Figure 2B), as detailed in the Supplemental Text.
Figure 2. Truncating de novo germline variant in the CACNA2D2 gene.
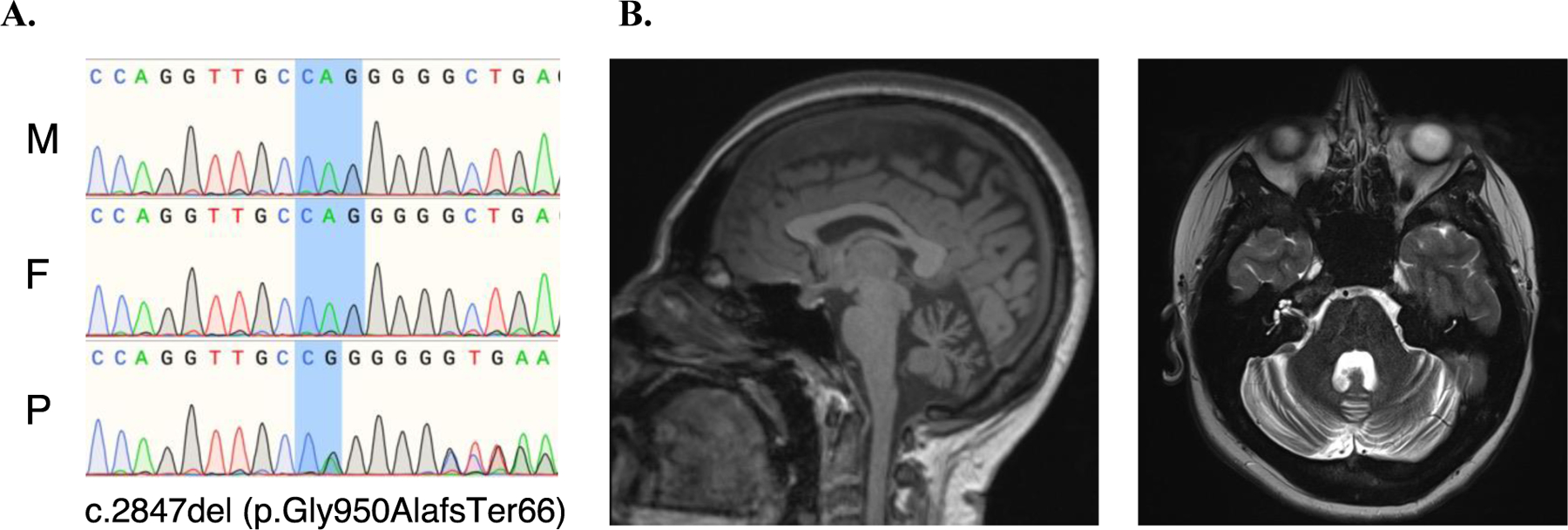
A. Sanger sequencing chromatograms showing the ENST00000479551.1:c.2874del (p.Gly950AlafsTer66) de novo variant in the affected individual. The chromatograms illustrate the genetic sequence of the mother (M), father (F), and proband (P). B. Structural brain magnetic resonance imaging of the patient with the de novo germline variant in the CACNA2D2 gene. T1-weighted sagittal (left panel) and T2-weighted axial (right panel) views show severe cerebellar atrophy.
Upon identifying this case, we evaluated all patients with clinically-obtained brain MRI (n=39) for cerebellar atrophy by reviewing their last available MRI reports. Five (12.8%) had evidence of cerebellar atrophy with median OMAS duration of 14.7 years (range 0.7 – 21.6). The rate of genetic variant detection in patients with and without cerebellar atrophy (4/5, 80% vs. 23/34, 67.6%; Fisher’s exact test, p=1.00) did not differ but was limited by small sample size. The four patients with cerebellar atrophy had DNVs in the CACNA2D2 and TUBA3E genes, a homozygous variant in the CLDN34 gene, and compound heterozygous variants in the XIRP2 gene.
Furthermore, during the study period, a 3-year-old patient previously diagnosed with OMAS at an outside institution was evaluated for a second opinion. Given the early onset of abnormal eye movements at 2 months and subsequent development of epilepsy, a monogenic mimic of OMAS was suspected. This was clinically confirmed as glucose transporter deficiency, attributed to a mutation in the SLC2A1 gene. (See Supplemental Text for additional details).
High HLA-DRB1*01 and HLA-DOB*01:01 frequency in OMAS
Autoimmune diseases, including neuroimmune phenotypes, exhibit a pronounced genetic predisposition linked to the HLA region. We performed high-resolution haplotyping of both class I and II HLA loci using phased genomic intervals derived from LR-WGS. The HLA-DRB1*01 haplotype was observed in 27 of 84 (32.1%) alleles in our cohort (Supplemental Table 1). This frequency is comparable to a previous report in patients with OMAS (30/86, 35%; Chi-square test p=0.69), and appears elevated relative to the control group in the same study (21/200, 10.5%; Chi-square test, p <0.0001)7. However, direct comparisons should be interpreted with caution due to differences in HLA typing methods (WGS-based in our study vs. PCR-based in the prior report), as well as the absence of an ancestry-matched control group and the potential influence of population stratification. Notably, our cohort included approximately 67% individuals of European ancestry, in whom the reported general population frequency of HLA-DRB101 is ~9.1% and lower in non-European populations (2–6%).25 The HLA-DOB*01:01 haplotype was present in 72 of 84 alleles (85.7%) in our cohort, with no significant difference between NB-positive and NB-negative subgroups. This frequency was higher than previously reported in OMAS patients (63%) and neuroblastoma patients without OMAS (36%) using RNA-sequencing–derived HLA calls.8
Discussion
In this first application of WGS to study potential genetic contributions to OMAS, we found putative disease-associated variants in 17 patients in different genes including 12 confirmed DNVs in protein-coding regions in nine patients (29%, out of 30 trios and 1 quartet). However, we did not identify any recurrent DNVs in the same gene, suggesting that there is not a monogenic cause for OMAS. The monozygotic twin pair which was discordant for OMAS further highlights this point. In addition, we did not identify any clear cases of true monogenic mimics of OMAS as clinical phenotypes associated with mutations in the genes with DNVs have either not been described or are associated with autosomal recessive inheritance. Therefore, we do not recommend routine clinical application of whole-exome sequencing (WES) or WGS in patients with typical presentations of OMAS.
However, over the same study period in our neuro-immunology clinic, we clinically identified one patient with a prior misdiagnosis of OMAS at an outside institution who actually had glucose transporter deficiency due to a DNV in the SLC2A1 gene encoding the glucose transporter 1 (GLUT1). Glucose transporter deficiency is known to cause abnormal eye movements, including opsoclonus or opsoclonus-like movements. One previously published patient with glucose transporter deficiency with onset of opsoclonus at 1.5 months old had improvement with initiation of high dose oral corticosteroids at 6 months old and worsening with weaning, prompting consideration of OMAS and diagnostic workup for NB13. These two cases highlight the need to consider glucose transporter deficiency in the differential diagnosis of OMAS. In both cases, the onset of opsoclonus occurred at a very early age (1.5 and 2 months respectively) and both patients had epilepsy, which are diagnostic red flags for OMAS, which has an average age of onset of 18 to 24 months and very rarely occurs before 6 months of age. Combined with other reported monogenic mimics of OMAS in the literature including the KCTD7 12 and SGCE 14 genes, we propose that the following features should prompt the clinician to consider sending WES or WGS in patients with possible OMAS: age of onset less than 6 months old, epilepsy, pre-existing developmental delay, and/or consanguinity.
Although we did not identify any other definitive monogenic mimics of OMAS, the protein-truncating DNV in the CACNA2D2 gene was notable given its high level of expression in the cerebellum and its association with ataxia and cerebellar atrophy with recessive mutations in CACNA2D226. Although cerebellar atrophy can occur in a subset of patients with OMAS, it is typically mild, whereas our patient with the DNV in CACNA2D2 had very severe cerebellar atrophy. It is not clear why only some patients with OMAS develop cerebellar atrophy. In our cohort, the rate of genetic variant detection in patients with (n=5) and without (n=34) cerebellar atrophy did not differ but comparison was limited by small sample size. Notably, of the four patients with cerebellar atrophy, two had variants in genes which are highly expressed in the cerebellum (CACNA2D2 and CLDN3427) and a third had a variant in a gene associated with structural brain malformation (TUBA3E28). Based on these findings, we speculate that the genetic background of cerebellar and brain structural genes may influence the risk of cerebellar atrophy in OMAS. Further research into whether there are functional effects of heterozygous protein-truncating DNV in CACNA2D2 may be warranted.
The de novo missense variant p.Pro397His in the ADRA2C gene is also noteworthy, as it is absent in the unaffected twin sibling. ADRA2C encodes Adrenoceptor Alpha 2C, one of three adrenoreceptor genes in humans, which is highly expressed in brain cells. Intriguingly, ADRA2C gene expression levels in NB tumor tissues have been significantly correlated with the severity of OMAS symptoms.8 Our patient with the de novo missense variant in ADRA2C was also diagnosed with NB and exhibited notably severe clinical manifestations being nonambulatory for five years from OMAS onset despite complete tumor resection, early multiagent immunotherapy including rituximab, and additional immunotherapies including cyclophosphamide, mycophenolate mofetil, and tacrolimus. AlphaMissense29 predicts this missense variant to be likely pathogenic, assigning it a high pathogenicity score of 0.99. Thus, further functional characterization is warranted to evaluate the precise contribution of this de novo variant in ADRA2C to OMAS.
Finally, we confirm two prior studies suggesting that the allele frequency of HLA-DRB1*01 may be higher in OMAS than the general population, although we are limited by the lack of a control population in our study. HLA-DRB1*01 has been implicated in other autoimmune disorders including rheumatoid arthritis30, juvenile idiopathic arthritis31, and Henoch-Schönlein purpura32. We also confirm a high allele frequency of HLA-DOB*01:018 in OMAS and extend this finding to include those without NB. Prior studies have also shown HLA associations in other paraneoplastic neurological disorders including anti-LGI1 encephalitis33. Further research should assess the allele frequency of HLA-DRB1*01 and HLA-DOB*01:01 in patients with OMAS both with and without NB compared to healthy and disease controls to determine if it is a genetic risk factor for OMAS.
Our study had some limitations. First, the cohort size was relatively small, consistent with the rarity of OMAS. As such, the limited sample size reduces statistical power to detect rare or modest-effect genetic associations and increases the likelihood of Type II error. Therefore, negative findings should be interpreted with caution. Additionally, we included only patients who met strict diagnostic criteria for definite OMAS as determined by pediatric neuroimmunology specialists. We did not include individuals for whom OMAS was considered a less likely diagnosis. These factors may have limited our ability to detect very rare monogenic mimics or phenotypic variants of OMAS. Future studies incorporating broader inclusion criteria and larger, multi-institutional cohorts may help identify additional monogenic contributors and improve sensitivity to genetically heterogeneous mechanisms. Second, we did not have a control population to determine whether the rate of DNVs or the allele frequencies of HLA-DRB1*01 and HLA-DOB*01:01 in our cohort was higher than expected. However, the comparable rate of the allele frequency of HLA-DRB1*01 in our cohort compared with one prior published cohort with OMAS (which was higher than the controls in the study) support further investigation into the role of HLA-DRB1*01 in OMAS. Lastly, we did not perform in vivo studies to determine whether the DNVs in our patients may have directly contributed to their phenotypes. Of the DNVs we identified, the DNV in CACNA2D2 was the most intriguing and may warrant further research. Lastly, we did not perform in vivo or in vitro studies to assess the functional consequences of the DNVs identified in our cohort. As such, the relevance of these variants to OMAS pathogenesis remains uncertain. Of the DNVs we identified, a truncating variant in CACNA2D2 was particularly notable due to its expression in the cerebellum and its role in calcium signaling. Although speculative at this stage, this variant may merit further investigation using disease-relevant cellular or animal models. Future studies incorporating functional validation will be critical to establish biological causality and mechanistic insight.
Strengths of our study include a carefully diagnosed cohort with OMAS and utilization of third-generation linked-reads sequencing technique that significantly improved coverage of the human reference genome, detection of large structural variations (> 30kbps), and reconstruction of haplotypes (phasing)16. As a result, this approach not only facilitated the identification of compound heterozygous variants but also enabled precise high-resolution HLA typing20.
In summary, we have conducted the first genome sequencing study to date on patients with OMAS, which represents a significant step forward in our understanding of the potential genetic basis of OMAS. Based on the current study, combined with our prior study showing higher rates of autoimmune disorders in parents of children with OMAS compared to controls10, we propose that the pathogenesis of OMAS likely involves polygenic predisposition, possibly including HLA-DRB1*01 and HLA-DOB*01:01, combined with as yet unidentified environmental risk factors. Confirming the role of HLA-DRB1*01 and HLA-DOB*01:01 is an important next step. Determining whether there are other genetic risk factors will likely require genome wide association studies (GWAS) which may be challenging given the typically small relative risk contributions of genes identified in GWAS, the large samples sizes required, and the rarity of OMAS. Global, collaborative research efforts as we are undertaking may help in further OMAS genetic research34.
Supplementary Material
Funding Statement
This study was supported by funding from Pfizer, Inc. and National Institute of Health (NINDS R01NS129188 and NCATS U01TR002623). Authors acknowledge material and data support from the PrecisionLink Biobank for Health Discovery at Boston Children’s Hospital and the Repository Core for Neurological Disorders within the Rosamund Stone Zander Translational Neuroscience Center at Boston Children’s Hospital, which is also supported by the IDDRC (NIH P50HD105351).
Footnotes
Ethics Declaration
The study was conducted in accordance with the declaration of Helsinki 1975 for studies involving human participants. The study was approved and reviewed by the Institutional Review Board of Boston Children’s Hospital (IRB-P00027605, IRB-P00041986). Written informed consents were obtained from all participants.
Conflicts of Interest
The authors declare no conflicts of interest in association with the present study.
Data Availability
The data supporting the current study have not been deposited in a public repository because of concerns regarding patient privacy but are available for collaborative sharing upon reasonable request to the corresponding authors.
REFERENCES
- 1.Gorman MP. Update on diagnosis, treatment, and prognosis in opsoclonus-myoclonus-ataxia syndrome. Curr Opin Pediatr 2010;22:745–750. [DOI] [PubMed] [Google Scholar]
- 2.Gambini C, Conte M, Bernini G, et al. Neuroblastic tumors associated with opsoclonus-myoclonus syndrome: histological, immunohistochemical and molecular features of 15 Italian cases. Virchows Arch 2003;442:555–562. [DOI] [PubMed] [Google Scholar]
- 3.Torres-Vega E, Duran-Moreno M, Sanchez Del Pino M, et al. Immunoproteomic studies on paediatric opsoclonus-myoclonus associated with neuroblastoma. J Neuroimmunol 2016;297:98–102. [DOI] [PubMed] [Google Scholar]
- 4.Graus F, Dalmau J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors. Nat Rev Clin Oncol 2019;16:535–548. [DOI] [PubMed] [Google Scholar]
- 5.Anand G, Bridge H, Rackstraw P, et al. Cerebellar and cortical abnormalities in paediatric opsoclonus-myoclonus syndrome. Dev Med Child Neurol 2015;57:265–272. [DOI] [PubMed] [Google Scholar]
- 6.Rossor T, Yeh EA, Khakoo Y, et al. Diagnosis and Management of Opsoclonus-Myoclonus-Ataxia Syndrome in Children: An International Perspective. Neurol Neuroimmunol Neuroinflamm 2022;9. [Google Scholar]
- 7.Hero B, Radojska S, Gathol B. Opsomyoclonus syndrome in infancy with or without neuroblastoma is associated with HLA DRB1*01. 37th Annual Conference of the Society for Industrial and Organizational Psychology 2022. Seattle, USA2022: 480. [Google Scholar]
- 8.Rosenberg MI, Greenstein E, Buchkovich M, et al. Polyclonal lymphoid expansion drives paraneoplastic autoimmunity in neuroblastoma. Cell Rep 2023;42:112879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krasenbrink I, Fuhlhuber V, Juhasz-Boess I, et al. Increased prevalence of autoimmune disorders and autoantibodies in parents of children with opsoclonus-myoclonus syndrome (OMS). Neuropediatrics 2007;38:114–116. [DOI] [PubMed] [Google Scholar]
- 10.Santoro JD, Kerr LM, Codden R, et al. Increased Prevalence of Familial Autoimmune Disease in Children With Opsoclonus-Myoclonus Syndrome. Neurol Neuroimmunol Neuroinflamm 2021;8. [Google Scholar]
- 11.Rossi M, van der Veen S, Merello M, Tijssen MAJ, van de Warrenburg B. Myoclonus-Ataxia Syndromes: A Diagnostic Approach. Mov Disord Clin Pract 2021;8:9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blumkin L, Kivity S, Lev D, et al. A compound heterozygous missense mutation and a large deletion in the KCTD7 gene presenting as an opsoclonus-myoclonus ataxia-like syndrome. J Neurol 2012;259:2590–2598. [DOI] [PubMed] [Google Scholar]
- 13.Appavu B, Mangum T, Obeid M. Glucose Transporter 1 Deficiency: A Treatable Cause of Opsoclonus and Epileptic Myoclonus. Pediatr Neurol 2015;53:364–366. [DOI] [PubMed] [Google Scholar]
- 14.Drivenes B, Born AP, Ek J, Dunoe M, Uldall PV. A child with myoclonus-dystonia (DYT11) misdiagnosed as atypical opsoclonus myoclonus syndrome. Eur J Paediatr Neurol 2015;19:719–721. [DOI] [PubMed] [Google Scholar]
- 15.Alburaiky S, Dale RC, Crow YJ, et al. Opsoclonus-myoclonus in Aicardi-Goutieres syndrome. Dev Med Child Neurol 2021;63:1483–1486. [DOI] [PubMed] [Google Scholar]
- 16.Marks P, Garcia S, Barrio AM, et al. Resolving the full spectrum of human genome variation using Linked-Reads. Genome Res 2019;29:635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matthay KK, Blaes F, Hero B, et al. Opsoclonus myoclonus syndrome in neuroblastoma a report from a workshop on the dancing eyes syndrome at the advances in neuroblastoma meeting in Genoa, Italy, 2004. Cancer Lett 2005;228:275–282. [DOI] [PubMed] [Google Scholar]
- 18.Bishara A, Liu Y, Weng Z, et al. Read clouds uncover variation in complex regions of the human genome. Genome Res 2015;25:1570–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawaguchi S, Matsuda F. High-Definition Genomic Analysis of HLA Genes Via Comprehensive HLA Allele Genotyping. Methods Mol Biol 2020;2131:31–38. [DOI] [PubMed] [Google Scholar]
- 21.Kong SW, Lee IH, Liu X, Hirschhorn JN, Mandl KD. Measuring coverage and accuracy of whole-exome sequencing in clinical context. Genet Med 2018;20:1617–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dennis G Jr., Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4:P3. [PubMed] [Google Scholar]
- 23.Miller JA, Guillozet-Bongaarts A, Gibbons LE, et al. Neuropathological and transcriptomic characteristics of the aged brain. Elife 2017;6. [Google Scholar]
- 24.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurley CK, Kempenich J, Wadsworth K, et al. Common, intermediate and well-documented HLA alleles in world populations: CIWD version 3.0.0. HLA 2020;95:516–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Punetha J, Karaca E, Gezdirici A, et al. Biallelic CACNA2D2 variants in epileptic encephalopathy and cerebellar atrophy. Ann Clin Transl Neurol 2019;6:1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackowski AP, Rando K, Maria de Araujo C, Del Cole CG, Silva I, Tavares de Lacerda AL. Brain abnormalities in Williams syndrome: a review of structural and functional magnetic resonance imaging findings. Eur J Paediatr Neurol 2009;13:305–316. [DOI] [PubMed] [Google Scholar]
- 28.Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep 2015;10:148–161. [DOI] [PubMed] [Google Scholar]
- 29.Cheng J, Novati G, Pan J, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023;381:eadg7492. [DOI] [PubMed] [Google Scholar]
- 30.Karami J, Aslani S, Jamshidi A, Garshasbi M, Mahmoudi M. Genetic implications in the pathogenesis of rheumatoid arthritis; an updated review. Gene 2019;702:8–16. [DOI] [PubMed] [Google Scholar]
- 31.De Silvestri A, Capittini C, Poddighe D, et al. HLA-DRB1 alleles and juvenile idiopathic arthritis: Diagnostic clues emerging from a meta-analysis. Autoimmun Rev 2017;16:1230–1236. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez-Gay MA, Lopez-Mejias R, Pina T, Blanco R, Castaneda S. IgA Vasculitis: Genetics and Clinical and Therapeutic Management. Curr Rheumatol Rep 2018;20:24. [DOI] [PubMed] [Google Scholar]
- 33.Muniz-Castrillo S, Honnorat J. Genetic predisposition to autoimmune encephalitis and paraneoplastic neurological syndromes. Curr Opin Neurol 2024;37:329–337. [DOI] [PubMed] [Google Scholar]
- 34.Kerr LM, Ryan ME, Lim M, et al. An International Pediatric-Onset Opsoclonus-Myoclonus Ataxia Syndrome Registry and Clinical Research Network: Development, Progress, and Vision. Pediatr Neurol 2023;148:145–147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the current study have not been deposited in a public repository because of concerns regarding patient privacy but are available for collaborative sharing upon reasonable request to the corresponding authors.