

Abstract
Establishment of the mammalian germ line is a prerequisite for fertility of the adult animal but we know surprisingly little about the molecular mechanisms regulating germ-line development in mammals. Signaling from the c-Kit receptor tyrosine kinase is essential for primordial germ cell (PGC) growth both in vivo and in vitro. Many downstream effectors of the c-Kit signaling pathway have been identified in other cell types but how these molecules control PGC survival and proliferation are unknown. Determination of the c-Kit effectors acting in PGCs has been hampered by the lack of effective methods to easily manipulate gene expression in these cells. We overcame this problem by testing the efficacy of retroviral-mediated gene transfer for manipulating gene expression in mammalian germ cells. We found that PGCs can be successfully infected with a variety of types of retroviruses. We used this method to demonstrate an important role for the AKT kinase in regulating PGC growth. Such technology for manipulating gene expression in PGCs will allow many of the molecular mechanisms regulating germ cell growth, behavior, and differentiation to be comprehensively analyzed.
Primordial germ cell (PGCs) are the embryonic precursors of the gametes of the adult animal. During embryonic development, PGC proliferation is strictly regulated to ensure that enough germ cells are present in the gonad at birth to generate functional gametes (1). Failure of PGCs to proliferate properly can result in reduced fertility or complete sterility (reviewed in ref. 2). Despite the importance of PGCs in the formation of the germ line the molecular mechanisms regulating their growth and survival remain ill-defined. Some of the genes required for PGC survival and proliferation have been identified through analysis of sterile mouse mutants. For example, mutations at the Dominant White Spotting (W) and Steel (Sl) loci identify a receptor tyrosine kinase (c-Kit) and its ligand (Kit ligand or KL) as key regulators of PGC growth and survival (3, 4). Mice carrying mutations at either of these loci can show defective PGC development and may be completely sterile. These data point to a critical role for the c-Kit/KL axis in PGC development. A large number of mutations at the Sl locus have helped define the relationship between KL structure and function in germ-line development. KL is produced as a membrane-bound growth factor that can undergo proteolytic cleavage to generate a soluble form. Importantly, mice lacking membrane-bound KL are severely deficient in PGC numbers and are sterile. The importance of c-Kit signaling in PGC survival is also seen in vitro. When PGCs are cultured in the absence of membrane-bound KL, most of the cells do not survive, mirroring the in vivo situation (5). Alternatively, when PGCs are cultured over cells producing the membrane-bound form of KL, PGC numbers rise for the first 3 days in culture and slowly decrease until they eventually disappear after 10 days (5, 6). Taken together these data suggest a critical role for membrane-bound forms of KL in PGC development. An important question is how KL binding to the c-Kit receptor mediates its effects on PGC growth.
In other cell types, binding of KL causes c-Kit receptor dimerization and receptor autophosphorylation. The activated receptor in turn phosphorylates different substrates, and thus activates distinct signaling cascades or pathways. These include the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR/p70S6K, Ras/mitogen-activated protein kinase kinase (MEK)/mitogen-activated protein kinase (MAPK), the Janus kinase (JAK)/signal transducer and activator of transcription (STAT), and the Src signaling pathways (for review, see refs. 7–9). In this way c-Kit mediates its different effects on cell survival, cell proliferation, cell differentiation, and chemotaxis. It remains to be resolved how the different signaling pathways activated by c-Kit mediate its different functions in different cell types. Although many signaling molecules have been shown to be downstream effectors of the c-Kit receptor in other cell types, which, if any, of these molecules act to effect PGC survival and proliferation remains unknown.
To dissect the role of the c-Kit signaling pathway in PGCs it will be necessary to manipulate components of those pathways within the cell. A major obstacle to studying signaling pathways in PGCs is the difficulty in manipulating the cells by using the tools of modern molecular biology. Introduction of genes into PGCs in vivo should be possible by targeted mutagenesis and transgenesis but such methods will be time consuming and laborious. Transfection of genes into PGCs in vitro has been carried out but gene transduction efficiency is low and the transfection procedures drastically affect PGC viability (10). No further studies using this technology have been reported. To overcome these problems we developed a facile method for introducing genes into murine PGCs by using retroviral vectors. To demonstrate the utility of this system we used retroviral-mediated gene transfer to dissect the intracellular c-Kit signaling pathway in PGCs.
We demonstrate here that one of the key downstream effectors of c-Kit in PGCs is the AKT kinase. Activation of AKT in PGCs is brought about by a c-Kit-dependent pathway. However, AKT activation does not seem to occur, as it does in other cell types, through activation of PI3K. Rather AKT may be activated downstream of c-Kit by other molecules including Src. Our data also suggest an important role for two molecules acting downstream of AKT, mammalian target of rapamycin/FK506-binding protein⋅rapamycin-associated protein (mTOR/FRAP) and p70S6K, in mediating PGC growth in vitro. mTOR/FRAP may be activated in PGCs by both AKT and the Ras/MEK/MAPK pathway. These studies provide a comprehensive dissection of the c-Kit signaling pathway in PGCs. Understanding how this signaling pathway mediates PGC growth should provide a critical insight into the regulation of early germ-line development. These studies also provide a demonstration of retroviral-mediated gene transduction in mammalian PGCs and provide a method for dissecting mechanisms regulating germ cell development in mammals.
Materials and Methods
Mice.
Embryos were derived from B6D2F1 mice purchased from The Jackson Laboratory. In experiments using avian leukosis virus (ALV)-derived vectors, embryos were derived from transgenic mice (β-AKE) expressing the receptor for the subgroup A ALVs (tva) from the constitutive chicken β-actin promoter (11). Most tissues in the β-AKE mice are susceptible to ALV infection, including the germ line, as the provirus can be transmitted as a transgene. Embryos were obtained from timed matings at 8.5 days postcoitum as described (6, 12). All animals were maintained and used under protocols approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University.
Cell Isolation and Culture.
PGC isolation and culture have been described (6, 12). We used two types of feeder cells: STO cells, an embryonic fibroblast cell line that produces membrane-bound KL (6), or bone marrow-derived stromal cells from Sl/Sld mice (BM-Sld) that produce only soluble KL (5). PGCs were plated on wells of 24- or 96-well plates containing mitotically inactive feeder cells. The day of plating was considered day 0. PGCs were cultured in DMEM (GIBCO) with 15% FCS (HyClone), penicillin/streptomycin (GIBCO), l-glutamine (GIBCO), and Na-pyruvate (Sigma). In some instances, cultures were subjected to osmotic shock on the first day of culture to remove PGCs but conserve embryonic somatic cells (13).
Analysis of PGC Growth.
PGCs were cultured in 96-well plates as described above. One day after plating, the cultures were incubated with viruses carrying the green fluorescent protein (GFP) gene (as controls) or viruses carrying the c-AKT and AKT-T308A/473A genes. After 3, 5, or 7 days, PGCs were labeled for the germ cell marker tissue-nonspecific alkaline phosphatase and counted. In some cases cultures were treated with specific pharmacological inhibitors every 24 h from the first day of culture, as follows: PI3K inhibitors, wortmannin (Sigma) at 100 nM for 30 min and LY294002 (Calbiochem) at 10 μm for 30 min; MEK inhibitors, PD98059 (Calbiochem) at 5 μM for 2 h and U0126 (Calbiochem) at 10 μm for 30 min; Src inhibitors, PP2 (Calbiochem) at 20 ng/ml for 1 h and SU6656 (Calbiochem) at 1 μm for 1 h, and mTOR/FRAP signaling inhibitor, rapamycin (Calbiochem) at 50 nM for 30 min. The choice of inhibitor concentration was based on previously published studies (9, 14–19). The experiments were repeated at least twice, and each experiment consisted of at least triplicate cultures. BrdUrd labeling and immunohistochemistry were carried out as detailed (6, 12). Terminal deoxynucleotidyltransferase-mediated dUTP end labeling assays (Oncogene) were carried out according to the manufacturer's instructions with microwave antigen retrieval modifications (20). Results were analyzed for statistical significance by Student's t test.
Retroviral Vectors.
The pBabe-GFP vector [a wild-type murine leukemia virus (MLV)-derived vector] was constructed by modifying the pBabe-puro vector (21, 22) by substituting the puromycin resistance gene (HindIII–ClaI fragment) with the GFP gene. For the construction of pBabe-GFP-c-AKT vector, c-AKT was derived from the Srα vector by EcoRI digestion and introduced into the EcoRI site of the multiple cloning site of pBabe-GFP. The MLV-derived vector LGIN and the murine stem cell virus (MSCV) vector MGIN have been described (23). The ALV-derived vector RCASBP(A)-GFP was derived from pBR plasmids. The Rous sarcoma virus src gene was replaced by a unique ClaI recognition site, where the GFP gene was inserted (11, 24). RCASBP(A)-c-AKT and RCASBP(A)-AKT-T308A/S473A have been described (25).
Production of Retrovirus Stocks.
For pBabe retroviral production, the 293T/17 packaging cell line was transfected with the pBabe vectors together with a vector (pγMLV) encoding the ecotropic MLV envelope. MGIN and LGIN retroviruses were produced in the PA317 amphotropic packaging cells (26). RCASBP(A) retroviruses were produced in the avian fibroblast cell line DF-1. Transient production of retroviruses was carried out as detailed in ref. 27.
Retroviral Infection.
One day after PGC plating, infection with the different viral supernatants was performed. The infection mixture consisted of viral supernatant whose serum concentrations had been adjusted for optimal PGC survival (see above). MLV and MSCV infection mixtures were supplemented with polybrene (5 μg/ml) to increase infection efficiency (28). Cultures were infected daily with viral supernatant mixed 1:1 with medium during the experimental time period (repeated infection method) or undiluted supernatant was centrifuged onto the cultures at 1,800 g for 2 h on the first day of culture (spinoculation method) (26, 28).
Assessment of Retroviral-Mediated Gene Expression.
PGCs were infected with viruses expressing the GFP reporter gene and identified by anti-SSEA-1 immunohistochemistry as detailed (6). GFP-expressing cells were identified and counted under an inverted microscope equipped with fluorescence optics (Nikon). To determine retroviral transduction efficiency at least 100 PGCs were identified and counted in each culture. In some experiments, cultures infected with the c-AKT viruses were subjected to triple indirect immunofluorescence with anti-c-Kit antibody (GIBCO) detected with a rhodamine isothiocyanite-conjugated anti-rat antibody (Santa Cruz Biotechnology) to identify PGCs, anti-hemagglutinin (HA) antibody (Covance, Richmond, CA) and an anti-mouse FITC-conjugated antibody (Sigma) to demonstrate viral transduction of the HA-tagged AKT molecule, and either anti-phospho-AKT (Cell Signaling, Beverly, MA) or anti-phospho-p70S6K (Sigma) detected with an 7-amino-4-methylcoumarin-3-acetic acid-conjugated anti-rabbit antibody (Dakopatts, Glostrup, Denmark) to show activation of AKT or p70S6K.
Results
Kit-Dependent PGC Growth Involves MEK/MAPK and Src, but Not PI3K, and Is Rapamycin-Sensitive.
To study the c-Kit signaling pathway in PGCs we used pharmacological inhibitors of known c-Kit effectors in other cell types. One of the main pathways activated downstream of c-Kit in other cell types is the PI3K pathway. Activation of PI3K can activate AKT, which in turn activates mTOR/FRAP. mTOR/FRAP then activates p70S6K. We found that two potent PI3K inhibitors, wortmannin and LY294002, had no effect on PGC numbers in vitro, suggesting that the PI3K pathway is not important for PGC survival (Fig. 1). However, rapamycin, an inhibitor of mTOR/FRAP signaling, significantly reduced PGC numbers, strongly suggesting that mTOR/FRAP is required for PGC growth (Fig. 1). These apparently contradictory results can be explained if mTOR/FRAP is activated in PGCs by another, PI3K-independent, pathway. In fact, mTOR/FRAP can also be activated by MAPK, a component of the Ras pathway downstream of c-Kit. Therefore, we tested the effect of the MEK/MAPK inhibitors PD98059 and U0126 on PGC growth. PD98059 and U0126 reduced PGC numbers (Fig. 1), suggesting that mTOR/FRAP can be activated by MEK/MAPK. Another molecule that has been described to activate mTOR/FRAP directly is AKT. AKT itself can be activated downstream of c-Kit by PI3K but also by PI3K-independent pathways. In fact, we found that the Src family inhibitors PP2 and SU6656 did reduce PGC numbers in the presence of KL (Fig. 1), suggesting that Src family kinases are involved in the signal transduction pathway downstream of c-Kit in PGCs. These data support the possibility that in PGCs AKT could be activated by PI3K-independent mechanisms including a pathway involving Src. These results also suggest that mTOR/FRAP may be activated downstream of the c-Kit receptor in PGCs by both AKT and MEK/MAPK. Further they indicate an important role for AKT and its downstream effector, mTOR/FRAP, in mediating PGC growth in response to c-Kit signaling.
Fig 1.

Effect of pharmacological inhibitors on PGC growth after 7 days in culture. Data represent the mean ± SEM of between two and six pooled experiments each comprising at least three, and sometimes 18, cultures. *, Difference is significant at the 0.05 level.
Combinations of inhibitors (PD98059, PP2, or rapamycin) did not further reduce PGC numbers, suggesting that MEK/MAPK, Src, and mTOR/FRAP are part of the same signal transduction pathway (data not shown). The fact that the pharmacological inhibitors never caused 100% growth inhibition suggests that there are other important growth signaling pathways in PGCs that have not been examined here. Using BrdUrd incorporation and terminal deoxynucleotidyltransferase-mediated dUTP end labeling we found that the effects of these inhibitors are caused by a 40% decrease in PGC proliferation rather than increased apoptosis (data not shown). In addition, none of the inhibitors affected PGC survival in the absence of membrane-bound KL (not shown), suggesting that Src, MEK/MAPK, and mTOR/FRAP are not activated in PGCs in the absence of c-Kit signaling.
Retroviral Infection of Murine PGCs.
Because inhibitors of AKT are unavailable, we sought other methods to investigate the role of this kinase in PGC growth. To confirm the involvement of AKT in mediating c-Kit-induced survival in PGCs, the direct approach would be to express a dominant-negative form of AKT in PGCs. One way to manipulate gene expression in PGCs is to infect them with retroviral vectors that express specific genes. As a first step to testing this idea, we compared the ability of several retroviral vectors to infect PGCs and drive gene expression. The vectors included those based on MLV, MSCV, and ALV. Because ALV vectors cannot infect normal mouse strains, in experiments using ALV we used a transgenic mouse strain expressing the ALV receptor in all cell types. Each of the vectors contained the enhanced GFP reporter gene. Gene transduction was assayed by monitoring GFP expression in PGCs identified by anti-SSEA-1 staining. PGCs were effectively infected with all of the retroviruses used as were accompanying embryonic somatic cells (Fig. 2, Table 1). GFP-positive PGCs were detectable within 24 h of infection and increased until reaching a maximum between 5 and 7 days after infection. This pattern of retroviral-mediated gene expression likely reflects the necessity for PGCs to transit through M phase for viral integration and for the integrated gene to be expressed. We tested the effects of retroviral envelope type, retroviral long terminal repeat, and method of viral delivery on the infection rate of PGCs. The most efficient method of PGC infection was achieved by repeated infection with ALV vectors or by spinoculation with MLV vectors with an ecotropic envelope (Table 1). As expected, the mitotically inactive feeder cells were not transduced with any of the vectors, because retroviral-mediated gene expression requires retroviral integration (which in turn requires cell cycle progression of the host cell) (not shown). To address potential differences in PGC viability and proliferative activity caused by retroviral infection, parallel-uninfected cultures were assayed simultaneously. Total numbers of PGCs and BrdUrd incorporating PGCs were counted for up to 7 days after retroviral infection. No statistically significant differences in PGC survival or proliferation were seen (Fig. 3). These data demonstrate that viral vectors can be used to express genes in PGCs and that viral infection and viral-mediated gene expression per se has no effect on PGC viability and proliferation.
Fig 2.

GFP expression in retrovirally infected cultures of PGCs 7 days after infection. (A and B) Cultures infected with the ALV-GFP vector. (C and D) Cultures infected with the ecotropic MLV-GFP vector. Infected cells are recognized under fluorescent light by using GFP excitation and emission filters. (B and D) Expression of GFP in PGCs. (B) The same field as A, in which a PGC can be recognized by SSEA-1 antigen staining. (D) The same field as C, labeled for SSEA-1. (A and C) Embryonic somatic cells (SSEA-1-negative) are also infected with both retroviral vectors. (Bar: 14 μm.)
Table 1.
Retroviral transduction efficiencies of mouse gonadal cells
|
|
ALV | MLV eco | MLV amph | MSCV | ||||
|---|---|---|---|---|---|---|---|---|
| Rep | Spin | Rep | Spin | Rep | Spin | Rep | Spin | |
| P | 11.7 | 8 | ND | 15.6 | 0 | 4.6 | 3 | 5.7 |
| S | 16 | 13.8 | ND | 76.7 | 2.8 | 17.9 | 3.1 | 7.4 |
Comparison of transduction efficiencies achieved in PGCs (P) and somatic cells (S) isolated from 8.5 days postcoitum embryos, by different retroviruses and methods of infection (see Materials and Methods). The percentage of infected cells was determined after 7 days of culture. The data are expressed as the mean percent of transduced cells. The experiments were repeated at least four times, and each experiment comprised at least triplicate cultures. ND, Not determined.
Rep, Repeated infection; Spin, spinoculation.
Fig 3.

Effect of retroviral gene delivery on PGC survival and proliferation. (A) Survival of PGCs in culture either uninfected or infected with MLV-derived vectors. (B) Proliferative rates of PGCs in culture either uninfected or infected with ALV-derived vectors. Data represent the mean ± SEM of two pooled experiments, each comprising at least triplicate cultures. No significant differences in PGC survival or proliferation were seen at any time point. P < 0.05.
AKT Can Substitute for KL Signaling in PGCs.
Having demonstrated that viruses can drive gene expression in PGCs and that viral infection per se does not affect PGC survival or proliferation, we tested the possible role of AKT in PGC survival. ALV and MLV vectors were used to express the wild-type form of AKT (c-AKT) in PGCs. Both vectors improved PGC survival either in the absence (Fig. 4 A and B) or the presence of membrane-bound KL (Fig. 4C and data not shown). To further dissect the role of AKT in PGC survival, PGCs were infected with an ALV vector expressing a dominant-negative form of AKT. Expression of the dominant-negative form of AKT (AKT-T308A/S473A) caused a statistically significant decrease in PGC numbers, even in the presence of KL (Fig. 4D). These data strongly suggest that AKT is required for PGC survival at least in vitro. Further, these data suggest that AKT acts downstream of the c-Kit receptor to promote PGC growth and confirm that mTOR/FRAP can be activated by both AKT and MEK/MAPK downstream of c-Kit signaling.
Fig 4.

Effect of AKT expression on PGC survival after 7 days in culture. c-AKT expressed from either an ecotropic MLV-derived vector (A) or an ALV-derived vector (B) improves PGC survival when c-Kit-mediated signaling is not activated (PGCs cultured on BM-Sld cells that do not express membrane-bound KL). (C) ALV-mediated expression of c-AKT also improves PGC survival when c-Kit signaling is activated (PGCs cultured on STO cells that express membrane-bound KL). (D) ALV-mediated expression of a dominant-negative AKT gene (AKT-T308A/S473A) in PGCs with an activated Kit signaling pathway (PGCs cultured on STO cells that express membrane-bound KL) significantly impairs survival. Data represent the mean ± SEM of three pooled experiments each comprising triplicate cultures. *, Difference is significant at the 0.05 level.
AKT Effect on PGC Growth Is Direct and Involves p70S6K.
PGC cultures naturally contain both PGCs and embryonic somatic cells that can be infected with retroviruses. Therefore, an important question is whether the effect of retrovirally expressed genes is direct or mediated via somatic cells. We noted that MLV and ALV infected the same percentage of PGCs but that MLV was much more effective than ALV in infecting somatic cells (MLV 77% vs. ALV 14%) (Table 1). Importantly, when cultures are infected with either MLV or ALV expressing AKT, the effect on PGC survival is equivalent. Therefore, the effect of virus is related to the PGC infection rate and not to the somatic cell infection rate. These data suggest that the effect of viruses on PGCs is direct. To test this notion directly we created cultures in which the PGCs were either susceptible or resistant to infection. We isolated PGCs and somatic cells from mice that can be infected with ALV and placed them into culture. In some of the cultures we killed the PGCs and reseeded the cultures with PGCs from animals that are resistant to ALV infection. We thereby created two types of cultures, one in which the germ cells could be infected with ALV and one in which they could not. In all cultures the somatic cells were susceptible to infection with ALV. When we infected both types of cultures with an ALV vector expressing AKT we observed an increase in PGC numbers only in cultures in which the PGCs were susceptible to infection, supporting the idea that the effect of viruses on PGCs is direct (Fig. 5). In addition, because the AKT in the ALV vector was HA-tagged we were also able to test whether AKT was expressed directly in the PGCs. We found that HA-tagged AKT was indeed expressed in PGCs infected with ALV containing c-AKT (Fig. 6). In addition, we examined whether the expressed AKT was phosphorylated in infected PGCs. We found that both AKT itself and a downstream component of the AKT signaling pathway, p70S6K, were phosphorylated only in the PGCs that were infected (Fig. 6). AKT and p70S6K are likely to be phosphorylated in all PGCs with an activated c-Kit receptor but the levels of expression may be too low for detection with immunocytochemistry. Most likely, only in cells where AKT is overexpressed and the levels of phospho-AKT and phospho-p70S6K are very high can we detect them by immunofluorescence. Taken together our data strongly suggest that the effect of viruses on PGCs in this system is direct. Further, our data demonstrate that p70S6K, a molecule activated by mTOR/FRAP, is activated in PGCs overexpressing AKT. This finding suggests that the AKT pathway in PGCs involves mTOR/FRAP and p70S6K. A summary of the proposed c-Kit signaling pathway in PGCs is shown in Fig. 7.
Fig 5.
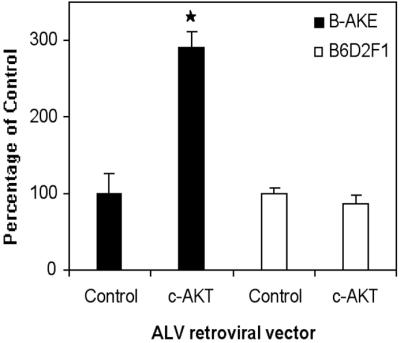
Effect of AKT expression on the survival of β-AKE or B6D2F1 PGCs after 7 days in culture. PGCs and somatic cells were isolated from β-AKE embryos and cultured for 24 h over irradiated STO cells. PGCs were then removed from half of the cultures by osmotic shock, and PGCs and somatic cells isolated from B6D2F1 embryos were plated together with the β-AKE-derived somatic cells in the shocked cultures (empty bars). Cultures were infected with ALV-c-AKT and PGC survival assessed after 7 days. c-AKT improved PGC growth in cultures containing β-AKE PGCs and β-AKE somatic cells as expected (filled bars) but not in the cultures containing B6D2F1 PGCs and both β-AKE and B6D2F1 somatic cells (empty bars). *, Difference is significant at the 0.05 level.
Fig 6.

Analysis of phosphorylation state of AKT (A–C) and p70S6K (D–F) in cultures infected with the avian c-AKT vector. PGCs are recognized by staining with a rat monoclonal anti-c-Kit antibody and a rhodamine isothiocyanite-coupled anti-rat antibody adsorbed with mouse Igs (A and D). Infected cells are recognized by staining for HA-tagged AKT with a mouse mAb and a FITC-coupled anti-mouse Ig adsorbed with rat serum proteins (B and E). Phosphorylated AKT is recognized by staining with a rabbit anti-phospho-AKT antiserum and an anti-rabbit Ig antiserum coupled to 7-amino-4-methylcoumarin-3-acetic acid (C). Phosphorylated p70S6K is recognized by staining with a rabbit anti-phospho-p70S6K and an anti-rabbit antiserum coupled to 7-amino-4-methylcoumarin-3-acetic acid (F). (Bar: 14 μm.)
Fig 7.

Schematic representation of the proposed c-Kit signal transduction pathway in PGCs. Molecules highlighted in bold represent those molecules demonstrated to act downstream of c-Kit in this study. KL produced by somatic cells binds to c-Kit receptor present in the PGC membrane, causing receptor dimerization and autophosphorylation. The activated receptor activates the Ras/MEK/MAPK pathway and Src. Src activates AKT, which together with MAPK activates mTOR/FRAP. mTOR/FRAP in turn activates p70S6K.
Discussion
Signaling from the c-Kit receptor has been demonstrated to be indispensable for PGC growth both in vivo (W and Sl mutants, for review see ref. 29) and in vitro (5, 30). Importantly, long-term PGC survival requires membrane-bound forms of KL. Sld mutant mice, which produce only a soluble form of KL, are sterile in both sexes. Consistent with these data, soluble KL is able to effect short-term PGC growth in culture but is ineffective in supporting long-term survival. To dissect the c-Kit signaling pathway in PGCs we designed experiments using pharmacological inhibitors of components of the c-Kit signaling pathway. Studies in other cells have shown that PI3K is one of the major downstream effectors of c-Kit responsible for mediating cell growth (8). But, in two recent studies, PGC numbers in vivo were shown to be unaffected by mutation of the PI3K binding site on c-Kit (31, 32), suggesting that PI3K is not an important effector of c-Kit signaling in PGCs. Our results showing that PGCs survive similarly in the presence or absence of PI3K inhibitors wortmannin and LY294002 are consistent with these studies. It has been suggested that in most cells that require c-Kit signals there are redundant signaling pathways and that the PI3K pathway is critical only in spermatogenesis and oogenesis. We found that treatment of PGCs with the mTOR/FRAP signaling inhibitor, rapamycin, greatly inhibited their growth. These data demonstrate that one of the known downstream effectors of AKT, mTOR/FRAP (33), is indeed important for PGC growth. Some studies have shown that mTOR/FRAP can be activated directly by other downstream effectors of the c-Kit receptor such as MEK/MAPK (34). Indeed, we found that the MEK inhibitors PD98059 and U0126 inhibited PGC growth in the presence of KL, suggesting that in PGCs activation of mTOR/FRAP downstream of c-Kit may be brought about by MEK/MAPK as well as by AKT (Fig. 7). In most cells AKT activation is brought about by a PI3K-dependent pathway but other studies indicate that AKT can be activated by PI3K-independent pathways (35–39). Our observation that PP2 and SU6656, potent inhibitors of the Src family, inhibit PGC growth suggests that in PGCs AKT may be activated by Src (Fig. 7), as occurs in other cell types (36, 40). Importantly, we found that pharmacological inhibition of MEK/MAPK, Src, and mTOR/FRAP caused a decrease in PGC proliferation rather than increased apoptosis (data not shown).
We tested whether downstream effectors of the c-Kit signaling pathway identified in other cell types could prevent the decrease in PGC numbers induced by loss of c-Kit signaling. Our results demonstrate that overexpression of AKT stimulates PGCs growth on cells that produce only soluble KL. Moreover, a dominant-negative form of AKT inhibited PGC growth on feeder cells expressing membrane-bound KL. Taken together these data strongly suggest that in PGCs AKT acts downstream of the c-Kit receptor and are consistent with studies on other cell types in which the c-Kit signaling pathway has been dissected. Significantly, these hypotheses can now be tested in PGCs by using the retroviral-mediated gene delivery system we describe here.
The observed effects of viruses on PGCs are most likely direct. The growth of PGCs in culture primarily, and possibly solely, depends on the preplated, mitotically inactive feeder cells (5, 6), which cannot be transduced by retroviruses. Moreover, in cultures in which only embryonic somatic cells were susceptible to infection, AKT overexpression gave no improvement in PGC survival whereas in cultures in which the PGCs were also susceptible to infection a dramatic effect on PGC growth was observed. Finally, we found that in PGCs infected with viruses expressing AKT, both AKT itself and some of its downstream effectors were phosphorylated. Taken together these data strongly suggest that viruses act directly in PGCs and not indirectly via somatic cells.
Few of the genes regulating PGC survival and proliferation have been identified in mammals, in part, because of the inability to manipulate PGCs by using many standard techniques of molecular biology that have been applied to other cells. Here we report retroviral-mediated gene delivery to mouse PGCs and further demonstrate the utility of this system for identifying genes involved in regulating PGC growth. This method provides a robust system for studying many aspects of PGC growth and behavior. A single report describing gene delivery to mouse PGCs made use of transfection techniques (10). However, no subsequent studies have made use of this technology presumably because of the drastic effect of transfection on PGC viability. In contrast, retroviral infection of PGCs per se has no effect either on their viability or proliferative activity and genes can be successfully expressed in PGCs after viral integration. Generally we found that higher levels of PGC infection were achieved by spinoculation of virus onto PGCs, the use of an ecotropic envelope, and the use of viruses containing the MSCV long terminal repeat. Therefore, our data suggest that many of the currently available retroviral delivery systems can be used to study PGC development. The ability to target ALV to a selected cell type adds a level of specificity currently unavailable with murine retroviruses. To this end we are currently generating mice in which the ALV receptor tva is expressed exclusively in the germ cell lineage from the promoter of the vasa gene (41) through a knock-in strategy.
Our data demonstrate that many types of retroviruses can now be used to manipulate gene expression in PGCs and to analyze any aspect of PGC behavior that can be studied in vitro. Both feeder layer and organ culture systems have been used to study many aspects of PGC development including cell survival (5, 30), proliferation (30, 42, 43), migration (6, 44), chemotaxis (45), and entry into meiosis (46, 47). The application of gene transfer technology to the study of germ cell development could accelerate efforts to define the molecules required for germ-line development in mammals and for the development of pluripotent stem cells from germ cells.
Acknowledgments
We gratefully acknowledge M. Aoki and P. K. Vogt for the ALV-AKT vectors, S. Hughes for the β−AKE mice, J. Pongubala and P. N. Tsichlis for pBabe vectors, T. Hunter, G. Romano, and U. Rodeck for helpful insights, L. F. Lock for critical reading of the manuscript, and all of the above for helpful advice. This work was sponsored in part by Cancer Core Grant P30CA56036 to the Kimmel Cancer Center.
Abbreviations
PGC, primordial germ cell
KL, Kit ligand
ALV, avian leukosis virus
MLV, murine leukemia virus
MSCV, murine stem cell virus
GFP, green fluorescent protein
Sl, Steel
PI3K, phosphatidylinositol 3-kinase
MAPK, mitogen-activated protein kinase
MEK, mitogen-activated protein kinase kinase
FRAP, FKBP-12-rapamycin-associated protein
HA, hemagglutinin
References
- 1.Wylie C. (1999) Cell 96, 165-174. [DOI] [PubMed] [Google Scholar]
- 2.Donovan P. J., De Miguel, M. P., Hirano, M. P., Parsons, M. S. & Lincoln, A. J. (2001) Int. J. Dev. Biol. 45, 523-531. [PubMed] [Google Scholar]
- 3.Besmer P., Manova, K., Duttlinger, R., Huang, E. J., Packer, A., Gyssler, C. & Bachvarova, R. F. (1993) Development (Cambridge, U.K.) 1993,Suppl., 125-137. [PubMed] [Google Scholar]
- 4.Lev S., Blechman, J. M., Givol, D. & Yarden, Y. (1994) Crit. Rev. Oncog. 5, 141-168. [DOI] [PubMed] [Google Scholar]
- 5.Dolci S., Williams, D. E., Ernst, M. K., Resnick, J. L., Brannan, C. I., Lock, L. F., Lyman, S. D., Boswell, H. S. & Donovan, P. J. (1991) Nature (London) 352, 809-811. [DOI] [PubMed] [Google Scholar]
- 6.Donovan P. J., Stott, D., Cairns, L. A., Heasman, J. & Wylie, C. C. (1986) Cell 44, 831-838. [DOI] [PubMed] [Google Scholar]
- 7.Blume-Jensen P., Janknecht, R. & Hunter, T. (1998) Curr. Biol. 8, 779-782. [DOI] [PubMed] [Google Scholar]
- 8.Rameh L. E. & Cantley, L. C. (1999) J. Biol. Chem. 274, 8347-8350. [DOI] [PubMed] [Google Scholar]
- 9.Ueda S., Mizuki, M., Ikeda, H., Tsujimura, T., Matsumura, I., Nakano, K., Daino, H., Hondaz, I, Z., Sonoyama, J., Shibayama, H., et al. (2002) Blood 99, 3342-3349. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe M., Shirayoshi, Y., Koshimizu, U., Hashimoto, S., Yonehara, S., Eguchi, Y., Tsujimoto, Y. & Nakatsuji, N. (1997) Exp. Cell. Res. 230, 76-83. [DOI] [PubMed] [Google Scholar]
- 11.Federspiel M. J., Bates, P., Young, J. A., Varmus, H. E. & Hughes, S. H. (1994) Proc. Natl. Acad. Sci. USA 91, 11241-11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Miguel M. P. & Donovan, P. J. (2000) Methods Mol. Biol. 137, 403-408. [DOI] [PubMed] [Google Scholar]
- 13.Galdieri M., Zani, B. M., Monaco, L., Ziparo, E. & Stefanini, M. (1983) Exp. Cell Res. 145, 191-198. [DOI] [PubMed] [Google Scholar]
- 14.Feng L. X., Ravindranath, N. & Dym, M. (2000) J. Biol. Chem. 275, 25572-25576. [DOI] [PubMed] [Google Scholar]
- 15.Akishita M., Ito, M., Lehtonen, J. Y., Daviet, L., Dzau, V. J. & Horiuchi, M. (1999) J. Clin. Invest. 103, 63-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah B. H. & Catt, K. J. (2002) Mol. Pharmacol. 61, 343-351. [DOI] [PubMed] [Google Scholar]
- 17.Bowman T., Broome, M. A., Sinibaldi, D., Wharton, W., Pledger, W. J., Sedivy, J. M., Irby, R., Yeatman, T., Courtneidge, S. A. & Jove, R. (2001) Proc. Natl. Acad. Sci. USA 98, 7319-7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vlahos C. J., Matter, W. F., Hui, K. Y. & Brown, R. F. (1994) J. Biol. Chem. 269, 5241-5248. [PubMed] [Google Scholar]
- 19.Favata M. F., Horiuchi, K. Y., Manos, E. J., Daulerio, A. J., Stradley, D. A., Feeser, W. S., Van Dyk, D. E., Pitts, W. J., Earl, R. A., Hobbs, F., et al. (1998) J. Biol. Chem. 273, 18623-18632. [DOI] [PubMed] [Google Scholar]
- 20.Labat-Moleur F., Guillermet, C., Lorimier, P., Robert, C., Lantuejoul, S., Brambilla, E. & Negoescu, A. (1998) J. Histochem. Cytochem. 46, 327-334. [DOI] [PubMed] [Google Scholar]
- 21.Morgenstern J. P. & Land, H. (1990) Nucleic Acids Res. 18, 3587-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitamura T., Onishi, M., Kinoshita, S., Shibuya, A., Miyajima, A. & Nolan, G. P. (1995) Proc. Natl. Acad. Sci. USA 92, 9146-9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng L., Du, C., Murray, D., Tong, X., Zhang, Y. A., Chen, B. P. & Hawley, R. G. (1997) Gene Ther. 4, 1013-1022. [DOI] [PubMed] [Google Scholar]
- 24.De Miguel M. P., Federspiel, M. J. & Donovan, P. J. (2000) in The Testis: From Stem Cell to Sperm Function, ed. Goldberg, E. (Springer, New York), pp. 55–70.
- 25.Aoki M., Batista, O., Bellacosa, A., Tsichlis, P. & Vogt, P. K. (1998) Proc. Natl. Acad. Sci. USA 95, 14950-14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young J. A., Bates, P. & Varmus, H. E. (1993) J. Virol. 67, 1811-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller A. D., Miller, D. G., Garcia, J. V. & Lynch, C. M. (1993) Methods Enzymol. 217, 581-599. [DOI] [PubMed] [Google Scholar]
- 28.Kotani H., Newton, P. B., 3rd, Zhang, S., Chiang, Y. L., Otto, E., Weaver, L., Blaese, R. M., Anderson, W. F. & McGarrity, G. J. (1994) Hum. Gene Ther. 5, 19-28. [DOI] [PubMed] [Google Scholar]
- 29.Donovan P. J. (1994) Curr. Top. Dev. Biol. 29, 189-225. [DOI] [PubMed] [Google Scholar]
- 30.Godin I., Deed, R., Cooke, J., Zsebo, K., Dexter, M. & Wylie, C. C. (1991) Nature (London) 352, 807-809. [DOI] [PubMed] [Google Scholar]
- 31.Blume-Jensen P., Jiang, G., Hyman, R., Lee, K. F., O'Gorman, S. & Hunter, T. (2000) Nat. Genet. 24, 157-162. [DOI] [PubMed] [Google Scholar]
- 32.Kissel H., Timokhina, I., Hardy, M. P., Rothschild, G., Tajima, Y., Soares, V., Angeles, M., Whitlow, S. R., Manova, K. & Besmer, P. (2000) EMBO J. 19, 1312-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nave B. T., Ouwens, M., Withers, D. J., Alessi, D. R. & Shepherd, P. R. (1999) Biochem. J. 344, 427-431. [PMC free article] [PubMed] [Google Scholar]
- 34.Abraham R. T. (1998) Curr. Opin. Immunol. 10, 330-336. [DOI] [PubMed] [Google Scholar]
- 35.Chan T. O., Rittenhouse, S. E. & Tsichlis, P. N. (1999) Annu. Rev. Biochem. 68, 965-1014. [DOI] [PubMed] [Google Scholar]
- 36.Downward J. (1998) Curr. Opin. Cell. Biol. 10, 262-267. [DOI] [PubMed] [Google Scholar]
- 37.Franke T. F., Kaplan, D. R., Cantley, L. C. & Toker, A. (1997) Science 275, 665-668. [DOI] [PubMed] [Google Scholar]
- 38.Virdee K., Xue, L., Hemmings, B. A., Goemans, C., Heumann, R. & Tolkovsky, A. M. (1999) Brain Res. 837, 127-142. [DOI] [PubMed] [Google Scholar]
- 39.Li Y., Dowbenko, D. & Lasky, L. A. (2001) J. Biol. Chem. 276, 20323-20329. [DOI] [PubMed] [Google Scholar]
- 40.Timokhina I., Kissel, H., Stella, G. & Besmer, P. (1998) EMBO J. 17, 6250-6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka S. S., Toyooka, Y., Akasu, R., Katoh-Fukui, Y., Nakahara, Y., Suzuki, R., Yokoyama, M. & Noce, T. (2000) Genes Dev. 14, 841-853. [PMC free article] [PubMed] [Google Scholar]
- 42.Matsui Y., Zsebo, K. & Hogan, B. L. (1992) Cell 70, 841-847. [DOI] [PubMed] [Google Scholar]
- 43.Resnick J. L., Bixler, L. S., Cheng, L. & Donovan, P. J. (1992) Nature (London) 359, 550-551. [DOI] [PubMed] [Google Scholar]
- 44.Stott D. & Wylie, C. C. (1986) J. Cell Sci. 86, 133-144. [DOI] [PubMed] [Google Scholar]
- 45.Godin I. & Wylie, C. C. (1991) Development (Cambridge, U.K.) 113, 1451-1457. [DOI] [PubMed] [Google Scholar]
- 46.McLaren A. & Southee, D. (1997) Dev. Biol. 187, 107-113. [DOI] [PubMed] [Google Scholar]
- 47.Chuma S. & Nakatsuji, N. (2001) Dev. Biol. 229, 468-479. [DOI] [PubMed] [Google Scholar]