

Abstract
Hypoxia-inducible factor-1 (HIF) is a transcription factor central to oxygen homeostasis. It is regulated via its α isoforms. In normoxia they are ubiquitinated by the von Hippel-Lindau E3 ligase complex and destroyed by the proteasome, thereby preventing the formation of an active transcriptional complex. Oxygen-dependent enzymatic hydroxylation of either of two critical prolyl residues in each HIFα chain has recently been identified as the modification necessary for targeting by the von Hippel-Lindau E3 ligase complex. Here we demonstrate that polypeptides bearing either of these prolyl residues interfere with the degradative pathway, resulting in stabilization of endogenous HIFα chains and consequent up-regulation of HIF target genes. Similar peptides in which the prolyl residues are mutated are inactive. Induction of peptide expression in cell cultures affects physiologically important functions such as glucose transport and leads cocultured endothelial cells to form tubules. Coupling of these HIFα sequences to the HIV tat translocation domain allows delivery of recombinant peptide to cells with resultant induction of HIF-dependent genes. Injection of tat-HIF polypeptides in a murine sponge angiogenesis assay causes a markedly accelerated local angiogenic response and induction of glucose transporter-1 gene expression. These results demonstrate the feasibility of using these polypeptides to enhance HIF activity, opening additional therapeutic avenues for ischemic diseases.
Ischemia is a major cause of morbidity and mortality, and effective molecular therapies are being intensively sought (1, 2). The transcription factor hypoxia-inducible factor-1 (HIF) is a master regulator of the hypoxic response, controlling genes involved in diverse processes that balance metabolic supply and demand within tissues (3, 4), making modulation of HIF activity an attractive approach for the treatment of ischemic disease.
Regulation of HIF is mediated at multiple levels via its α chains (5–9). Analysis of the HIFα oxygen-dependent degradation domains (ODD) by transient transfection studies (7, 8, 10–13) suggested a possible specific, alternative approach to HIF stabilization. We have used peptides containing the sites of oxygen-regulated prolyl hydroxylation necessary for proteasomal destruction mediated by the von Hippel-Lindau E3 ubiquitin ligase complex (VHL E3) (14–18). Despite the multiple steps involved in HIF activation, we demonstrate that peptides from two regions of the ODD not only stabilize HIFα but produce a transcriptional response that modulates angiogenesis and metabolism in vivo, suggesting that the peptides affect mechanisms that are common to all activation steps, or that when HIFα chains are present in large excess mechanisms inhibiting transcriptional activation (9, 19) become saturated. These results indicate that these polypeptides, or molecules based on them, are useful tools for studying HIF-mediated responses and ultimately may provide a viable therapeutic approach for ischemic tissues.
Methods
Plasmids, Transient, and Stable Transfections.
Plasmid constructs.
For reporter gene assays DNA encoding the indicated HIF-1α amino acids were generated by PCR and inserted into pCMV/myc/nuc (Invitrogen). pUHD10 (20) and ptat-hemagglutinin (HA) (21) were used to make tet-operator-dependent plasmids and plasmids expressing HIV tat-HIF fusion proteins. P402A and P564G mutants were generated by using a QuikChange kit (Stratagene). All constructs were confirmed by DNA sequencing.
Reporter gene assays.
Cells were cotransfected with an hypoxia response element (HRE) containing reporter gene, pCMV/myc/nuc constructs, and a cytomegalovirus (CMV)-promoted β-galactosidase gene by using Fugene6 (Roche Molecular Biochemicals) (22). Luciferase and β-galactosidase activities were determined after culture in normoxia for 24 hr or in hypoxia for the final 16 hr.
Stably transfected cell lines.
U2OS cells bearing the reverse tetracycline responsive transactivator (20) and a tetKRAB silencer construct (23) were transfected with pUHD/HIF plasmids and individual colonies picked after selection in G418 (1 mg/ml). DoxNODD, doxCODD, and empty vector clones expressed pUHD/HIF-1aa343–417/NLS/c-myc, pUHD/HIF-1aa549–82/NLS/c-myc and pUHD/NLS/c-myc, respectively.
mRNA and Protein Detection.
RNA analysis.
Total RNA was extracted by using RNAzol B (Biogenesis, Poole, U.K.) and analyzed by ribonuclease protection with glucose transporter-1 (Glut-1), carbonic anhydrase IX (CAIX), and small nuclear RNA (internal control) probes (6, 24).
Immunoblotting.
Cell extracts were prepared in buffer (8 M urea/10% glycerol/1% SDS/5 mM DTT/10 mM Tris, pH 6.8), separated by SDS/PAGE and transferred to Immobilon-P membrane (Millipore). Abs against HIF-1α, c-myc tag, and HA tag were from Transduction Laboratories (Lexington, KY), Innogenex (San Ramon, CA), and Roche Molecular Biochemicals, respectively.
Ubiquitination and Interaction Assays.
Ubiquitination assays (25) were performed by using cytoplasmic extracts from empty vector and dox CODD cells induced with 0.8 μg/ml doxycycline (dox) for 48 hr.
VHL E3 interaction assays (16) used 35S-methionine-labeled HIF-1α substrates synthesized in the presence of 100 μM desferrioxamine (DFO) to suppress the prolyl hydroxylase activity of the TnT7 rabbit reticulocyte lysate (Promega). Substrate modification was achieved by incubation with RCC4 cell lysate in the presence of ferrous chloride (100 μM).
Glucose Uptake.
Empty vector and doxCODD cells were exposed to 1% oxygen, 21% oxygen, or 0.8 μg/ml dox for 16 h, washed with glucose-free DMEM, incubated for 10 min with 1 μCi/ml 2-deoxy-d 3H-glucose (Amersham Pharmacia), and uptake was determined by liquid scintillation counting (22).
tat-HIF Protein Synthesis and Purification.
Tat HIF fusion proteins bearing HA and HIS tags to facilitate detection and purification were synthesized in BL21pLysS (Novagen) and purified from sonicates in lysis buffer (0.5% Tween-20/50 mM NaH2PO4/300 mM NaCl/5 mM imidazole) on nickel-nitrilotriacetic acid columns (Qiagen, Chatsworth, CA). Proteins were eluted with 100 mM imidazole, desalted on a PD10 column (Amersham Pharmacia) in 10 mM Tris (pH 7.0) or 10 mM Tris (pH 8.0) and 30 mM KCl, and snap-frozen in liquid nitrogen. Tat-HIF proteins were added to cultures in DMEM/1% FCS at the beginning and 8 hr after starting the experiments and cells harvested after 24 hr.
Angiogenesis Assays.
Tubule formation assay.
The doxCODD or empty vector cells were cocultivated with human microvascular endothelial cells in a ratio of 2:1. DMEM with or without dox (0.8 μg/ml) or epidermal growth factor (5 ng/ml) (Sigma) was renewed every second day. On day 5 the distribution of endothelial cells was determined by using Abs to von Willebrand factor (Dako).
Murine sponge model.
Sterile 8-mm polyurethane sponges were inserted under the dorsal skin of anaesthetized black C57 female mice on day 0. Tat-fusion protein (100 μl of 1 μM) was injected into the sponges on days 1, 2, 4, and 5. Mice were killed on day 7, and sponges with surrounding tissue were excised and fixed in 3.5% paraformaldehyde.
Immunohistochemistry.
Sections (6 μm) of paraffin-embedded sponges were stained with von Willebrand factor (Dako), Glut-1 (Alpha Labs, Eastleigh, U.K.), vascular endothelial growth factor (Santa Cruz Biotechnology), and smooth muscle cell actin (Dako) Abs by using Dako Envision System and Vector Laboratories ABC Vectastain kits.
Results
Overexpression of CODD and NODD Polypeptides Induces HRE-Dependent Reporter Gene Expression.
Because normoxic HIFα degradation is saturable (12) and depends on subregions within the ODD, we tested whether peptides encoding the HIF-1α amino-terminal ODD (NODD) and CODD (16) could affect HIF activity (proposed model outlined in Fig. 1a). Plasmids encoding NODD or CODD linked to nuclear localization sequences and a c-myc epitope tag were transiently cotransfected into U2OS and Hep3B cells with an HRE-dependent luciferase reporter plasmid. Under normoxic conditions expression of either ODD-derived polypeptide increased relative luciferase activity after 24 hr to levels comparable to those induced by hypoxia in the absence of peptide (Fig. 1b). That polypeptide action was mediated via the endogenous HIF pathway and was confirmed by repeating this experiment by using reporter plasmids lacking HREs or in a mutant Chinese hamster ovary cell line lacking HIFα chains (Ka13) and a HIF-1α-complemented transfectant (KH-1) (26). Transfection of the NODD and CODD plasmids led to enhanced luciferase activity only when both HIF and an HRE were present (not shown). We tested shorter fragments of NODD and CODD peptides, defining amino acids 390–417 and amino acids 556–74 as minimal domains capable of HRE-dependent luciferase activation. (Fig. 1 c and d).
Fig 1.

NODD and CODD expression plasmids enhance HRE-reporter gene activity. (a) Proposed model for peptide effects on HIFα–VHL interaction. Degradation is prevented by the NODD and CODD polypeptides competing for prolyl hydroxylation and/or VHL binding, thereby blocking subsequent ubiquitination. (b) Transfections of the NODD or CODD (HIF-1α amino acids 343–417 or amino acids 549–82) led to increased HRE-dependent luciferase activity comparable to hypoxic levels. No induction was seen after transfections with corresponding sequences bearing P402A or P564G mutations. RLU, relative light units. (c and d) Amino acids 390–417 for the NODD and amino acids 556–574 for the CODD were the shortest active domains defined.
HIFα chain degradation depends on recognition by the VHL E3 ubiquitin ligase after oxygen-dependent enzymatic hydroxylation of prolyl residues at positions 402 and 564 (14–16). Using mutated expression plasmids (P402A or P564G), we demonstrated complete ablation of HRE-dependent induction of luciferase activity (Fig. 1b), showing the critical role of these residues for polypeptide function. This finding suggested that expression of the NODD and CODD fusion proteins interfered with degradation of endogenous HIFα chains, most likely by interfering with VHL recognition or prolyl hydroxylation in normoxic cells.
Stable NODD and CODD Polypeptide Expression Results in Endogenous HIF-1α Accumulation.
To explore further the effects of NODD and CODD polypeptides, we stably transfected U2OS cells with dox-inducible NODD (doxNODD) and CODD (doxCODD) constructs encoding identical sequences to those used for transient transfections. Sixteen hours after dox administration, endogenous HIF-1α protein accumulation was detectable after immunoblotting of extracts from both the doxNODD and doxCODD cell lines but not in empty vector transfected U2OS cells (Fig. 2a). The endogenous HIF-1α induction was dox dose (0.2–3.2 μg/ml) and time dependent, peaking after 48 hr. Maximal levels were ≈20% and ≈70% of HIF-1α levels seen after hypoxic or DFO treatment of the doxNODD and doxCODD cells, respectively. Dox did not induce HIF-1α protein in cells transfected with empty vector despite its weak ability to chelate iron.
Fig 2.
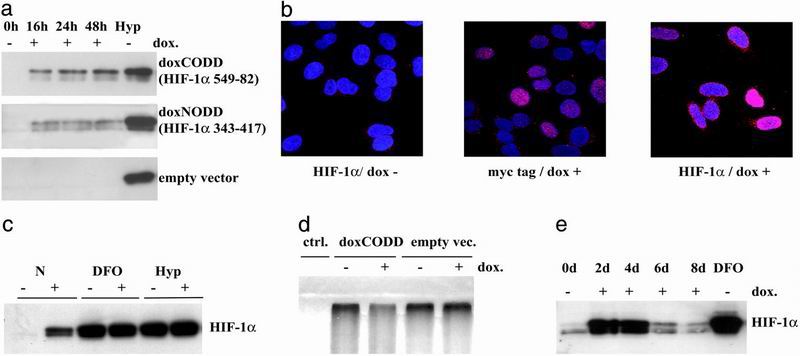
NODD or CODD expression affects endogenous HIF-1α protein levels in stably transfected cells. (a) Extracts of doxNODD (HIF-1α amino acids 343–417), doxCODD (HIF-1α amino acids 549–82), and control cells (empty vector) were immunoblotted for HIF-1α protein. Increased HIF-1α signals were detected from 16 to 48 hr after dox administration in doxNODD and doxCODD cells but not in empty vector cells. Levels of HIF-1α induced by hypoxia are shown for comparison (Hyp). (b) Confocal immunofluorescence analysis of HIF-1α and polypeptides in stable transfectants (doxCODD). The c-myc tag of polypeptides and endogenous HIF-1α signals were both localized in the nuclei. HIF-1α staining intensity varied from cell to cell. (c) Polypeptides had no additional effects on DFO (75 μM) or hypoxia (Hyp; 1% O2)-induced HIF-1α levels. (d) Ubiquitination assays demonstrated reduced ubiquitination of HIF-1α in the presence of dox-treated doxCODD but not of empty vector transfectant cell extracts. (e) Immunoblotting for HIF-1α of doxCODD cells exposed to dox for up to 8 d showed a sharp decline of signals after 4 d. HIF-1α levels induced by 16-hr exposure to DFO (75 μM) are shown for comparison.
Immunofluorescence microscopy allowed visualization of both the c-myc tag of the expressed fusion proteins and endogenous HIF-1α. In dox-activated doxCODD cells both were located in nuclei (Fig. 2b). HIF-1α expression varied considerably from cell to cell.
Combined treatment of doxCODD cells with dox and optimal DFO or hypoxic stimuli did not lead to further increases in HIF-1α signals on immunoblots (Fig. 2c), confirming that the peptides had no additional action when endogenous HIFα chains were fully induced by physiological stimuli.
To test directly whether the polypeptides prevented cellular HIF-1α targeting by the VHL-ubiquitin-proteasome system, we showed that ubiquitination of exogenous 35S-methionine-labeled HIF-1α was markedly reduced in the presence of doxCODD extracts compared to control cell extracts lacking the peptide (Fig. 2d).
HIF and HIF-dependent target gene expression is subject to a number of negative feedback controls. To investigate the consequences of continuous activation of the system, we exposed doxCODD cells to dox for 8 d. HIF-1α protein levels were significantly elevated on days 2 and 4 but decreased thereafter (Fig. 2e). Switching off the system by removing dox from the medium for 48 hr before re-exposure resulted in reinduction of elevated HIF-1α protein levels (data not shown), indicating that the suppressive effects were reversible. Such down-regulation is not seen with all genes expressed under dox control in this cell background, but similar effects also have been observed in cells expressing HIF-1α in a dox-inducible manner (C. C. Wykoff, personal communication). This phenomenon will need to be considered when using the HIF system to modulate complex physiological downstream effects.
NODD and CODD Fusion Proteins Induce Target Gene mRNA and Protein Levels.
Results thus far presented indicate that under normoxic conditions NODD and CODD polypeptide expression results in stabilization of endogenous HIF-1α chains and consequent activation of transiently transfected artificial HRE-dependent promoters. Because expression of natural HIF target genes in chromosomal DNA may be constrained by other factors, we investigated peptide modulation of endogenous genes known to be HIF targets.
Carbonic anhydrase IX (CAIX) is transcriptionally up-regulated under hypoxic conditions (24). We measured CAIX mRNA at intervals after dox treatment in doxCODD and empty vector-transfected cells by ribonuclease protection assay. Dox markedly induced mRNA levels in doxCODD cells after 24 and 48 hr to levels similar to those obtained by hypoxic incubation. Immunoblots demonstrated an associated increase of CAIX protein, paralleling detection of the CODD peptide, visualized by using the c-myc tag (Fig. 3b). To test the generality of this effect, we performed comparable experiments on Glut-1 mRNA expression, obtaining similar results (Fig. 3c). To test for the physiological relevance of increased Glut-1 expression, we conducted glucose uptake experiments. In contrast to control cells, expression of the CODD polypeptides mimicked the effect of hypoxia by inducing glucose uptake in stably transfected cells.
Fig 3.

Induction of HIF-1α target genes. (a) Ribonuclease protection assay. In doxCODD, but not empty vector control cells, CAIX mRNA was induced after 24 hr and peaked after 48-hr dox exposure (0.8 μg/ml). CAIX mRNA levels induced by hypoxic exposure are shown for comparison (Hyp). snRNA, small nuclear RNA-loading control. (b) Immunoblotting for CAIX protein. CAIX protein levels were increased concordant with detection of the c-myc tag of the CODD polypeptide. Hypoxic induction of CAIX protein is shown for comparison. (c) When dox was repeatedly added to cell culture medium (+), Glut-1 mRNA, detected by ribonuclease protection, increased for the first 4 d and then declined in parallel with the HIF-1α protein levels (compare Fig. 2e). Maximal Glut-1 mRNA levels were comparable to those induced after exposure to 75 μM desferrioxamine. (d) 3H-glucose uptake. An enhanced uptake of 3H-glucose was measured in doxCODD cells compared to empty vector cells after 24-hr induction with dox (dox+). In contrast, basal levels (dox−) and hypoxically induced levels (hypoxia) of 3H-glucose uptake were comparable between cell lines. *, P < 0.01; error bars represent the SEM of three replicates.
tat-NODD and tat-CODD Fusion Proteins Enter Cultured Cells and Induce HIF-1α Under Normoxic Conditions.
The experiments presented show that oxygen-dependent gene expression can be modulated in normoxia by plasmid-based expression of NODD and CODD polypeptides. To extend this approach, we chose to study the effects of delivering comparable peptides into cells by using the transduction domain of HIV tat-protein (21).
We performed VHL E3 interaction assays (16) with tat-NODD and tat-CODD, demonstrating their ability to undergo the necessary modifications for interaction with VHL. The 35S-methionine-labeled recombinant polypeptides interacted with VHL (Fig. 4a), supporting the results of the ubiquitination experiments (Fig. 2d). The interaction was enhanced by the presence of cell extracts that promote hydroxylation of the prolines at positions 402 and 564 (14). In contrast, no binding occurred by using peptides in which prolines were mutated (Fig. 4a).
Fig 4.

tat-NODD and tat-CODD polypeptides enter cells and induce HIF-1α protein. (a) VHL E3 ubiquitin ligase interaction assay. 35S-methionine labeled IVTT products of tat-ODD expression vectors were tested for their ability to bind to VHL E3 ligase. Concordant with the ubiquitination assays (Fig. 2d) tat-NODD and tat-CODD polypeptides, but not their corresponding proline mutants [tat-NODDmut(P402A) and tat-CODDmut(P564G)], bound to VHL E3 ubiquitin ligase after modification by cell extracts. Inputs, loading control; c.e., cytoplasmic extract. (b) Immunoblotting for HIF-1α in extracts from tat-polypeptide-treated cells. HIF-1α protein was induced by the tat-NODD and the tat-CODD polypeptides (0.5 μM), but not by the corresponding proline mutants. Maximal levels were comparable to those induced after exposure to 75 μM DFO. (Lower) HIF induction by the indicated doses of tat-CODD and tat-CODDmut and the amounts of surviving intracellular polypeptides detected by the HA tag.
We next tested whether these tat-fusion proteins traversed cell membranes and induced HIF-1α. Two hours after the addition of tat-NODD or tat-CODD fusion proteins to cell cultures, intact peptide was detectable in whole-cell protein extracts by immunoblotting for the HA tag (data not shown). In experiments with repetitive polypeptide administration, endogenous HIF-1α was detectable in normoxia 20 hr after initial exposure of the cells to wild-type fusion proteins but not by using the mutant peptides lacking the prolines (Fig. 4b). We tested effects of different doses of tat-CODD and tat-CODDmut (Fig. 4c). The efficacy of tat-CODD increased sharply at doses between 0.2 and 1 μM whereas tat-CODDmut had little effect at any dose. Blotting for the HA tag showed that at each dose the tat-CODDmut surviving in the cells exceeded tat-CODD, confirming that the lack of action of tat-CODDmut was not a quantitative effect and suggesting that tat-CODD was less stable.
It has been reported that denaturation enhances uptake of tat-fused proteins (21). Denatured tat-NODD and tat-CODD peptides were still able to enter cells but were inactive in mediating HIF-1α up-regulation (not shown), perhaps because they were no longer capable of being hydroxylated.
Endothelial Activation and in Vivo Angiogenesis Assays.
Artificial activation of the HIF-signaling pathway would be predicted to induce angiogenesis. We tested the effect of polypeptide-induced HIF stabilization in an in vitro angiogenesis assay, coculturing human microvascular endothelial cells with empty vector transfected or doxCODD cells. In view of the possibility of sustained activation inducing a negative feedback loop, we opted to test the effects of intermittent induction. In doxCODD, but not in empty vector-transfected cells, intermittent exposure to dox over 5 d led to assembly of endothelial cells into complex tubular structures visualized by immunostaining for von Willebrand factor (Fig. 5a). To extend these observations into an in vivo model, we assayed the effects of injecting tat-fusion proteins into polyurethane sponges implanted s.c. in mice. Intermittent injections on days 1, 2, 4, and 5 produced vessels of increasing density and complexity over days 5–9 (not shown). Further experiments on day 7 (Fig. 5b) showed activity of tat-CODD compared to tat-CODDmut excluding a contribution from the tat component (27). The vessel endothelium within sponges was surrounded by cells expressing smooth muscle actin (results not shown), compatible with the generation of more mature and less leaky vessels than those generated by individual growth factors (28–31). Immunostaining for vascular endothelial growth factor and Glut-1 also showed increased gene expression in tissues surrounding the NODD- or CODD-treated sponges.
Fig 5.
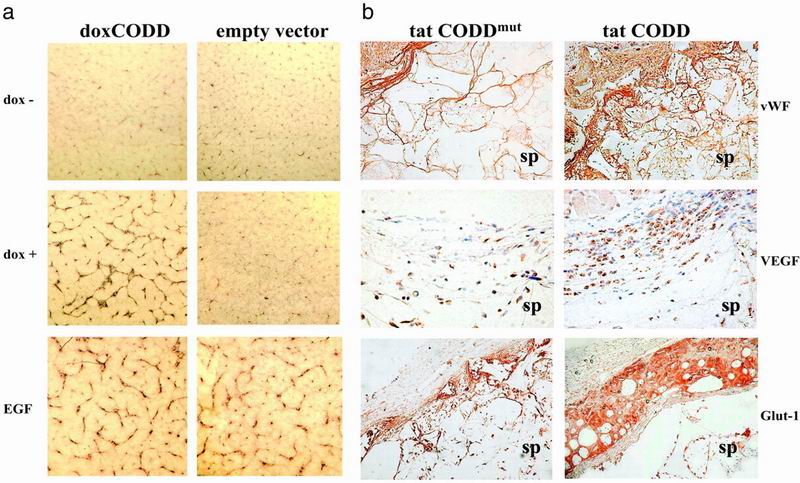
Angiogenesis assays. (a) Tubule assay. Coculture of human microvascular endothelial cells with doxCODD or empty vector transfectants led to activation and alignment of endothelial cells into tubular structures in doxCODD cells, but not in control cells, after 5 d of dox exposure, revealed by immunostaining for von Willebrand factor. Epidermal growth factor (EGF; 5 ng/ml), which is known to induce growth of human microvascular endothelial cells, was used for control assays. (Magnification: a, ×40). (b) Mouse sponge assay. Comparison of tat-CODD and tat-CODDmut treatment in sponges explanted on day 7 revealed increased vessel density (detected by immunohistochemistry for von Willebrand factor, ×100), staining for vascular endothelial growth factor (×400), and Glut-1 (×100) in tat-CODD-treated animals compared to tat-CODDmut controls. Cells surrounding the sponge showed particularly intense staining. sp, sponge. Similar results were obtained with tat-NODD (data not shown).
Discussion
In this study, we describe the use of polypeptides, which stabilize the hypoxia-regulated transcription factor HIF-1α. We provide evidence that complex physiological systems like glucose uptake and angiogenesis can be induced strongly, even under normoxic conditions by local administration. Related molecular approaches to treating ischemic disease include use of single growth factors (2) or gene therapy with HIF-based sequences lacking the degradation domains (28, 32). The approach reported here has advantages over the former in that it co-opts the entire physiological response, resulting in metabolic adaptation as well as angiogenesis, and provides an alternative to gene therapy that should be easier to apply.
Influences of HIF on cancer growth and apoptosis (33, 34) lead to concerns that long-term HIF activation might have deleterious effects, including proneoplastic actions. However, these processes probably require additional events beyond HIF activation and are likely to have a much longer time course than that required for therapeutic angiogenesis. Furthermore, the peptides used here are inherently unstable and act locally, and their effects autoregulate when continuously applied, allowing circumscribed dosing schedules that avoid continued and general activation of the HIF system.
The NODD and CODD polypeptides were effective alone and in combination. Mechanisms of polypeptide action within cells include competition for HIF prolyl hydroxylase activity or VHL-binding capacity. Three lines of evidence suggest the latter is more probable. First, we demonstrate that these NODD and CODD fusion proteins bind to VHL, presumably after their own hydroxylation. Second, the action of either peptide is sufficient to stabilize HIFα even though it contains both prolyl residues, which can be targeted by different hydroxylase isoforms (17). Third, concentrations of synthetic peptide necessary to quench HIF prolyl hydroxylase activity in cell extracts are unlikely to be produced in cells (N.M., unpublished data).
Comparison of the NODD and CODD sequences in the light of recently published structural studies clarifying the nature of the CODD–VHL interaction (35, 36) and perhaps the structures of different prolyl hydroxylase isoforms may allow further refinements to these agents. Use of other protein transduction domains and/or tissue-specific targeting sequences may lead to new formulations with lower risks of side effects (37, 38). However, the polypeptides reported here allow controlled activation of HIF with consequent angiogenic and metabolic effects in normoxia after local administration.
Acknowledgments
We thank S. Dowdy, S. Geley, and C. Wykoff for reagents and S. Peak, D. Watling, L. Nicholls, and K. Yeates for technical assistance. The work was financed by the Wellcome Trust, the Charité, Berlin, and the Medical Research Council.
Abbreviations
HIF, hypoxia-inducible factor
VHL E3, von Hippel-Lindau E3 ubiquitin ligase
ODD, oxygen-dependent degradation domain
CODD, carboxyl-terminal ODD
CAIX, carbonic anhydrase IX
dox, doxycycline
DFO, desferrioxamine
Glut-1, glucose transporter-1
HA, hemagglutinin
HRE, hypoxia response element
NODD, amino-terminal ODD
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Marti H. H. & Risau, W. (1999) Thromb. Haemostasis 82, Suppl. 1, 44-52. [PubMed] [Google Scholar]
- 2.Ferrara N. & Alitalo, K. (1999) Nat. Med. 5, 1359-1364. [DOI] [PubMed] [Google Scholar]
- 3.Semenza G. L. (1998) Curr. Opin. Genet. Dev. 8, 588-594. [DOI] [PubMed] [Google Scholar]
- 4.Li J., Post, M., Volk, R., Gao, Y., Li, M., Metais, C., Sato, K., Tsai, J., Aird, W., Rosenberg, R. D., et al. (2000) Nat. Med. 6, 49-55. [DOI] [PubMed] [Google Scholar]
- 5.Kallio P. J., Okamoto, K., O'Brien, S., Carrero, P., Makino, Y., Tanaka, H. & Poellinger, L. (1998) EMBO J. 17, 6573-6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maxwell P. H., Wiesener, M. S., Chang, G. W., Clifford, S. C., Vaux, E. C., Cockman, M. E., Wykoff, C. C., Pugh, C. W., Maher, E. R. & Ratcliffe, P. J. (1999) Nature (London) 399, 271-275. [DOI] [PubMed] [Google Scholar]
- 7.Salceda S. & Caro, J. (1997) J. Biol. Chem. 272, 22642-22647. [DOI] [PubMed] [Google Scholar]
- 8.Huang L. E., Gu, J., Schau, M. & Bunn, H. F. (1998) Proc. Natl. Acad. Sci. USA 95, 7987-7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lando D., Peet, D. J., Whelan, D. A., Gorman, J. J. & Whitelaw, M. L. (2002) Science 295, 858-861. [DOI] [PubMed] [Google Scholar]
- 10.Srinivas V., Zhang, L. P., Zhu, X. H. & Caro, J. (1999) Biochem. Biophys. Res. Commun. 260, 557-561. [DOI] [PubMed] [Google Scholar]
- 11.Yu F., White, S. B., Zhao, Q. & Lee, F. S. (2001) Cancer Res. 61, 4136-4142. [PubMed] [Google Scholar]
- 12.O'Rourke J. F., Tian, Y. M., Ratcliffe, P. J. & Pugh, C. W. (1999) J. Biol. Chem. 274, 2060-2071. [DOI] [PubMed] [Google Scholar]
- 13.Pugh C. W., O'Rourke, J. F., Nagao, M., Gleadle, J. M. & Ratcliffe, P. J. (1997) J. Biol. Chem. 272, 11205-11214. [DOI] [PubMed] [Google Scholar]
- 14.Jaakkola P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J., Kriegsheim, A., Hebestreit, H. F., Mukherji, M., Schofield, C. J., et al. (2001) Science 292, 468-472. [DOI] [PubMed] [Google Scholar]
- 15.Ivan M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara, J. M., Lane, W. S. & Kaelin, W. G., Jr. (2001) Science 292, 464-468. [DOI] [PubMed] [Google Scholar]
- 16.Masson N., Willam, C., Maxwell, P. H., Pugh, C. W. & Ratcliffe, P. J. (2001) EMBO J. 20, 5197-5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epstein A. C., Gleadle, J. M., McNeill, L. A., Hewitson, K. S., O'Rourke, J., Mole, D. R., Mukherji, M., Metzen, E., Wilson, M. I., Dhanda, A., et al. (2001) Cell 107, 43-54. [DOI] [PubMed] [Google Scholar]
- 18.Bruick R. K. & McKnight, S. L. (2001) Science 294, 1337-1340. [DOI] [PubMed] [Google Scholar]
- 19.Mahon P. C., Hirota, K. & Semenza, G. L. (2001) Genes Dev. 15, 2675-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gossen M., Freundlieb, S., Bender, G., Muller, G., Hillen, W. & Bujard, H. (1995) Science 268, 1766-1769. [DOI] [PubMed] [Google Scholar]
- 21.Nagahara H., Vocero-Akbani, A. M., Snyder, E. L., Ho, A., Latham, D. G., Lissy, N. A., Becker-Hapak, M., Ezhevsky, S. A. & Dowdy, S. F. (1998) Nat. Med. 4, 1449-1452. [DOI] [PubMed] [Google Scholar]
- 22.Vaux E. C., Wood, S. M., Cockman, M. E., Nicholls, L. G., Yeates, K. M., Pugh, C. W., Maxwell, P. H. & Ratcliffe, P. J. (2001) J. Biol. Chem. 276, 44323-44330. [DOI] [PubMed] [Google Scholar]
- 23.Deuschle U., Meyer, W. K. & Thiesen, H. J. (1995) Mol. Cell. Biol. 15, 1907-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wykoff C. C., Beasley, N. J., Watson, P. H., Turner, K. J., Pastorek, J., Sibtain, A., Wilson, G. D., Turley, H., Talks, K. L., Maxwell, P. H., et al. (2000) Cancer Res. 60, 7075-7083. [PubMed] [Google Scholar]
- 25.Cockman M. E., Masson, N., Mole, D. R., Jaakkola, P., Chang, G. W., Clifford, S. C., Maher, E. R., Pugh, C. W., Ratcliffe, P. J. & Maxwell, P. H. (2000) J. Biol. Chem. 275, 25733-25741. [DOI] [PubMed] [Google Scholar]
- 26.Wood S. M., Wiesener, M. S., Yeates, K. M., Okada, N., Pugh, C. W., Maxwell, P. H. & Ratcliffe, P. J. (1998) J. Biol. Chem. 273, 8360-8368. [DOI] [PubMed] [Google Scholar]
- 27.Albini A., Soldi, R., Giunciuglio, D., Giraudo, E., Benelli, R., Primo, L., Noonan, D., Salio, M., Camussi, G., Rockl, W., et al. (1996) Nat. Med. 2, 1371-1375. [DOI] [PubMed] [Google Scholar]
- 28.Elson D. A., Thurston, G., Huang, L. E., Ginzinger, D. G., McDonald, D. M., Johnson, R. S. & Arbeit, J. M. (2001) Genes Dev. 15, 2520-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yancopoulos G. D., Davis, S., Gale, N. W., Rudge, J. S., Wiegand, S. J. & Holash, J. (2000) Nature (London) 407, 242-248. [DOI] [PubMed] [Google Scholar]
- 30.Carmeliet P. & Jain, R. K. (2000) Nature (London) 407, 249-257. [DOI] [PubMed] [Google Scholar]
- 31.Bruick R. K. & McKnight, S. L. (2001) Genes Dev. 15, 2497-2502. [DOI] [PubMed] [Google Scholar]
- 32.Vincent K. A., Shyu, K. G., Luo, Y., Magner, M., Tio, R. A., Jiang, C., Goldberg, M. A., Akita, G. Y., Gregory, R. J. & Isner, J. M. (2000) Circulation 102, 2255-2261. [DOI] [PubMed] [Google Scholar]
- 33.Maxwell P. H., Pugh, C. W. & Ratcliffe, P. J. (2001) Curr. Opin. Genet. Dev. 11, 293-299. [DOI] [PubMed] [Google Scholar]
- 34.Carmeliet P., Dor, Y., Herbert, J. M., Fukumura, D., Brusselmans, K., Dewerchin, M., Neeman, M., Bono, F., Abramovitch, R., Maxwell, P., et al. (1998) Nature (London) 394, 485-490. [DOI] [PubMed] [Google Scholar]
- 35.Hon W.-C., Wilson, M. I., Harlos, K., Claridge, T. D. W., Schofield, C. J., Pugh, C. W., Maxwell, P. H., Ratcliffe, P. J., Stuart, D. I. & Jones, E. Y. (2002) Nature (London) 417, 975-978. [DOI] [PubMed] [Google Scholar]
- 36.Min J. H., Yang, H., Ivan, M., Gertler, F., Kaelin, W. G. & Pavletich, N. P. (2002) Science 296, 1886-1889. [DOI] [PubMed] [Google Scholar]
- 37.Ho A., Schwarze, S. R., Mermelstein, S. J., Waksman, G. & Dowdy, S. F. (2001) Cancer Res. 61, 474-477. [PubMed] [Google Scholar]
- 38.Rajotte D., Arap, W., Hagedorn, M., Koivunen, E., Pasqualini, R. & Ruoslahti, E. (1998) J. Clin. Invest. 102, 430-437. [DOI] [PMC free article] [PubMed] [Google Scholar]