

Abstract
Although irreversible reaction of NO with the oxyheme of hemoglobin (producing nitrate and methemoglobin) is extremely rapid, it has been proposed that, under normoxic conditions, NO binds preferentially to the minority deoxyheme to subsequently form S-nitrosohemoglobin (SNOHb). Thus, the primary reaction would be conservation, rather than consumption, of nitrogen oxide. Data supporting this conclusion were generated by using addition of a small volume of a concentrated aqueous solution of NO to a normoxic hemoglobin solution. Under these conditions, however, extremely rapid reactions can occur before mixing. We have thus compared bolus NO addition to NO generated homogeneously throughout solution by using NO donors, a more physiologically relevant condition. With bolus addition, multiple hemoglobin species are formed (as judged by visible spectroscopy) as well as both nitrite and nitrate. With donor, only nitrate and methemoglobin are formed, stoichiometric with the amount of NO liberated from the donor. Studies with increasing hemoglobin concentrations reveal that the nitrite-forming reaction (which may be NO autoxidation under these conditions) competes with reaction with hemoglobin. SNOHb formation is detectable with either bolus or donor; however, the amounts formed are much smaller than the amount of NO added (less than 1%). We conclude that the reaction of NO with hemoglobin under normoxic conditions results in consumption, rather than conservation, of NO.
One of the most important experimental findings that led to the postulate that the endothelium-derived relaxing factor (EDRF) is identical to nitric oxide (nitrogen monoxide, NO) was the demonstration that endothelium-dependent relaxation is exquisitely sensitive to inhibition by hemoglobin, which rapidly reacts with NO (1). Curiously, however, even though very small concentrations of hemoglobin are quite potent in preventing EDRF-dependent relaxation, little attention was initially paid to the problem this raises with the NO/EDRF hypothesis, namely, that in vivo NO is produced immediately adjacent to a pool of very high (mM) concentrations of hemoglobin, which will act as a potent sink for the NO, thus decreasing its concentration at all locations and preventing it from reaching a physiologically functional level. Mathematical modeling has illustrated the validity of this concept (2–4).
There have emerged two hypotheses to explain how NO might still function as EDRF. According to one (5–10), rather than irreversibly consuming NO, hemoglobin actually conserves it, in the form of a nitrosothiol moiety, and the linkage of the accessibility/reactivity of the thiol group involved (βcys93) to the oxygenation-dependent allosteric transitions in hemoglobin establishes a respiratory cycle whereby NO and O2 are simultaneously taken up in the lung and then delivered to the tissue during the arterial/venous transit. Thus, hemoglobin would deliver to oxygen-deficient vascular beds not only the oxygen required for sustained metabolism but also a vasodilator (NO).
The alternative hypothesis (3, 4, 11–17), is that the consumption of NO by hemoglobin in the blood is greatly decelerated relative to free hemoglobin (by a factor of approximately 1,000-fold) because of two phenomena, the limitation of reaction by the rate at which NO enters the erythrocyte (which is substantially slower than the reaction per se; refs. 11 and 14–16) and existence of an erythrocyte-free zone immediately adjacent to the endothelial cell layer of the vessel (3, 4, 13). Thus, although free hemoglobin is an avid scavenger of NO, when packaged within erythrocytes, this reaction is restricted enough to allow free NO to act effectively as the endothelium-derived vasodilator (13).
A crucial question is the precise nature of the reaction(s) of NO with hemoglobin under physiological conditions. The irreversible consumption of NO occurs by reaction of NO with oxyhemoglobin to produce methemoglobin and nitrate: NO + Hb(Fe2+)O2 → NO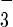 + Hb(Fe3+).
+ Hb(Fe3+).
The rate constant of this reaction has been measured as high as k = 8.9 × 107 M−1⋅s−1 (on a molar heme basis; refs. 18 and 19), an exceedingly rapid number, which raises a serious challenge to the postulate that, under physiological conditions, hemoglobin preserves NO by formation of S-nitrosothiol. However, as pointed out by Gow et al. (8), what will determine the partitioning of NO between the two pathways (irreversible oxidation vs. SNOHb formation) is the relative magnitudes of the rate of oxidation compared with the rate of reaction of NO with the small proportion of deoxyheme (≈1%) with the hemoglobin in the R or high affinity state. Thus, Gow et al. (8) measured the rates of SNOHb formation under normoxic conditions with addition of NO solution and presented data to suggest that indeed the rate of this reaction (which could lead to SNOHb formation) is more rapid than irreversible consumption. We present here a reexamination of these studies, comparing bolus addition of NO (as performed by Gow et al. using their conditions) with more slowly releasing NO donors, which deliver NO homogeneously throughout the solution. These conditions avoid complications from reactions that occur during the incomplete mixing before agitation (which become critically important when the formation of only minority species are measured) and more closely resemble the physiological situation. We show that, under these more physiologically relevant conditions (compared with bolus addition of high concentrations of NO in a stock solution), we can detect only minute quantities of SNOHb (compared with the NO added) and all of the detectable reaction products are the result of irreversible consumption.
Materials and Methods
Hemoglobin Preparation.
Fresh human blood was collected in heparinized (10 units/ml) tubes. The plasma and buffy coat were removed after centrifugation at 800 × g for 10 min. The cells were resuspended and purified by filtration through a mixture of alpha and microcrystalline cellulose. The purified RBCs were washed three times in 10 mM phosphate buffer adjusted to 290 mOsm with NaCl and lysed at 1:4 with a 5 mM phosphate buffer. The lysed RBC solution was then centrifuged at 22,000 × g for 30 min. OxyHb was isolated by collecting the upper half of the centrifuged lysate and passing it through a G-25 Sephadex column. Stock solutions of oxyHb at 10, 5, and 1 μM were prepared. From this set of oxyHb solutions, three sets of metHb, deoxyHb, and HbNO solutions were prepared in sealed cuvettes. MetHb was prepared by addition of slight molar (per heme) excess ferricyanide. This preparation was filtered through a Sephadex G-25 column to remove the excess ferricyanide. DeoxyHb was prepared by removal of oxygen from oxyHb solution with argon. The HbNO samples were prepared from the deoxyHb samples by the addition of small amounts of NO gas. Difference spectra (vs. oxyHb) were recorded from 500 to 700 nm. The saturation of oxyhemoglobin was >95% as judged by comparison of the standard spectra (Fig. 1A) to published spectra (8, 20).
Fig 1.

Spectroscopic characteristics of reaction of oxyhemoglobin with bolus vs. donor NO. (A) Standard spectra. Difference spectra vs. oxyHb were recorded by using preparations of metHb, deoxyHb, and nitrosylHb as described in Materials and Methods. (B) Difference spectra (vs. oxyHb) for bolus compared with donor NO. Hemoglobin (33 μM; 132 μM heme) in 10 mM phosphate buffer containing 100 μM diethylenetriamine pentaacetic acid was treated with either bolus or donor (MAHMA/NO) NO (final NO concentration 2.2 μM concentration in both cases) as described in Results. For bolus addition, NO was added as described by Gow et al. (8). UV/Vis spectra recorded as described in Materials and Methods.
Spectrophotometric Analysis of the NO/OxyHb Reaction.
Solutions of oxyhemoglobin in 10 mM or 100 mM potassium phosphate, pH 7.4, containing 100 μM diethylenetriamine pentaacetic acid (DTPA) were treated with either bolus NO [added from an anaerobic solution prepared as described (21)] or donor as described in Results. After correction for dilution by addition of equivalent amounts of buffer to the reference cuvette, difference absorption spectra (vs. oxyHb solution before treatment) were recorded from 500 to 700 nm wavelength.
MetHb Assay.
For Fig. 4, metHb formation was measured by the method of Rodkey et al. (22), where spectral formation of cyanmethemoglobin is measured, which will be specific for methemoglobin over other forms of hemoglobin. Addition of cyanide to nitrosylheme hemoglobin under these conditions does not displace NO (data not shown).
Fig 4.

Formation of nitrate, MetHb, and nitrite with bolus vs. donor NO. Bolus or NO donor (MAHMA/NO; final NO concentration 5 μM) was added to 125 μM hemoglobin (500 μM heme) in 10 mM phosphate buffer + diethylenetriamine pentaacetic acid. Nitrate, metHb, and nitrite were measured as described in Materials and Methods.
Quantification of Nitrogen Oxides and SNOHb.
Nitrate was measured by the Griess method, after conversion to nitrite as described (23). Nitrite was measured either by the Griess method (23) or by chemiluminescence as described below.
The extent of nitros(yl)ation of oxyHb exposed to either authentic NO or an NO donor was measured by reductive denitrosation of samples with subsequent detection of the liberated NO by its gas phase chemiluminescence reaction with ozone (24). NO was quantified by using an ultrasensitive chemiluminescence detector (CLD 77 AM sp, Eco Physics, Ann Arbor, MI; detection limit 20 parts per trillion), the sample inlet of which was directly connected to the outlet of a water-jacketed, septum-sealed reaction chamber containing a potassium iodide/triiodide mixture in glacial acetic acid. A stream of nitrogen gas (110 ml/min) was bubbled continuously through the reaction mixture, which was kept at 60°C. Sample aliquots were injected into the reaction mixture by means of a gas-tight Hamilton syringe, and NO signal output was captured by using SpiroWare (Eco Physics). Signal integration was performed by using chromprocessor software (version 4.5; ACD Labs, Toronto, Canada). Nitrite concentrations in the sample were determined by the difference in signal of aliquots with and without preincubation with 0.1% sulfanilamide/HCl (25), with the NO signal remaining after preincubation with sulfanilamide representing total nitrosated species. Discrimination between —SNO and —NNO/NO—heme species was achieved by preincubation of sample aliquots with mercuric chloride (HgCl2; 0.2% for 30 min), which selectively cleaves S—NO bonds while preserving N—NO and NO—heme moieties. The chemiluminescence analyzer was calibrated weekly by using a 100-parts per billion mixture of NO in N2 (H.P. Gas Products, Baytown, TX), and calculated NO amounts were validated by injection of freshly prepared nitrite standards into the reaction mixture. At an injection volume of 250 μl, the detection limit corresponded to 100 fmol NO (S/N ratio > 3:1).
Results
With a rate constant of 6–9 × 107 M−1⋅s−1 (18, 19), it can be calculated that the half-life of NO when added to a 50-μM oxyhemoglobin solution is 58–39 μs. This will actually be the maximum half-life of NO because it represents the oxidation reaction, which, according to Gow et al., will be approximately 200-fold slower than reaction with deoxyheme. This reaction is far more rapid than the time required for agitation to disperse the bolus volume added. Thus, the NO concentration in these regions (before uniform dispersion) will be quite high, and initially in fact it will equal that of the NO stock solution, which is 1.8 mM for a saturated solution. At this exceptionally high unphysiological concentration, in theory a number of possible reactions may occur, both with the hemoglobin species and also with dioxygen. As described in more detail in Discussion, these reactions might be capable of forming potent nitrosating species that could result in SNOHb formation. We reasoned that a more physiologically relevant condition would be where the rate of NO appearance will be such that the NO concentration will be homogeneously distributed throughout the solution. Thus, we compared bolus NO addition to results with NO donors that release NO at defined rates.
We began by examining the formation of species detected by visible absorbance spectroscopy. Fig. 1A shows our standard difference spectra (vs. oxyhemoglobin) for each of the three spectrally detectable species (methemoglobin, deoxyhemoglobin, and nitrosylhemoglobin). Of particular importance is the relatively broad absorptive excursion for methemoglobin in the wavelength range 620–650 nm and the maximum intensities of the troughs at 540 and 575 nm (compared with those of deoxyHb and nitrosylHb). Also distinctive of the methemoglobin spectrum (compared with the deoxyHb and nitrosylHb) is the peak at 560 nM between the two troughs at 540 and 575 nm; this peak is much more pronounced in the deoxyHb and nitrosylHb spectra than in the methemoglobin spectra. In addition, as also was pointed out by Gow et al. (8), both deoxyHb and nitrosylHb exhibit a small peak at 595 nm.
Fig. 1B shows the difference spectra (vs. oxyhemoglobin solution with no NO) with 10 mM phosphate, with 33 μM hemoglobin (132 μM heme) at a constant temperature of 20°C. Either bolus NO (2.2 μM final concentration) or 1.1 μM (Z)-1-{N-methyl-N- [6-(N-methylammoniohexyl)amino]}diazenium 1,2-diolate (MAHMA/NO; t1/2 2.3 min, liberating 2 NO per mole) was added (each experiment performed in triplicate), and the spectra were recorded 15 min later. The spectra with bolus NO display substantial variability in intensity, and are of less intensity than the spectra with donor. The spectra from bolus addition demonstrate substantial amounts of methemoglobin, but closer examination of the spectra reveals the absence of clean isosbestic points, indicating the existence of multiple species. When NO is added by generation homogeneously throughout the solution, the spectrum is virtually entirely methemoglobin. It is important to point out that we used low NO/heme molar ratio (2.2/132) and 10 mM phosphate buffer, conditions that according to Gow et al. afford maximum SNOHb formation (8).
We also used another NO donor, which liberates NO more slowly, so that we could closely monitor the gradual spectral changes with time. For this experiment, the donor spermine-NO (SPER/NO) was used (100 μM), which breaks down and liberates NO with a half-life of 230 min at 24°C (26), and we recorded spectra at 1-min intervals for a total of 6 min. The theoretical maximal NO concentration achieved amounted to 2–3 μM (total heme concentration 100 μM). Fig. 2A shows the results with 100 mM phosphate buffer, and Fig. 2B shows the results with 10 mM phosphate. In both cases, the gradual changes exhibit pure isosbestic points, which are indicative of a gradual conversion between only two species. The resulting spectrum is identical to the spectrum for methemoglobin, allowing the conclusion that liberation of NO results in only the oxidation of oxyhemoglobin to methemoglobin. These results are essentially identical to those reported previously by using other NO donors (27–29). Knowing the rate of NO production (via donor release) and the rate of NO reaction (via reaction with oxyhemoglobin), it can be calculated that the steady-state NO concentration is 0.14 pM, which clearly satisfies the requirement of Gow et al. (8) that the concentration of NO should be substantially less than the heme concentration, which for Fig. 2 is 100 μM.
Fig 2.

Time course for spectral changes with NO donor in 100 mM (A) or 10 mM (B) phosphate. At zero time, 100 μM spermine-NO was added to the reference cuvette of a solution containing 25 μM oxyhemoglobin (100 μM heme), and spectra were recorded at 1-min intervals. Arrows denote direction of excursion movement. Over this time period, 2–3 μM NO was released.
If the reaction of NO with the hemoglobin species in solution is due only to an interaction of NO with the protein, the products of the reaction should be independent of hemoglobin concentration. However, as shown in Fig. 3A, increasing hemoglobin concentration with bolus NO results in increased methemoglobin formation, and the result is relatively insensitive to whether the buffer is 10 mM or 100 mM phosphate. This result indicates that hemoglobin competes with another reaction of NO. Evidence for the presence of another reaction is shown in Fig. 3B, which shows the formation of appreciable amounts of nitrite with bolus NO; the decrease in nitrite with increasing hemoglobin indicates that the nitrite is a product of this competing reaction. In contrast to the results with bolus NO, liberation of NO with donor (MAHMA/NO) results in stoichiometric metHb formation (Fig. 3A) and constant low levels of nitrite (Fig. 3B). As described in Discussion, the formation of nitrite indicates the existence of nitrosative chemistry with bolus NO addition. Gow et al. (8) also suggested the existence of other reaction pathways with bolus NO addition, because the amount of metHb and nitrosylheme did not total the amount of NO added.
Fig 3.

MetHb (A) and nitrite (B) formation as a function of hemoglobin concentration with bolus and donor NO. Bolus or donor NO (10 μM NO final concentration) was added to solutions containing the indicated concentrations of oxyhemoglobin (on a heme basis), and methemoglobin and nitrite were measured as described in Materials and Methods.
Finally, we measured the formation of nitrate, nitrite, methemoglobin, and SNOHb for bolus compared with donor NO (5 μM NO total for both). As shown in Fig. 4, with bolus NO there is only partial oxyhemoglobin oxidation, producing metHb (1.99 ± 0.70 μM) and nitrate (2.7 ± 0.77 μM), with substantial amounts of nitrite (2.51 ± 0.19 μM). However, with NO donor, all NO can be accounted for stoichiometrically as nitrate (5.19 ± 0.49 μM) and metHb (4.61 ± 0.57 μM), and nitrite is virtually undetectable (0.29 ± 0.04 μM). Under these conditions, we observe the formation of only very small amounts of SNOHb, 51 ± 1.9 nM with bolus and 39.8 ± 5.9 nM with donor (compared with 11.3 ± 3.3 nM with no treatment). Thus, although there does appear to be a slight increase in SNOHb with bolus compared with donor (consistent with a nitrosative reaction(s) occurring with bolus), the total amount of NO that produces SNOHb (30–40 nM) is much smaller than either the amount that reacts with oxyHb (2–5 μM) or the amount that is responsible for nitrite formation (2.5 μM for bolus NO). Similarly, low nitrosation yields were obtained for other sources of hemoglobin—e.g., lyophilized human Hb from Sigma, recombinant HbA0 (a generous gift from Chris Privalle, Apex Biosciences)—albeit with some of these preparations part of the signal was Hg2+ resistant, indicating the presence of nitrosamine or nitrosylheme species in addition to the formation of nitrosothiol (data not shown).
Discussion
Early studies by Riggs demonstrated that covalent modification of thiols in human and horse hemoglobin results in increased affinity of hemoglobin to O2 and that oxygen binding results in increased thiol reactivity, which in human hemoglobin occurs on the β chains (30, 31). Nitrosation is another form of covalent thiol modification, and so it is not surprising that, like alkylation, nitrosative reactivity of the cysβ93 would be affected by the allosterically controlled exposure of the thiol residue, as demonstrated by Jia et al. (5). However, additional studies by Stamler's group have suggested a more specific and detailed mechanism of nitrosation in hemoglobin that, in addition to thiol accessibility, involves participation of the heme (7–9).
According to this hypothesis, upon transit through the lungs, hemoglobin not only absorbs the O2 required for mitochondrial respiration of tissues but also binds comparatively (to O2) smaller amounts of NO, which subsequently is oxidized and migrates to the cysβ93 thiol (through a yet undefined mechanism), forming the nitrosated species SNOHb. The “capture” of NO is, according to this hypothesis, cooperatively linked to the capture of O2. After transit to relatively hypoxic areas, this process is reversed, with the release of O2 and delivery of NO [presumably in the form of a nitrosothiol (5, 6, 9, 10)] from the SNOHb. Thus, relatively hypoxic tissue beds would be delivered both O2 and NO. In this way, hemoglobin would perform a function as carrier of both O2 and NO, and the major problem of scavenging of NO by its extremely rapid reaction with oxyhemoglobin would be avoided.
Recent evidence has appeared challenging several aspects of this overall idea, including the mechanism(s) of hemoglobin nitrosation and its donation of nitrosonium to acceptor (32, 33). Huang et al. (34) have shown that the rate of nitrosylHb formation is independent of oxygen saturation and that the rate of NO binding to R-state hemoglobin is similar to that for binding to T-state (35), contrary to the conclusions of Gow et al. (8). Gladwin et al. (36) demonstrated that, in contrast to studies with rats exposed to hyperbaric oxygen (6), in the human forearm there is no detectable change in SNOHb levels during the A/V transit. In subsequent work, measurements of intravascular nitrogen oxide levels under basal conditions in humans reveal that, although SNOHb is measurable, A/V gradients were not significant (37). Cannon et al. (38) reported that, during l-N-monomethyl arginine (l-NMMA) infusion, NO inhalation significantly lowered forearm resistance in human volunteers, suggesting transmission of a vasodilator substance through the blood from the lung to the peripheral vasculature. However, neither SNOHb nor plasma nitrosothiol levels changed with NO inhalation. Han et al. (29) have presented evidence that the formation of HbNO is evident only in NO bolus addition and not from NO donors. Finally, Doherty et al. (39) demonstrated that the hypertensive actions of free hemoglobin with various recombinant mutant hemoglobins correlate with their in vitro rate of NO oxidation.
Here, we address another important issue in this controversy—i.e., whether significant amounts of SNOHb are formed in the reaction of NO with hemoglobin under physiological conditions. As pointed out by Gow et al. (8), previous studies examining the very rapid and irreversible reaction of NO with oxyhemoglobin were performed with high ratios of NO/heme. Under more physiological NO/heme ratios, Gow et al. claimed that NO reacts preferentially with the minority unliganded heme, thus preserving NO rather than destroying it. We have reexamined this question, using the conditions of Gow et al. This reexamination was prompted by our realization that, under the conditions of Gow et al., namely, bolus addition of a stock anaerobic NO solution to a solution of, for example, 50 μM oxyhemoglobin, the half-life of the reaction will be so fast (t1/2 = ln 2/(8.9 × 107 M−1⋅s−1 × 20 × 10−5 M) = 39 μs) that substantial reactions may occur before uniform dispersion of the bolus NO solution by agitation. In addition, with an NO concentration of 1.8 mM (which is the concentration in a saturated solution) the pseudo half-life of the autoxidation reaction** (4NO + O2 + 2H2O → 4 H+ + 4NO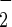 ) will be t1/2 = ln 2/(8 × 106 M−2⋅s−1 × 1.8 × 10−3 M × 2.2 × 10−4 M) = 0.22 s (40), during which time mM concentrations of highly reactive and nitrosative species will be formed in the vicinity of the bolus (29). These calculations reveal the likelihood that highly reactive species are generated during the mixing time of the reaction, and these reactions (including nitrosative species such as N2O3) may account for the formation of products such as SNOHb. We tested this possibility by comparing the results with such bolus addition of NO solution with results obtained with an NO donor that liberates NO uniformly throughout the solution. In our hands, bolus NO addition to an aerobic solution of hemoglobin generates spectral species of uncertain origin, whereas NO donors yield only methemoglobin (Fig. 1). In addition, monitoring of the gradual spectral changes that occur with donor reveals only gradual formation of methemoglobin, with no indication of other species (Fig. 2), as has been reported using other NO donors (27, 28). Also, with bolus NO, substantial amounts of nitrite are formed, and this formation is decreased with increasing hemoglobin (Fig. 3), which indicates that the reaction with hemoglobin competes with the reaction that produces nitrite. The most obvious explanation for this result is that substantial reaction of NO with oxygen occurs with bolus NO; such a reaction will produce nitrosative species such as N2O3, which could nitrosate thiols, including the thiol groups of hemoglobin. Finally, we find that only part of the NO added as a bolus reacts with oxyhemoglobin to produce nitrate and methemoglobin (with substantial amounts of nitrite being formed), but with donor almost all NO added reacts with the oxyhemoglobin (Fig. 4). In either case, the amount of SNOHb that is produced is very small (40–50 nM for 50 μM hemoglobin) and is not appreciably different for bolus compared with donor. We thus are unable to verify the apparently high levels of SNOHb reported by Gow et al. (8) under similar conditions (400 nM for 48 μM hemoglobin). Similar results are obtained with higher concentrations of hemoglobin and NO (29). We suggest that the extent of nitrosative chemistry that takes place with bolus NO addition will be very much influenced by the exact geometry of the solution and how rapidly the solution is agitated after NO addition. With bolus addition, several possible mechanisms may exist to produce nitrosative reactive species, including formation of nitrous anhydride (N2O3) via NO autoxidation, and also formation of nitrite, which reacts with hemoglobin in a complex series of reactions (41). Variabilities in mixing and agitation may well explain differences in the amounts of SNOHb detected in different laboratories, as exemplified by the variabilities we observe in replicate experiments (Fig. 1B).
) will be t1/2 = ln 2/(8 × 106 M−2⋅s−1 × 1.8 × 10−3 M × 2.2 × 10−4 M) = 0.22 s (40), during which time mM concentrations of highly reactive and nitrosative species will be formed in the vicinity of the bolus (29). These calculations reveal the likelihood that highly reactive species are generated during the mixing time of the reaction, and these reactions (including nitrosative species such as N2O3) may account for the formation of products such as SNOHb. We tested this possibility by comparing the results with such bolus addition of NO solution with results obtained with an NO donor that liberates NO uniformly throughout the solution. In our hands, bolus NO addition to an aerobic solution of hemoglobin generates spectral species of uncertain origin, whereas NO donors yield only methemoglobin (Fig. 1). In addition, monitoring of the gradual spectral changes that occur with donor reveals only gradual formation of methemoglobin, with no indication of other species (Fig. 2), as has been reported using other NO donors (27, 28). Also, with bolus NO, substantial amounts of nitrite are formed, and this formation is decreased with increasing hemoglobin (Fig. 3), which indicates that the reaction with hemoglobin competes with the reaction that produces nitrite. The most obvious explanation for this result is that substantial reaction of NO with oxygen occurs with bolus NO; such a reaction will produce nitrosative species such as N2O3, which could nitrosate thiols, including the thiol groups of hemoglobin. Finally, we find that only part of the NO added as a bolus reacts with oxyhemoglobin to produce nitrate and methemoglobin (with substantial amounts of nitrite being formed), but with donor almost all NO added reacts with the oxyhemoglobin (Fig. 4). In either case, the amount of SNOHb that is produced is very small (40–50 nM for 50 μM hemoglobin) and is not appreciably different for bolus compared with donor. We thus are unable to verify the apparently high levels of SNOHb reported by Gow et al. (8) under similar conditions (400 nM for 48 μM hemoglobin). Similar results are obtained with higher concentrations of hemoglobin and NO (29). We suggest that the extent of nitrosative chemistry that takes place with bolus NO addition will be very much influenced by the exact geometry of the solution and how rapidly the solution is agitated after NO addition. With bolus addition, several possible mechanisms may exist to produce nitrosative reactive species, including formation of nitrous anhydride (N2O3) via NO autoxidation, and also formation of nitrite, which reacts with hemoglobin in a complex series of reactions (41). Variabilities in mixing and agitation may well explain differences in the amounts of SNOHb detected in different laboratories, as exemplified by the variabilities we observe in replicate experiments (Fig. 1B).
Even if the major reaction of NO with hemoglobin under aerobic conditions is irreversible oxidation, as we claim, it could be argued that the small minority of NO that is converted to SNOHb could be competent to induce dilation, and this amount is generally believed to be in the low nanomolar range. However, we point out that it is not the amount of SNOHb present in the erythrocyte that is functionally important, but the amount of NO produced from this SNOHb that is able to stimulate soluble guanylate cyclase in the smooth muscle cell, and this points to the importance of the mechanism(s) of transit of the nitrogen oxide group from SNOHb to the abluminal compartment. This mechanism is illustrated schematically in Fig. 5. Because it is only free NO that stimulates guanylate cyclase (42), the determining factor for vasodilation is the concentration of free NO at the smooth muscle cell, and this NO can originate from two sources, the endogenous formation by adjacent endothelial cells or from erythrocytic SNOHb. Stimulation of guanylate cyclase in the smooth muscle cell will be accomplished by the sum of the NO from SNOHb and NO produced by the endothelial cell. Thus, in order for NO originating from SNOHb to have an effect, it must move into the smooth muscle cell at a rate that is competitive with the rate of free NO movement from the adjacent endothelial cell. NO produced by the endothelial cell is freely diffusible, and will have no difficulty in reaching the smooth muscle cell because it is freely permeable to membranes (17). Bound form(s) of NO (denoted “xNO” in Fig. 5) originating from SNOHb, however, must cross four distinct membrane systems (denoted by the solid circles), as well as the red cell-free zone between the red cell layer and the endothelial cell. We consider this scenario highly unlikely. Along these lines, Pawloski et al. (10) have recently reported the presence of vasodilatory species present in erythrocytes and isolated membranes from NO-treated cells. However, erythrocytes were treated with NO under anoxic conditions, and the results we present here would suggest that, under physiologically relevant (aerobic) conditions, NO will be irreversibly consumed when added to erythrocytes, as we (11, 13, 14, 29) have demonstrated previously.
Fig 5.

Scheme depicting free and bound NO movement in the vasculature. RBC, red blood cell; EC, endothelial cell; SMC, smooth muscle cell. xNO denotes the various bound forms of NO; filled circles denote putative transmembrane movement of the bound forms. For details see Discussion.
Although not presented here, it can be shown with a more complete kinetic analysis similar to that in Fig. 5 that, regardless of the rate of movement of xNO, an absolute requirement for effective preservation of NO bioactivity by formation of SNOHb is that the rate of SNOHb must be competitive with the rate of free NO consumption by the irreversible reaction with oxyhemoglobin. We show here that, under physiologically relevant conditions (low levels of NO) within experimental error, all NO reacts with oxyhemoglobin and only extremely small amounts of SNOHb are detectable. There is also previous evidence that this result is true in vivo. Gladwin et al. (36) demonstrated that, with inhaled NO in humans, by far the predominant reaction product is nitrate plus metHb, which is more than 300-fold higher than SNOHb. Deem et al. (43) showed that both oxyhemoglobin and SNOHb perfused through ventilated isolated lung augments hypoxic pulmonary vasoconstriction and results in an 80% fall in exhaled NO. Thus, the predominant action of SNOHb in an intact organ is similar to oxyhemoglobin, namely, consumption rather than production of NO.
There has emerged an alternative explanation to the SNOHb hypothesis for how NO avoids rapid scavenging by oxyhemoglobin. We (11, 13, 14) have shown that, as was demonstrated for oxygen 75 years ago (44), reaction of NO with oxyhemoglobin is nearly 1,000-fold slower when hemoglobin is inside the erythrocyte compared with when the same amount is free in solution. In addition, the existence of an erythrocyte-free zone adjacent to the vessel wall during flow also attenuates NO reaction (3, 4, 13). These two phenomena limit NO consumption by intraluminal erythrocytes, as has been demonstrated experimentally (13, 45). Thus, we feel there is no need to postulate additional phenomena to preserve NO bioactivity. Indeed, there is evidence that irreversible consumption of NO by hemoproteins may perform a protective function against deleterious actions of NO in certain conditions (46–48).
Note Added in Proof.
After submission of this manuscript, we became aware of a paper reporting similar results involving myoglobin (49).
Acknowledgments
We thank Dr. Sanie Mnaimneh for excellent technical assistance. This work was supported, in part, by National Institutes of Health (NIH) Grant HL60302 (to T.B.F.), funds from the Feist Endowment (to M.F.), NIH Grant HL69029 (to M.F.), NIH Grant HL65741 (to J.C.L.), and NIH Grant DK46935 (to J.R.L.). T.R. is a Research Fellow, funded by the Deutsche Forschungsgemeinschaft (DFG; Ra-969 1/1).
Abbreviations
EDRF, endothelium-derived relaxing factor
MAHMA/NO, (Z)-1-{N-methyl-N-[6-(N-methylammoniohexyl)amino]}diazenium 1,2-diolate
This paper was submitted directly (Track II) to the PNAS office.
The rate of NO autoxidation increases exponentially with NO concentration (rate ∝ [NO]2) and thus can be quite rapid at relatively high concentrations while very slow at low concentrations.
References
- 1.Ignarro L. J., Byrns, R. E., Buga, G. M. & Wood, K. S. (1987) Circ. Res. 61, 866-879. [DOI] [PubMed] [Google Scholar]
- 2.Lancaster J. R. (1994) Proc. Natl. Acad. Sci. USA 91, 8137-8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaughn M. W., Kuo, L. & Liao, J. C. (1998) Am. J. Physiol. 274, H1705-H1714. [DOI] [PubMed] [Google Scholar]
- 4.Butler A. R., Megson, I. L. & Wright, P. G. (1998) Biochim. Biophys. Acta 1425, 168-176. [DOI] [PubMed] [Google Scholar]
- 5.Jia L., Bonaventura, J. & Stamler, J. S. (1996) Nature (London) 380, 221-226. [DOI] [PubMed] [Google Scholar]
- 6.Stamler J. S., Jia, L., Eu, J. P., McMahon, T. J., Demchenko, I. T., Bonaventura, J., Gernert, K. & Piantadosi, C. A. (1997) Science 276, 2034-2037. [DOI] [PubMed] [Google Scholar]
- 7.Gow A. J. & Stamler, J. S. (1998) Nature (London) 391, 169-173. [DOI] [PubMed] [Google Scholar]
- 8.Gow A. J., Luchsinger, B. P., Pawloski, J. R., Singel, D. J. & Stamler, J. S. (1999) Proc. Natl. Acad. Sci. USA 96, 9027-9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McMahon T. J., Stone, A. E., Bonaventura, J., Singel, D. J. & Stamler, J. S. (2000) J. Biol. Chem. 275, 16738-16745. [DOI] [PubMed] [Google Scholar]
- 10.Pawloski J. R., Hess, D. T. & Stamler, J. S. (2001) Nature (London) 409, 622-626. [DOI] [PubMed] [Google Scholar]
- 11.Liu X., Miller, M. J., Joshi, M. S., Sadowska-Krowicka, H., Clark, D. A. & Lancaster, J. R., Jr. (1998) J. Biol. Chem. 273, 18709-18713. [DOI] [PubMed] [Google Scholar]
- 12.Vaughn M. W., Kuo, L. & Liao, J. C. (1998) Am. J. Physiol. 274, H2163-H2176. [DOI] [PubMed] [Google Scholar]
- 13.Liao J. C., Hein, W., Vaughn, M. W., Huang, K. T. & Kuo, L. (1999) Proc. Natl. Acad. Sci. USA 96, 8757-8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaughn M. W., Huang, K. T., Kuo, L. & Liao, J. C. (2000) J. Biol. Chem. 275, 2342-2348. [DOI] [PubMed] [Google Scholar]
- 15.Vaughn M. W., Huang, K. T., Kuo, L. & Liao, J. C. (2001) Nitric Oxide 5, 18-31. [DOI] [PubMed] [Google Scholar]
- 16.Huang K. T., Vaughn, M. W., Hyduke, D. R., Van Herle, H., Hein, T. W., Zhang, C., Kuo, L. & Liao, J. C. (2001) Proc. Natl. Acad. Sci. USA 98, 11771-11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lancaster J. R. (2000) in Nitric Oxide: Biology and Pathobiology, ed. Ignarro, L. J. (Academic, San Diego), pp. 209–224.
- 18.Eich R. F., Li, T., Lemon, D. D., Doherty, D. H., Curry, S. R., Aitken, J. F., Mathews, A. J., Johnson, K. A., Smith, R. D., Phillips, G. N. J. & Olson, J. S. (1996) Biochemistry 35, 6976-6983. [DOI] [PubMed] [Google Scholar]
- 19.Herold S., Exner, M. & Nauser, T. (2001) Biochemistry 40, 3385-3395. [DOI] [PubMed] [Google Scholar]
- 20.Antonini E. & Brunori, M., (1971) Hemoglobin and Myoglobin in Their Reactions with Ligands (North–Holland, Amsterdam).
- 21.Thomas D. D., Liu, X., Kantrow, S. P. & Lancaster, J. R., Jr. (2001) Proc. Natl. Acad. Sci. USA 98, 355-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodkey F. L., Hill, T. A., Pitts, L. L. & Robertson, R. F. (1979) Clin. Chem. 25, 1388-1393. [PubMed] [Google Scholar]
- 23.Grisham M. B., Johnson, G. G. & Lancaster, J. R., Jr. (1996) Methods Enzymol. 268, 237-246. [DOI] [PubMed] [Google Scholar]
- 24.Hampl V., Walter, C. L. & Archer, S. L. (1996) in Methods in Nitric Oxide Research, eds. Feelisch, M. & Stamler, J. S. (Wiley, Chichester, U.K.), pp. 309–318.
- 25.Marley R., Feelisch, M., Holt, S. & Moore, K. (2000) Free Radical Res. 32, 1-9. [DOI] [PubMed] [Google Scholar]
- 26.Keefer L. K., Nims, R. W., Davies, K. M. & Wink, D. A. (1996) Methods Enzymol. 268, 281-293. [DOI] [PubMed] [Google Scholar]
- 27.Feelisch M. & Noack, E. A. (1987) Eur. J. Pharmacol. 139, 19-30. [DOI] [PubMed] [Google Scholar]
- 28.Kelm M., Dahmann, R., Wink, D. & Feelisch, M. (1997) J. Biol. Chem. 272, 9922-9932. [DOI] [PubMed] [Google Scholar]
- 29.Han, T. H., Hyduke, D. R., Vaughn, M. W., Fukuto, J. M. & Liao, J. C. (2002) Proc. Natl. Acad. Sci. USA, in press.
- 30.Riggs A. F. (1952) J. Gen. Physiol. 36, 1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riggs A. F. (1961) J. Biol. Chem. 236, 1948-1954. [PubMed] [Google Scholar]
- 32.Patel R. P., Hogg, N., Spencer, N. Y., Kalyanaraman, B., Matalon, S. & Darley-Usmar, V. M. (1999) J. Biol. Chem. 274, 15487-15492. [DOI] [PubMed] [Google Scholar]
- 33.Wolzt M., MacAllister, R. J., Davis, D., Feelisch, M., Moncada, S., Vallance, P. & Hobbs, A. J. (1999) J. Biol. Chem. 274, 28983-28990. [DOI] [PubMed] [Google Scholar]
- 34.Huang Z., Louderback, J. G., Goyal, M., Azzizi, F., King, S. B. & Kim-Shapiro, D. B. (2001) Biochim. Biophys. Acta 1568, 252-260. [DOI] [PubMed] [Google Scholar]
- 35.Huang Z., Ucer, K. B., Murphy, T., Williams, R. T., King, S. B. & Kim-Shapiro, D. B. (2002) Biochem. Biophys. Res. Commun. 292, 812-818. [DOI] [PubMed] [Google Scholar]
- 36.Gladwin M. T., Ognibene, F. P., Pannell, L. K., Nichols, J. S., Pease-Fye, M. E., Shelhamer, J. H. & Schechter, A. N. (2000) Proc. Natl. Acad. Sci. USA 97, 9943-9948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gladwin M. T., Shelhamer, J. H., Schechter, A. N., Pease-Fye, M. E., Waclawiw, M. A., Panza, J. A., Ognibene, F. P. & Cannon, R. O. (2000) Proc. Natl. Acad. Sci. USA 97, 11482-11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cannon R. O., Schechter, A. N., Panza, J. A., Ognibene, F. P., Pease-Fye, M. E., Waclawiw, M. A., Shelhamer, J. H. & Gladwin, M. T. (2001) J. Clin. Invest. 108, 279-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doherty D. H., Doyle, M. P., Curry, S. R., Vali, R. J., Fattor, T. J., Olson, J. S. & Lemon, D. D. (1998) Nat. Biotechnol. 16, 672-676. [DOI] [PubMed] [Google Scholar]
- 40.Ford P. C., Wink, D. A. & Stanbury, D. M. (1993) FEBS Lett. 326, 1-3. [DOI] [PubMed] [Google Scholar]
- 41.Spagnuolo C., Rinelli, P., Coletta, M., Chiancone, E. & Ascoli, F. (1987) Biochim. Biophys. Acta 911, 59-65. [DOI] [PubMed] [Google Scholar]
- 42.Dierks E. A. & Burstyn, J. N. (1996) Biochem. Pharmacol. 51, 1593-1600. [DOI] [PubMed] [Google Scholar]
- 43.Deem S., Gladwin, M. T., Berg, J. T., Kerr, M. E. & Swenson, E. R. (2001) Am. J. Respir. Crit. Care Med. 163, 1164-1170. [DOI] [PubMed] [Google Scholar]
- 44.Hartridge H. & Roughton, F. J. W. (1927) J. Physiol. 62, 232-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lauer T., Preik, M., Rassaf, T., Strauer, B. E., Deussen, A., Feelisch, M. & Kelm, M. (2001) Proc. Natl. Acad. Sci. USA 98, 12814-12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gardner P. R., Gardner, A. M., Martin, L. A. & Salzman, A. L. (1998) Proc. Natl. Acad. Sci. USA 95, 10378-10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hausladen A., Gow, A. J. & Stamler, J. S. (1998) Proc. Natl. Acad. Sci. USA 95, 14100-14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brunori M. (2001) Trends Biochem. Sci. 26, 209-210. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y. & Hogg, N. (2002) Free Radical Biol. Med. 32, 1212-1219. [DOI] [PubMed] [Google Scholar]