

Abstract
A classical peptide inhibitor of serine proteases that is hydrolyzed ≈107 times more slowly than a good substrate is shown to form an acyl–enzyme intermediate rapidly. Despite this quick first step, further reaction is slowed dramatically because of tight and oriented binding of the cleaved peptide, preventing acyl–enzyme hydrolysis and favoring the reverse reaction. Moreover, this mechanism appears to be common to a large class of tight-binding serine protease inhibitors that mimic good substrates. The arrest of enzymatic reaction at the intermediate stage allowed us to determine that the consensus nucleophilic attack angle is close to 90° in the reactive Michaelis complexes.
The protein inhibitors of serine-type proteases pose a classic biological puzzle that confronts the key phenomenon of enzyme specificity. These inhibitors, reviewed by Laskowski and others (1–4), feature peptide sequences that bind in a substrate-like manner to specific proteases, and, based on sequence, would be expected to be rapidly proteolyzed (Table 1). However, the inhibitors are bound more tightly than good substrates of these enzymes (with association constants up to 1014 M−1), yet are hydrolyzed more slowly by factors of 106-1010. The inhibitors comprise at least 18 convergently evolved families (2) that display a strikingly similar conformation of the peptide backbone surrounding the reactive site, despite an absence of similarity in sequence or topology (1, 3, 5–8) (Fig. 1).
Table 1.
Representative examples of protease inhibitors
| Inhibitor | Family | Reactive site sequence | KD, M | Ref. |
|---|---|---|---|---|
| CI2 | Potato I | IVTM↓EYRI | 2 × 10−12/subtilisin | 12 |
| BPTI | Kunitz-BPTI | GPCK↓ARII | 6 × 10−14/trypsin | 41 |
| STI | Kunitz-STI | PSYR↓IRFI | 1 × 10−11/trypsin | 42 |
| OMTKY3 | Kazal | ACTL↓EYRP | 3 × 10−12/chymotrypsin | 43 |
| PSTI | Kazal | GCPR↓IYNP | 3 × 10−11/trypsin | 41 |
| BBI | Bowman–Birk | ACTK↓SNPP | 8 × 10−10/trypsin | 44 |
| BBI | Bowman–Birk | ICAL↓SYPA | 6 × 10−10/chymotrypsin | 44 |
| SSI | SSI | MCPM↓VYDP | 2 × 10−11/subtilisin | 18 |
| CMTI I | Squash seed | VCPR↓ILEM | 2 × 10−12/trypsin | 45 |
| SGTI | Grasshopper | ACTR↓KGCP | 3 × 10−12/trypsin | 46 |
| SFTI-1 | Sunflower | RCTK↓SIPP | 1 × 10−10/trypsin | 47 |
Amino acid sequences flanking the cleavage site (indicated by the arrow) are shown; in each case, the amino acid preceding the scissile bond (the major specificity residue) is typical of a good substrate for the respective enzyme. BPTI, bovine pancreatic trypsin inhibitor; STI, soybean trypsin inhibitor; OMTKY3, turkey ovomucoid third domain; PSTI, pancreatic secretory trypsin inhibitor; BBI, Bowman–Birk inhibitor; SSI, Streptomyces subtilisin inhibitor; CMTI I, Cucurbita maxima trypsin inhibitor I; SGTI, Schistocerca gregaria trypsin inhibitor; SFTI-1, sunflower trypsin inhibitor.
Fig 1.

Superposition of reactive site loop backbones of 23 different protease inhibitors in complex with proteases. Structures are from 12 different inhibitor families unrelated in sequence or fold, in complex with 9 serine proteases including members of both chymotrypsin and subtilisin families. The white arrow indicates the cleavage site. Superpositioning was based on all of the backbone atoms of the 6 residues shown for each inhibitor.
The mechanism by which serine proteases cleave peptides is outlined in Eqs. 1–3:
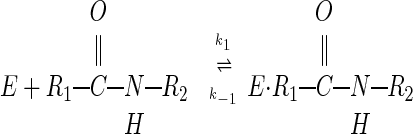 |
 |
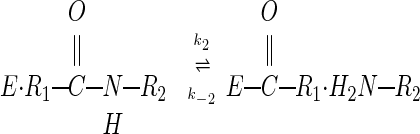 |
 |
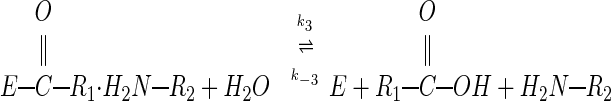 |
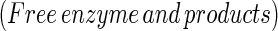 |
Reasons postulated for the inhibitors' surprising lack of reactivity include (i) that the extreme rigidity of the complex prevents productive nucleophilic attack (9–15), (ii) that poor orientation of the reacting groups results in a nonproductive complex (16–18), and (iii) that positioning of the leaving group H2N-R2 in the acyl–enzyme complex favors the back reaction toward the Michaelis complex (12, 14, 19). To clarify this fundamental anomaly of enzyme catalysis, we initiated studies involving incubation of the classical serine protease subtilisin (EC 3.4.21.62) with a classical inhibitor, chymotrypsin inhibitor 2 (CI2).
Materials and Methods
Subtilisin.
Recombinant subtilisin BPN′ (20), containing an added C-terminal 6-His tag, was expressed in protease-deficient Bacillus subtilis strain BG2036 (21) as described (22). The enzyme was recovered from the media by ethanol precipitation (22), purified by Ni-nitrilotriacetic acid (NTA) agarose affinity chromatography (Qiagen), dialyzed into 10 mM NH4OAc (pH 5.8), lyophilized, and stored at −80°C until use. Mutant subtilisin BPN′ with Asn155Leu and Met222Ala substitutions was similarly prepared.
CI2.
CI2 is an 83-aa protein originally identified in Hiproly barley; we studied a 63-aa recombinant protein with truncation of the first 19 (disordered) amino acids and replacement of Leu-20 with a new starting Met; this protein sequence has been shown to retain the complete structure and function of full-length CI2 (23). Throughout the text, we use the amino acid numbering of the original full-length protein sequence. A synthetic gene encoding the truncated version of CI2, with codon usage optimized for Escherichia coli, was assembled from 6 oligonucleotides by PCR, ligated into the NdeI/BamHI sites of expression vector pET27b(+) (Novagen), and sequenced. CI2 was expressed in E. coli strain BL21(DE3) (Stratagene), recovered from the periplasm by osmotic shock, and purified by SP Sepharose ion exchange chromatography (Amersham Pharmacia). Design of both the expression construct and the purification were based on strategies previously used with eglin, a CI2 homolog (24). CI2 was subsequently dialyzed into 10 mM NH4OAc (pH 5.8), lyophilized, and stored at −80°C until use.
SDS/PAGE, Protein Elution, and Mass Spectrometry.
Time courses involving incubation of subtilisin with CI2 were carried out in 100 mM Tris, pH 8.6, at 25°C, unless otherwise specified. For 0-min time points, enzyme was prequenched with acid before mixing with CI2, whereas for subsequent time points, active enzyme and CI2 were mixed and incubated, then aliquots of the mixture were withdrawn and quenched at defined intervals. All gel samples were acidified to pH 1 to inactivate subtilisin before denaturation by heating in loading buffer. SDS gels (15% acrylamide) were run under standard conditions by using a Bio-Rad Minigel apparatus, and Coomassie-stained using standard procedures. The band of interest was excised from an unstained, heavily overloaded 15% acrylamide gel; the band position was estimated from a Coomassie-stained section of the same gel containing the same sample. The excised gel piece was crushed and vortexed for 12 hours at 16°C in 6 M urea, 0.1 M NaOAc (pH 4.5) to extract the protein. The protein was then chromatographically purified on a 50 × 4.6 mm Jupiter 5μC4 300-Å column (Phenomenex, Belmont, CA), and analyzed on a Hewlett Packard 5989A electrospray mass spectrometer.
Crystallography.
Subtilisin and CI2 were each dissolved in 10 mM NaOAc (pH 5.8), mixed in a 1:1.2 stoichiometric molar ratio, and diluted to a combined concentration of 6.5 mg/ml of protein. Crystals were grown at 4°C in hanging drops over a reservoir of 0.2 M (NH4)2SO4, 0.1 M NaCacodylate (pH 6.5), and 30% PEG 8000; drops were prepared by mixing 2 μl of protein solution with 2 μl from the reservoir. Crystals (space group P212121) were flash-frozen in liquid N2 without additional cryoprotectant. Synchrotron x-ray data were collected from a single crystal at 100 K by using an Area Detector Systems (Poway, CA) Quantum 4 charge-coupled device detector at Advanced Light Source beam line 5.0.1, Lawrence Berkeley National Laboratory. The automation package elves (J. Holton, manuscript in preparation) was used to direct the programs molflm (25) for indexing and integration, and scala (26) for scaling and merging the reflections. The structure was solved by molecular replacement using cns (27), by using as the search model the 2.1 Å resolution subtilisin/CI2 structure in space group C2 of McPhalen and James (PDB ID code ) (11). The model was then rebuilt by using the automatic model building capacity of ARP/WARP (28), manually improved by using the interactive graphics program o (29), and further refined by using refmac (30). The free-R factor was calculated with 5% of the data. The final model, refined to 1.5-Å resolution, contains 345 amino acid residues (2,527 protein atoms), 505 water molecules, 1 calcium ion, and 4 sulfate ions. Ten of the amino acid side chains were refined with partial occupancies in multiple conformations. Fig. 3 was prepared with insightii (Biosym Technologies, Skokie, IL).
Fig 3.

(A) Ribbon diagram of subtilisin/CI2 complex structure. Subtilisin is shown in red, the N-terminal section of CI2 is shown in dark blue, and the C-terminal section of CI2 is shown in light blue. The reactive site peptide bond is at the junction of the dark and light blue segments. (B) Closer view of reactive site loop. CI2 side chains (labeled in white) and hydrogen bonds (yellow dotted lines) proposed to stabilize the positioning of the light blue (leaving group) side of the loop after acyl–enzyme formation (see text) are shown in detail. The serine, histidine, and aspartate of the subtilisin catalytic triad are also shown (labeled in yellow).
Comparative Structural Analysis.
The dataset used for structural comparisons consisted of all entries classified as serine endopeptidases (EC 3.4.21) in the Protein Data Bank (www.rcsb.org) in which peptide or protein inhibitors were noncovalently bound at the active site. Further criteria were that the inhibitor peptide bond positioned at the active site be intact, and that the catalytic serine of the enzyme not be mutated or chemically modified. These criteria were met by 79 structures, including the earlier structure of subtilisin BPN′/CI2, which was replaced in the dataset with the higher-resolution structure reported here. Angles and bond lengths for all structures were measured with the molecular modeling package SYBYL V. 6.7 (Tripos Associates, St. Louis), and superpositions comparing active site geometry were generated with insightii, which was also used to prepare Figs. 1 and 4 C and D.
Fig 4.

Nucleophilic attack trajectories for protease/inhibitor complexes. (A) The geometric parameters describing the nucleophilic attack trajectory are diagrammatically defined. θy is the angle defined by the enzyme serine γ-oxygen, the inhibitor carbonyl carbon, and the inhibitor carbonyl oxygen. θx is the angle between (i) the plane defined by the enzyme serine γ-oxygen, the inhibitor carbonyl carbon, and the inhibitor carbonyl oxygen, and (ii) the plane defined by the peptide bond. γO—C represents the distance between the enzyme serine γ-oxygen and the inhibitor carbonyl carbon. (B) Plot of θy vs. θx. Blue triangles represent the structures of 78 protease/inhibitor complexes, the orange circle represents the subtilisin/CI2 complex, and the red square represents the thrombin/fibrinogen analog structure (16). The peptide bond diagrammed in the background is for illustrative purposes. (C and D) Two views of the superposition of 79 protease/inhibitor complexes, including subtilisin/CI2. Superpositioning was based on the α-carbon and carbonyl oxygen of the P1 residue, and the amide nitrogen of the P residue, which overlay closely for all structures. The red spheres represent the relative positions of the enzyme serine γ-oxygen for each structure. The outlying structure apparent in B, C, and D is that of an ecotin mutant complexed with trypsin.
residue, which overlay closely for all structures. The red spheres represent the relative positions of the enzyme serine γ-oxygen for each structure. The outlying structure apparent in B, C, and D is that of an ecotin mutant complexed with trypsin.
Results and Discussion
As expected, subtilisin cleaved CI2 extremely slowly, with an enzymatic hydrolysis rate of 3.8 × 10−6 s−1 (data not shown). However, we observed that incubating subtilisin with CI2 consistently produced a gel band with a molecular weight considerably larger than either subtilisin or CI2 (Fig. 2A). Hypothesizing that this species could be a stable acyl–enzyme intermediate, we carried out the incubation in the presence of 0.1% SDS, which destabilizes CI2 but does not affect subtilisin stability or activity; CI2 was completely hydrolyzed, and the new band disappeared (Fig. 2B). The band did not form when subtilisin was inactivated by low pH before incubation with CI2 (the 0-min time points in Fig. 2 B and C), nor did it form upon incubation of CI2 with a catalytically compromised mutant subtilisin (Fig. 2C). When CI2 and subtilisin were incubated together, quenched, and then denatured with urea, SDS, or by boiling, the new band was not disrupted. However, after the denatured sample was subjected to conditions that hydrolyze ester linkages (31), the new band was absent (Fig. 2C). The new band had a mass of 32,730 Da, identical to the expected mass for the acyl–enzyme intermediate that would form in cleavage of CI2 at the Met-59–Glu-60 reactive site bond (Fig. 2D).
Fig 2.

Formation of acyl–enzyme upon incubation of subtilisin with CI2. (A) A new species (indicated by the arrow) appeared when purified, active subtilisin was mixed with purified CI2 and incubated at pH 5.0 at 25°C for 15 min. (B) SDS at 0.1% destabilized CI2 to subtilisin proteolysis; complete proteolysis of CI2 coincided with disappearance of the new band. Two-hour time courses with and without SDS are shown; both contained identical initial concentrations of subtilisin and CI2. (C) Time courses to detect rapid formation of the new species were carried out with wild-type subtilisin and with a mutant, Asn155Leu/Met222Ala, that has a catalytic constant reduced by a factor of 103 with standard small peptide substrates, whereas substrate binding is less dramatically affected (data not shown). We verified that both wild-type and mutant subtilisin were completely inhibited by complex formation with CI2 within 10 s of mixing under the conditions used in the time courses. Additional aliquots of the wild-type subtilisin/CI2 mixture were withdrawn and quenched after 15 min, then adjusted to 4% SDS, 6 M urea, or 0.2M KOH/75% methanol, and incubated at 37°C for 30 min before SDS/PAGE resolution. An additional aliquot, quenched after the initial 15 min incubation, was boiled for 15 min after addition of SDS/PAGE loading buffer. (D) The new protein species was isolated from an SDS/PAGE gel, purified by reversed-phase HPLC, and analyzed by electrospray ionization MS.
When we studied the acyl–enzyme band as a function of time, it formed within seconds (Fig. 2C), then remained as a constant fraction of the mixture over a 2-h time course (Fig. 2B). The acyl–enzyme represented roughly 10% of the total enzyme. This observation suggests that an equilibrium between the Michaelis complex and the acyl–enzyme is quickly established, in which the Michaelis complex is thermodynamically favored relative to the acyl–enzyme. The rapidity with which equilibrium is reached indicates the absence of a large energy barrier to acylation. These results disprove the hypotheses that either rigidity or poor orientation prevents productive nucleophilic attack, and show that the slow step in hydrolysis of CI2 is deacylation, illustrated in Eq. 3 above.
Then, the question as to why CI2 inhibits subtilisin becomes more specific: why does deacylation proceed so slowly? One hypothesis suggested by Fersht and coworkers (12, 19) is that the leaving group amine is poised for nucleophilic attack on the acyl–enzyme, resulting in the back reaction illustrated by k−2 in Eq. 2. If this is so, there must be a reason why the back reaction is favored for the inhibitor, but not for a normal substrate. The answer is suggested by the crystal structure of the subtilisin/CI2 complex (Fig. 3A). We have refined a structure of the subtilisin/CI2 complex to 1.5 Å, by using as a starting model the lower resolution structure of McPhalen and James (11); crystallographic statistics are summarized in Table 2. Although interactions between subtilisin and the R2 leaving group residues are minimal (Tyr-61 is involved in a backbone hydrogen bond and a side chain offset ring-stacking interaction; ref. 32, see Fig. 3B), there are extensive interactions between the R1 (residues 1–59) and R2 (residues 60–83) chains. The R1 and R2 chains each contribute two strands to a β-sheet that makes up part of the hydrophobic core of CI2. The reactive site loop sits on the platform of this β-sheet, held in place by a dense hydrogen bonding network involving residues Thr-58, Glu-60, Arg-62, Arg-65, Arg-67, and Gly-83 (Fig. 3B). There has been substantial NMR evidence that this network remains intact in the cleaved inhibitor (examined in the absence of enzyme), and stabilizes the newly formed N terminus (19). Similar interactions have been identified in cleaved Cucurbita maxima trypsin inhibitor V, an inhibitor with sequence and structural homology to CI2 (33–35). Mutational studies of CI2 and another homolog, eglin c, also support the functional importance of the hydrogen bonding network; mutation of Thr-58, Glu-60, Arg-65, or Gly-83 (or the corresponding residues in eglin) result in accelerated inhibitor hydrolysis (15, 36, 37).
Table 2.
Data collection and refinement statistics for the subtilisin/CI2 complex
| Data set | Statistics |
|---|---|
| Space group | P212121 |
| Unit cell, Å | a = 54.33, b = 56.60, c = 118 |
| Resolution range, Å | 29.7–1.5 |
| Unique reflections | 57,374 |
| Completeness, % | 96.8 (88.9) |
| Multiplicity | 6.5 (5.0) |
| I/SD | 15.3 (4.3) |
| Rsym, % | 7.2 (34) |
| Rcryst/Rfree, % | 16.9/18.8 |
| rmsd bonds, Å/angles, ° | 0.007/1.4 |
| Average B factor for protein, Å2 | 10.4 |
| Average B factor for water, Å2 | 24.3 |
Outer shell (1.58–1.50 Å) values are given in parentheses.
I, intensity; SD, standard deviation.
Rsym = Σ|I − 〈I〉|ΣI × 100%.
Rcryst = Σ|Fobs − Fcalc|/ΣFobs × 100%.
rms deviations from ideal geometry.
The extensive intramolecular hydrogen bonding network within the cleaved inhibitor would not exist in a usual peptide or protein substrate vulnerable to proteolysis. Evidently, the combination of interactions of the R2 leaving group of CI2 (residues 60–83) with R1 (residues 1–59) and with subtilisin, in concert with covalent attachment of the R1 chain to subtilisin in the acyl–enzyme, greatly slows the dissociation of H2N–R2 from the active site. These numerous stabilizing contacts maintain the newly formed N terminus in an optimal orientation for nucleophilic attack on the acyl–enzyme, favoring religation. The positioning of the amine also sterically hinders the hydrolytic water molecule from achieving the necessary proximity to the histidine base for nucleophilic activation.
In light of the finding that subtilisin rapidly attacks CI2 to form an acyl–enzyme intermediate, yet the reaction goes no further, we asked two interrelated questions: (i) is this mechanism of inhibition common to other protein protease inhibitors, and (ii) what is the nucleophilic attack angle of the catalytic serine on the reactive acyl bond that results in rapid progression to the acyl–enzyme? Because the subtilisin/CI2 complex displays the tight binding typical of this class of inhibitors (Table 1), and because tight binding does not pose a barrier to acylation in this complex, we reasoned that it was unlikely to do so in other complexes. We used structural data from the Protein Data Bank, and superimposed 79 serine protease complexes with inhibitors to examine the trajectories of the nucleophilic serine oxygen atoms. Observed geometries were compared with each other, and with that of thrombin complexed with a noncleavable peptide substrate analog in which the nitrogen of the scissile amide bond was replaced with a methylene moiety (16). A table of all structures used, including references and measured angles and distances, is available in Table 4, which is published as supporting information on the PNAS web site, www.pnas.org.
When the structures of the complexes were superposed based only on the atoms of the inhibitor reactive site bond, we found that the nucleophilic serine γ-oxygens were tightly clustered around a consensus orientation of about 90° for attack on the carbonyl carbon (Table 3). Parameters that describe the nucleophilic attack trajectory are defined in Fig. 4A and plotted in Fig. 4B, and Fig. 4 C and D display the superposition of all 79 inhibitor complexes. The near-perpendicular attack on the scissile amide bond displayed by the subtilisin/CI2 complex is typical of other inhibitor complexes, and close to the trajectory in the substrate analog complex, all of which feature both angles, θx and θy (see Fig. 4A), consistently close to 90°. It is intriguing that the consensus angle of attack θy differs considerably from the value of 105° ± 5° obtained from small molecule structure correlations and theoretical calculations by Bürgi et al. (38–40). Previously, it had not been possible to evaluate angles of nucleophilic attack leading to enzymatic proteolysis, because good substrates react too quickly with native enzymes to allow collection of structural data, whereas slowly reacting poor substrates or poor enzymes are suspect models. The recognition that the geometry found in these protease/inhibitor complexes is conducive to rapid acylation establishes them as good models for the Michaelis complex—a common speculation, but one not previously supported by experimental data.
Table 3.
Geometric parameters that define the approach of the catalytic serine γ-oxygen towards the reactive site peptide carbonyl
| Structure(s) | Angle θx, ° | Angle θy, ° | Distance γ O—C, Å |
|---|---|---|---|
| Subtilisin/CI2 | 97.9 | 88.3 | 2.86 |
| Average of inhibitor complexes | 94 ± 6 | 89 ± 7 | 2.7 ± 0.2 |
| Thrombin/substrate analog | 84.4 | 90.8 | 2.62 |
The parameters used are defined in Fig. 4A.
We therefore propose that CI2 is representative of other inhibitors in its mechanism of inhibition: an otherwise appropriate substrate becomes an extremely effective inhibitor through retention of the H2N–R2 leaving group, favoring the religation reaction. It appears that previous postulates of reaction reversal due to tightly bound leaving amine were correct, whereas proposals of lack of reactivity due to rigidity or orientation were not. Thus, one could summarize the effect of this class of inhibitor as analogous to the clogging of a gutter drain by a combination of twigs and leaves. Neither twigs alone nor leaves alone can clog a gutter drain, but a combination of twigs and leaves can do so very effectively. The combination of a hydrogen bond network, an acyl–enzyme, and the correct orientation of the religating amide can arrest the reaction, whereas each element individually would be insufficient.
Supplementary Material
Acknowledgments
We thank D. King for mass spectrometric analysis, R. Rose, J. Holton, and T. Alber for assistance with crystallography, and G. Toth for assistance with structural comparisons. Funding was provided by National Institutes of Health Grant DK09765 (to D.E.K.); E.S.R. was supported by a National Institutes of Health postdoctoral fellowship.
Abbreviations
CI2, chymotrypsin inhibitor 2
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code ).
References
- 1.Laskowski M. & Kato, I. (1980) Annu. Rev. Biochem. 49, 593-626. [DOI] [PubMed] [Google Scholar]
- 2.Laskowski M. & Qasim, M. A. (2000) Biochim. Biophys. Acta 1477, 324-337. [DOI] [PubMed] [Google Scholar]
- 3.Bode W. & Huber, R. (1992) Eur. J. Biochem. 204, 433-451. [DOI] [PubMed] [Google Scholar]
- 4.Read R. J. & James, M. N. G. (1986) in Proteinase Inhibitors, eds. Barrett, A. J. & Salvesen, G. (Elsevier, Amsterdam), pp. 301–336.
- 5.Hubbard S. J., Campbell, S. F. & Thornton, J. M. (1991) J. Mol. Biol. 220, 507-530. [DOI] [PubMed] [Google Scholar]
- 6.Apostoluk W. & Otlewski, J. (1998) Proteins Struct. Funct. Genet. 32, 459-474. [PubMed] [Google Scholar]
- 7.Tyndall J. D. A. & Fairlie, D. P. (1999) J. Mol. Recognit. 12, 363-370. [DOI] [PubMed] [Google Scholar]
- 8.Jackson R. M. & Russell, R. B. (2000) J. Mol. Biol. 296, 325-334. [DOI] [PubMed] [Google Scholar]
- 9.Fujinaga M., Read, R. J., Sielecki, A., Ardelt, W., Laskowski, M., Jr. & James, M. N. G. (1982) Proc. Natl. Acad. Sci. USA 79, 4868-4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Read R. J., Fujinaga, M., Sielecki, A. R. & James, M. N. G. (1983) Biochemistry 22, 4420-4433. [DOI] [PubMed] [Google Scholar]
- 11.McPhalen C. A. & James, M. N. G. (1988) Biochemistry 27, 6582-6598. [PubMed] [Google Scholar]
- 12.Longstaff C., Campbell, A. F. & Fersht, A. R. (1990) Biochemistry 29, 7339-7347. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi Y., Satow, Y., Nakamura, K. T. & Mitsui, Y. (1991) J. Mol. Biol. 221, 309-325. [PubMed] [Google Scholar]
- 14.Peräkylä M. & Kollman, P. A. (2000) J. Am. Chem. Soc. 122, 3436-3444. [Google Scholar]
- 15.Lu W.-Y., Starovasnik, M. A., Dwyer, J. J., Kossiakoff, A. A., Kent, S. B. H. & Lu, W. (2000) Biochemistry 39, 3575-3584. [DOI] [PubMed] [Google Scholar]
- 16.Martin P. D., Malkowski, M. G., DiMaio, J., Konishi, Y., Ni, F. & Edwards, B. F. P. (1996) Biochemistry 35, 13030-13039. [DOI] [PubMed] [Google Scholar]
- 17.Coombs G. S., Rao, M. S., Olson, A. J., Dawson, P. E. & Madison, E. L. (1999) J. Biol. Chem. 274, 24074-24079. [DOI] [PubMed] [Google Scholar]
- 18.Tamura A., Kanaori, K., Kojima, S., Kumagai, I., Miura, K. & Akasaka, K. (1991) Biochemistry 30, 5275-5286. [DOI] [PubMed] [Google Scholar]
- 19.Shaw G. L., Davis, B., Keeler, J. & Fersht, A. R. (1995) Biochemistry 34, 2225-2233. [DOI] [PubMed] [Google Scholar]
- 20.Wells J. A., Ferrari, E., Henner, D. J., Estell, D. A. & Chen, E. Y. (1983) Nucleic Acids Res. 11, 7911-7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang M. Y., Ferrari, E. & Henner, D. J. (1984) J. Bacteriol. 160, 15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter P. & Wells, J. (1987) Science 237, 394-399. [DOI] [PubMed] [Google Scholar]
- 23.Jackson S. E., Moracci, M., elMasry, N., Johnson, C. M. & Fersht, A. R. (1993) Biochemistry 32, 11259-11269. [DOI] [PubMed] [Google Scholar]
- 24.Komiyama T. & Fuller, R. S. (2000) Biochemistry 39, 15156-15165. [DOI] [PubMed] [Google Scholar]
- 25.Leslie A. G. W., Brick, P. & Wonacott, A. (1986) Daresbury Lab. Info. Q. Protein Crystallogr. 18, 33-39. [Google Scholar]
- 26.Collaborative Computational Project No. 4 (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 27.Brünger A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 28.Perrakis A., Morris, R. & Lamzin, V. S. (1999) Nat. Struct. Biol. 6, 458-463. [DOI] [PubMed] [Google Scholar]
- 29.Jones T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 30.Murdushov G. N., Vagin, A. A. & Dodson, E. J. (1997) Acta Crystallogr. D 53, 240-255. [DOI] [PubMed] [Google Scholar]
- 31.Bizzozero O. A. (1995) Methods Enzymol. 250, 361-379. [DOI] [PubMed] [Google Scholar]
- 32.Otzen D. E. & Fersht, A. R. (1999) Protein Eng. 12, 41-45. [DOI] [PubMed] [Google Scholar]
- 33.Cai M., Gong, Y. X., Prakash, O. & Krishnamoorthi, R. (1995) Biochemistry 34, 12087-12094. [DOI] [PubMed] [Google Scholar]
- 34.Liu J., Prakash, O., Huang, Y., Wen, L., Wen, J. J., Huang, J.-K. & Krishnamoorthi, R. (1996) Biochemistry 35, 12503-12510. [DOI] [PubMed] [Google Scholar]
- 35.Cai M., Huang, Y., Prakash, O., Wen, L., Dunkelbarger, S. P., Huang, J.-K., Liu, J. & Krishnamoorthi, R. (1996) Biochemistry 35, 4784-4794. [DOI] [PubMed] [Google Scholar]
- 36.Jackson S. E. & Fersht, A. R. (1994) Biochemistry 33, 13880-13887. [DOI] [PubMed] [Google Scholar]
- 37.Heinz D. W., Hyberts, S. G., Peng, J. W., Priestle, J. P., Wagner, G. & Grütter, M. G. (1992) Biochemistry 31, 8755-8766. [DOI] [PubMed] [Google Scholar]
- 38.Bürgi H. B., Dunitz, J. D. & Shefter, E. (1973) J. Am. Chem. Soc. 95, 5065-5067. [Google Scholar]
- 39.Bürgi H. B., Lehn, J. M. & Wipff, G. (1974) J. Am. Chem. Soc. 96, 1956-1957. [DOI] [PubMed] [Google Scholar]
- 40.Bürgi H. B., Dunitz, J. D., Lehn, J. M. & Wipff, G. (1974) Tetrahedron 30, 1563-1572. [Google Scholar]
- 41.Vincent J.-P., Peron-Renner, M., Pudles, J. & Ladzunski, M. (1974) Biochemistry 13, 4205-4211. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski M. & Finkenstadt, W. R. (1972) Methods Enzymol. 26, 193-227. [DOI] [PubMed] [Google Scholar]
- 43.Empie M. W. & Laskowski, M., Jr. (1982) Biochemistry 21, 2274-2284. [DOI] [PubMed] [Google Scholar]
- 44.Ascenzi P., Amiconi, G., Bolognesi, M., Menegatti, E. & Guarneri, M. (1990) J. Mol. Recognit. 3, 192-196. [DOI] [PubMed] [Google Scholar]
- 45.Otlewski J. & Zbyryt, Z. (1994) Biochemistry 33, 200-207. [DOI] [PubMed] [Google Scholar]
- 46.Patthy A., Amir, S., Malik, Z., Bódi, Á., Kardos, J., Asbóth, B. & Gráf, L. (2002) Arch. Biochem. Biophys. 398, 179-187. [DOI] [PubMed] [Google Scholar]
- 47.Luckett S., Garcia, R. S., Barker, J. J., Konarev, Al. V., Shewry, P. R., Clarke, A. R. & Brady, R. L. (1999) J. Mol. Biol. 290, 525-533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.