

Abstract
Study Objectives
Extensive evidence suggests that histaminergic neurons promote wakefulness. Histaminergic neurons are found exclusively in the tuberomammillary nucleus (TMN), and electrolytic lesions of the posterior hypothalamus, where the TMN resides, produce intense hypersomnolence. However, electrolytic lesions disrupt fibers of passage, and the effects of fiber-sparing, cell-specific TMN lesions on sleep and wakefulness are unknown. Hence, we placed cell-specific lesions in the TMN to determine its role in spontaneous wakefulness.
Design
TMN neurons in rats are relatively resistant to excitotoxins. Hence, we ablated them using saporin conjugated to hypocretin 2, which ablates hypocretin receptor-bearing neurons such as TMN neurons. One to 2 weeks after bilateral injections of Hcrt2-SAP into Sprague-Dawley rats, we correlated loss of TMN neurons with changes in sleep.
Setting
N/A
Participants
N/A
Interventions
N/A
Measurements and Results
Four days after injections with hypocretin-2–saporin, the number of TMN neurons was markedly decreased, and most were lost after 12 days, as determined by immunohistochemistry for adenosine deaminase, a marker of TMN neurons. Nearby nonhistaminergic neurons were similarly ablated. Rats with an average 82.5% loss of TMN cells (determined 2 weeks after injection) did not have marked changes in total sleep amounts compared to saline-treated rats 1 or 2 weeks following the injection, except for a slight decrease in rapid eye movement sleep during the lights-on period for the first week only. The percentage of remaining TMN neurons positively correlated with the average duration of wake bouts during the lights-off period.
Conclusion
The absence of gross changes in sleep after extensive loss of histaminergic neurons suggests that this system is not critical for spontaneous wakefulness.
Keywords: Histamine, tuberomammillary nucleus, hypocretin-2—saporin, lesions, rats
INTRODUCTION
HISTAMINE NEURONS IN THE BRAIN ARE LOCATED EXCLUSIVELY IN THE TUBEROMAMMILLARY NUCLEUS (TMN) OF THE POSTERIOR HYPOTHALAMUS, FROM WHICH THEY PROJECT WIDELY WITHIN THE CENTRAL NERVOUS SYSTEM.1–3 Extensive evidence indicates that histamine has a potent arousing function. Injection of histamine either intracerebroventricularly or directly into various brain sites increases wakefulness.2,3 Posterior hypothalamic neurons in and near the TMN are most active during waking and relatively silent during sleep.4,5 Consistent with this pattern of activity, histamine levels are higher in the brain during wakefulness than during non-rapid eye movement (REM) or REM sleep.6
Although administration of histamine promotes wakefulness, it is not clear whether TMN neurons are necessary for wakefulness. Early lesion studies found dramatic long-lasting hypersomnolence after lesions of the posterior hypothalamus, where the TMN neurons reside.7–9 However, the lesions in these studies probably damaged fibers of passage, as well as hypothalamic areas outside the TMN. To avoid damaging fibers of passage, a more recent lesion study used ibotenic acid to ablate posterior hypothalamic neurons; these cytotoxic lesions produced little change in spontaneous sleep.10 However, this study did not verify whether histochemically identified histaminergic neurons were lost, a critical issue because TMN neurons in rats are relatively resistant to ablation by conventional excitotoxins.11 Most recently, sleep was monitored in mice with low levels of histamine as a result of deletion of the histidine decarboxylase (HDC) gene.12 These mice have a deficit of wakefulness at the beginning of lights-off periods and markedly decreased sleep latencies after novel behavioral stimuli, but they do not have any gross changes in overall levels of spontaneous sleep and wakefulness. 13 Histamine H1-receptor knockout mice also do not have significant changes in the amounts of spontaneous sleep and wakefulness.14 These genetic studies suggest that although the histaminergic system may modulate wakefulness, it may not be critically required for its homeostatic regulation. However, TMN neurons contain a number of other neurotransmitters besides histamine, such as γ-aminobutyric acid and galanin,15 that were not genetically deleted and that might have compensated for the loss of histamine signaling in addition to the developmental compensations commonly seen in knockout animals. Hence, in the present study, we lesioned TMN neurons using a novel saporin-based neurotoxin that binds to the hypocretin receptors that are abundantly expressed on TMN neurons.16 In lesioned rats, we correlated the loss of histaminergic neurons with sleep-wake measurements, to investigate the role of these neurons in regulating spontaneous sleep and wakefulness.
MATERIALS AND METHODS
Animals
The studies were conducted in accordance with the principles and procedures described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals, as well as protocols approved by the Harvard Medical School and Beth Israel Deaconess Medical Center Animal Care and Use committees. Twenty-three male Sprague-Dawley rats (300–400 g) were housed in separate cages in a room with a 12-hour dark/12-hour light cycle, with lights on at 7:00 AM. The temperature in the room was maintained at 21°C to 23°C. Food and water were provided ad libitum.
Experiment 1: Time course of the Effects of Hypocretin-2–Saporin on Adenosine Deaminase-Immunoreactive Neurons
Each rat was anesthetized with chloral hydrate (250 mg/kg, intraperitoneal [IP]) and placed in a stereotaxic frame. To determine the time course of TMN cell loss, we used the toxin hypocretin-2–saporin (Hcrt2-SAP) (50 ng in 0.25 μL of saline; n = 11), made by conjugating the ribosomal toxin saporin to hypocretin-2 (also termed orexin-B). This toxin has previously been shown using fluorescence-activated cell-sorting analysis to bind strongly to cells expressing hypocretin receptors but not to the cells lacking them.17 The Hcrt2-SAP was injected unilaterally into the posterior hypothalamus at the following coordinates: AP = −4.2 mm, ML = 0.7 mm, and DV = 9.3 mm below the dura.18 Saline solution (0.25 μL) was injected contralaterally at the same coordinates. The rats were killed 1 to 3 (n = 5), 4 (n = 3), or 12 (n = 3) days later, and their brains were processed for adenosine deaminase (ADA) and neuron-specific nuclear protein (NeuN) immunostaining.
Experiment 2: Effects of Bilateral Injection of Hcrt2-SAP on Sleep and Wakefulness
To study the effects of TMN lesions on amounts of sleep and wakefulness, Hcrt2-SAP (n = 5) or saline (n = 7) were injected under anesthesia (250 mg/kg chloral hydrate, IP) bilaterally into the posterior hypothalamus in rats that were then implanted with electrodes to record the electroencephalogram (EEG) and electromyogram (EMG). The EEG signals were recorded from 4 miniature stainless-steel screw electrodes (Plastics One, Roanoke, Virg) positioned in the skull to sit on the surface of the cortex. Two of these screws were inserted 2 mm on either side of the midline and 3 mm anterior to bregma (frontal cortex). The other 2 screws were located 2 mm on either side of the midline and 6 mm behind bregma (occipital cortex). The cortical EEG was recorded from 2 screws contralateral to each other (frontal-occipital). To record muscle activity (EMG), 2 flexible multi-stranded wires were inserted in the nuchal muscles. The electrodes were placed in a plastic plug and secured onto the skull using dental cement. The EEG and EMG recordings started 2 days after the surgery and continued for more than 14 days. These rats were killed within 1 month after the surgery.
Antibodies
Rabbit anti-ADA polyclonal (1:10,000, Cat. No. AB1815; Chemicon, Temecula, Calif) and mouse anti-NeuN monoclonal (1:1000, Cat. No. MAB377; Chemicon) primary antibodies were used. The following secondary antibodies were used: biotin-conjugated donkey anti-rabbit IgG (1:500, Cat. No. AP182B; Chemicon) and biotin-conjugated donkey anti-mouse IgG (1:500, Cat. No. AP192B; Chemicon).
Immunohistochemistry
Animals were deeply anesthetized with pentobarbital (150 mg/kg, IP) or chloral hydrate (750mg/kg, IP) and perfused transcardially with 0.9% saline (50 mL) followed by 500 mL of phosphate-buffered 4% paraformaldehyde, pH 7.0 (Formalin solution, Cat. No. HT50-1-128; Sigma, St Louis, MO). The brains were postfixed overnight, equilibrated in 20% to 30% sucrose, and stored at 4°C. Five series of brain sections were cut at 40 μm on a freezing microtome, and 1 series was processed for visualization of ADA. Sections were incubated overnight at room temperature in rabbit anti-ADA polyclonal antibody diluted 1:10,000 in phosphate-buffered saline (PBS) with 0.25% sodium azide. After incubation, sections were rinsed in PBS and transferred into secondary antibody for 1 hour (donkey anti-rabbit IgG, 1:1000 in PBS). After washing, the sections were incubated with an avidin-biotin complex (Vector Laboratories, Burlingame, Calif) for 1 hour, washed again, and reacted in a 1% solution of 3,3′-diaminobenzidine with 0.01% hydrogen peroxide, as well as 0.05% nickel ammonium sulfate to produce a gray-black stain. Some sections were subsequently stained for NeuN. During the following night, these sections were incubated with mouse anti-NeuN monoclonal antibody at room temperature, washed in PBS, and placed in the solution of the secondary antibody for 1 hour (donkey anti-mouse IgG). Then, the sections were washed in PBS, incubated with biotin-conjugated alkaline phosphatase for 1 hour (Vectastain ABC-AP Kit, Cat. No. AK-5000; Vector Laboratories), washed again, and reacted in a working solution of Vector Red substrate (Vector® Red Alkaline Phosphatase Substrate Kit I, Cat. No. SK-5100; Vector Laboratories) to produce a red reaction product. Sections were mounted onto gelatin subbed slides, dried, dehydrated in 100% ethanol, and delipidated in xylenes. Slides were Nissl counterstained and coverslipped using Cytoseal™ XYL mounting medium (Richard-Allan Scientific, Kalamazoo, Mich).
To better visualize ADA-immunoreactive fibers, ADA staining was enhanced in some brain sections using a silver intensification method described previously.19
Analysis of Sleep-Wake States
The EEG and EMG signals were amplified and band-pass filtered (0.3–70 Hz for EEG, 10–70 Hz for EMG) using a Grass polygraph (Astro-Med Inc., West Warwick, RI), and then sampled and digitized at 128 Hz using an analog-digital converter (National Instruments, Austin, Tex). Twenty-four hour EEG and EMG recordings obtained on the sixth and 14th day after injection were scored manually on a computer (Icelus software, Mark Opp, Ann Arbor, Mich) in 12-second epochs for wakefulness, REM sleep, and non-REM sleep. Wakefulness was identified by the presence of a desynchronized EEG and high EMG activity. Non-REM sleep consisted of high-amplitude slow waves together with a low EMG tone relative to waking. REM sleep was identified by a desynchronized EEG and/or theta activity coupled with a near-absence of EMG activity. The amount of time spent in wakefulness, non-REM, and REM sleep was determined for each hour. Mean bout durations for wakefulness, NREM, and REM sleep were determined by dividing the total time spent in each state by the number of episodes of that state.
Cell counts
TMN cell loss was determined by counting the number of ADA-immunoreactive neurons throughout the entire normal hypothalamic range of these neurons. In particular, we counted cells in coronal sections ranging from the level of the dorsomedial hypothalamic nucleus to the caudal edge of the hypothalamus, thereby counting both the dorsal and ventral subgroups of TMN neurons.
Statistical Analysis
Analysis of variance and t tests with Bonferroni correction (where appropriate) were used to compare changes in sleep parameters (SYSTAT, Version 8.0, SPSS Inc., Chicago, Ill, 1998). The same statistical methods were also used to compare counts of ADA-immunoreactive cells. Statistical significance was evaluated at the P < .05 level.
RESULTS
Hcrt2-SAP Lesions
Eleven rats received unilateral injections of Hcrt2-SAP into the TMN, to determine the time course of TMN cell loss, while 5 rats received bilateral injections of Hcrt2-SAP, to determine the effects of cell loss on sleep-wake behaviors. In normal rats, nearly all TMN neurons are immunoreactive for ADA,20 and hence we used ADA-immunoreactivity to identify histaminergic neurons.
Time Course of the Effects of Hcrt2-SAP on ADA-immunoreactive Neurons
Twelve days after Hcrt2-SAP injections, most rats showed greatly reduced numbers of ADA-expressing TMN neurons (Figure 1). Nissl and NeuN stains in these cases also showed very few neuronal profiles remaining in and around the injection site, confirming that TMN neurons were indeed ablated and not merely failing to produce ADA protein. Neurons just outside the TMN were also gone, possibly because these neurons also express hypocretin receptors21; hence, Hcrt2-SAP does not exhibit specificity for the TMN neurons, relative to other nearby cell groups. The loss of ADA-immunoreactive neurons was time dependent (Figure 2): rats killed on day 1, 2, or 3 after injection had little loss of ADA-immunoreactive neurons, while there was a significant decrease (63%) in the number of ADA-immunoreactive neurons by day 4 after the injection (paired t test with contralateral side, t = −2.82; df = 4; P < .05). By 12 days, there was an 89.5% loss of ADA-immunoreactive neurons (paired t test, t = −13.74; df = 4; P < .001). Results of the present study as well as our previous report17 indicate that Hcrt2-SAP destroys most ADA-positive neurons within 12 days after being injected.
Figure 1.
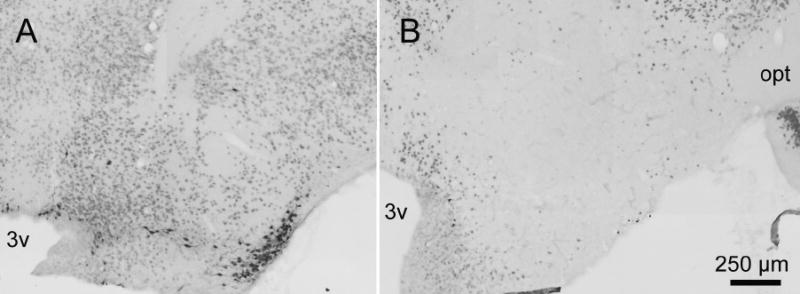
Effects of hypocretin-2–saporin injection into the posterior hypothalamus on adenosine deaminase and neuron-specific nuclear protein immunoreactive neurons. Panel A represents a section through the tuberomammillary nucleus of a rat injected with saline; panel B represents a similar section from a rat treated with hypocretin-2–saporin. Most of the neurons containing adenosine deaminase (black staining) as well as most containing neuron-specific nuclear protein-immunoreactive neurons (red staining) were destroyed by hypocretin-2–saporin. 3v refers to the third ventricle; opt, optic tract.
Figure 2.
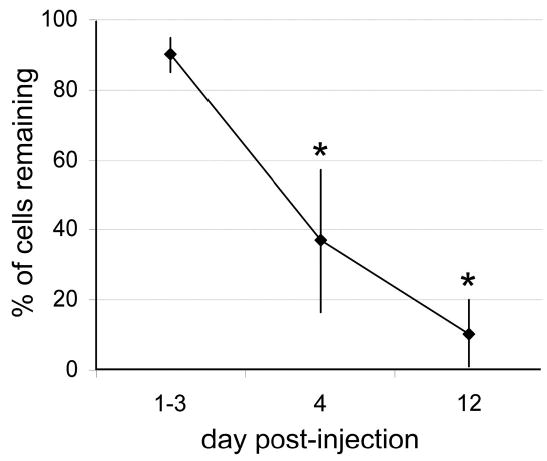
Time course of the effects of hypocretin-2–saporin on immunoreactive neurons in the posterior hypothalamus. A unilateral injection of hypocretin-2 saporin was made to the posterior hypothalamus, and rats were killed 1 to 3, 4, or 12 days later. The contralateral side injected with saline served as a control. There was a time-dependent loss of adenosine deaminase-immunoreactive neurons in the injected tuberomammillary nucleus. *P < .05.
Effects of Bilateral Injection of Hcrt2-SAP on Sleep and Wakefulness
Immunostaining for ADA performed at the end of experiment demonstrated that bilateral injections of Hcrt2-SAP produced an average loss of 82.5% ± 4.1% (n = 5%, range 72%–93% loss) of ADA-immunoreactive neurons 2 weeks after the injection, compared to rats injected with saline (n = 7). ADA-immunoreactive neurons were almost equally lesioned in all histaminergic subnuclei (Figure 3). The lesions did not extend far beyond the distribution of histaminergic neurons in the posterior hypothalamus. In 2 rats (rats 220 and 227), the lesions extended rostrally to a level where a few hypocretin-containing neurons are located. The density of ADA-immunoreactive fibers and varicosities in the hypothalamus was also markedly reduced (data not shown). Sleep data were obtained 1 and 2 weeks after the injections were made. Both Hcrt2-SAP and saline-injected rats demonstrated a clear diurnal distribution of sleep amounts, exhibiting more sleep during the lights-on period than during the lights-off period (Figure 4). The only significant difference between the groups was a decreased amount of REM sleep during the lights-on period in Hcrt2-SAP-treated rats 1 week following the injection (25.0% ± 7.2% decrease, P = 0.04). There were no significant differences in NREM or REM sleep amounts 2 weeks after injection between the saline- and Hcrt2-SAP-treated rats.
Figure 3.
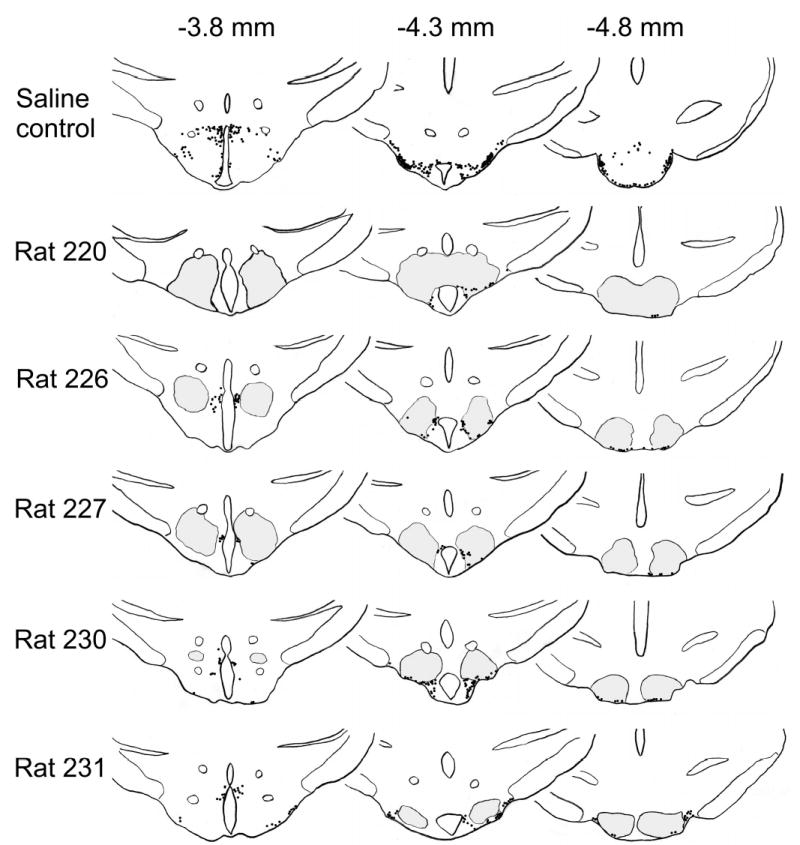
Schematic drawings demonstrating extent of the lesion areas (gray) as well as distribution of adenosine deaminase-immunoreactive neurons (black dots) in the rats treated with hypocretin-2–saporin. The sections are shown at 3 levels: 3.8 mm, 4.3 mm, and 4.8 mm posterior to bregma.18 Lesion brain areas were identified using Nissl stain. Distribution of adenosine deaminase-immunoreactive neurons in a control rat injected with saline is shown at the top.
Figure 4.
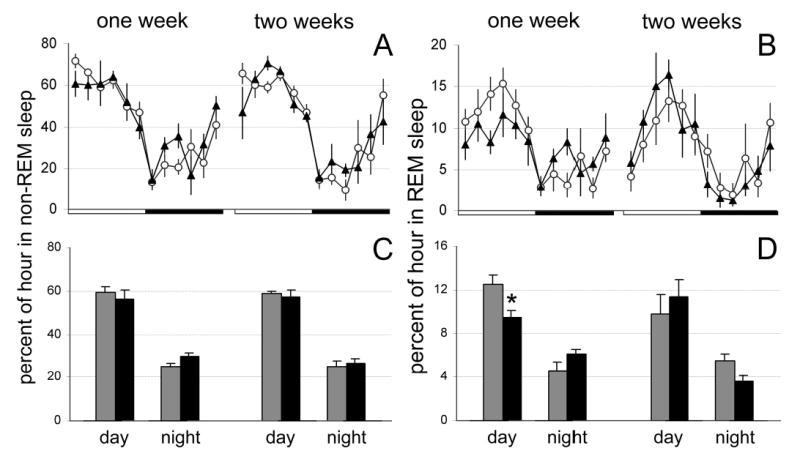
Mean (± SEM) percentage of non-rapid eye movement and rapid eye movement sleep during 24 hours in rats administered hypocretin-2–saporin (closed triangles) or saline (open circles) in the posterior hypothalamus (A and B). The 24 hours are represented in 2-hour blocks. The dark bar represents the 12-hour lights-off period. C and D show average amounts of sleep calculated during 12-hour lights-on or lights-off periods in rats administered hypocretin-2–saporin (black bars) or saline (gray bars). *P < .05. REM refers to rapid eye movement.
The Hcrt2-SAP-treated rats also did not have changes in wake-bout durations compared to saline-treated rats (1 week after surgery, Hcrt2-SAP = 7.9 ± 1.0 min, saline = 10.8 ± 1.9 min, i = −1.14, P = 28; 2 weeks after surgery Hcrt2-SAP = 9.9 ± 1.0 min; saline = 10.1 ± 2.1 min, t = −0.07, P = .9). However, there was a significant positive correlation between the percentage of remaining ADA-positive neurons and the duration of wake bouts during the lights-off period in Hcrt2-SAP-lesioned rats (r = 0.891, P < .05, n = 5) (Figure 5). The correlation was not significant when the control group was included in the analysis.
Figure 5.
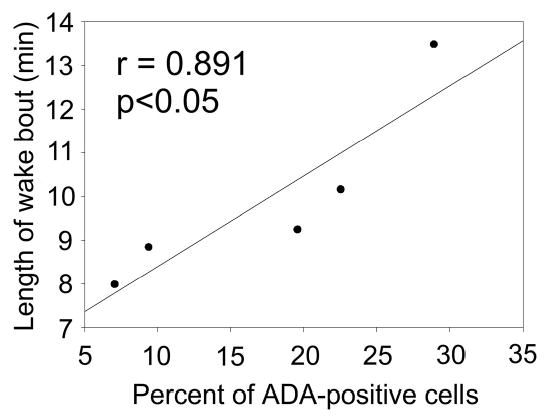
Positive correlation between the percentage of remaining adenosine deaminase-positive neurons and the duration of wake bouts during the lights-off period in rats lesioned with hypocretin-2–saporin.
Although the percentage of remaining ADA neurons ranged from 7% to 28% in Hcrt2-SAP-treated rats, we found little difference in total sleep amounts between rats in this group. Moreover, the daytime and nighttime levels of sleep in 2 rats with a greater than 90% loss of ADA-immunoreactive neurons were similar to sleep levels in control rats.
DISCUSSION
TMN neurons have been difficult to destroy using conventional excitotoxins, which typically ablate less than 40% of TMN neurons even at high doses.11 In the present study, we used the neurotoxin Hcrt2-SAP to lesion up to 82.5% of TMN histamine neurons. TMN neurons represent a very good target for Hcrt2-SAP because hypocretin-2 receptors are strongly expressed in the TMN.16 The same lesions, however, also ablate most of the neurons in adjacent cell groups in the posterior hypothalamus. Because injections of saporin alone, without a binding ligand, are not effective in killing neurons,22 we hypothesize that orexin receptors may be widespread in this region.
Rats with an average loss of 82.5% of histaminergic neurons showed no significant reductions in the total amount of wakefulness 2 weeks following injections of Hcrt2-SAP (Figure 2). Although seemingly at odds with early fiber-disrupting lesion studies, these findings are consistent with the lack of gross changes in spontaneous sleep-wake amounts in HDC-null mice, which show a significant reduction in brain histamine levels.12 Moreover, rats treated with the histamine H3 receptor agonist, immepip, which produces a rapid decrease in cortical histamine levels, do not have any marked changes in spontaneous amounts of sleep and wakefulness.23 When taken together, these results suggest that homeostatic regulation of sleep does not require the histaminergic neurons. However, because neurons take several days to die in the current experiment, our results do not rule out a major role for TMN neurons in moment-to-moment regulation of spontaneous arousal. On the other hand, these findings do suggest that the marked hypersomnolence seen in early hypothalamic lesion studies7–9 was due to additional damage outside of the TMN—either to fibers of passage, other cell groups, or both.
Although TMN neurons may not be critically required for spontaneous arousal, they may nevertheless play other important roles in promoting wakefulness. They may awaken the animal at specific times of the day, since HDC-null mice show decreased wakefulness during the early part of the lights-off period. In the present study, such an effect was not found, possibly because our sample size was not large enough to detect this relatively mild effect and possibly also because not all of the TMN neurons were destroyed in our study. The TMN might also critically promote arousal in response to environmental challenges, such as novelty, because HDC-null mice lack the normally robust increase in wakefulness seen when animals are placed into novel cages.12 In addition, it is possible that the TMN may be more important in the control of spontaneous wakefulness in species other than rats. For example, inactivation of posterior hypothalamic neurons by muscimol microinjections, which do not destroy fibers of passage, causes hypersomnia in cats.24 Alternately, the hypersomnolence seen in cats after muscimol injections could be due to the rapid action of muscimol, which inactivates neurons within minutes, whereas in the current study, cell death occurred over several days. Hence, it is possible the compensatory effects emerged in the present study that would not have been possible in the earlier study in cats.
TMN neurons receive a heavy projection from sleep-active neurons in the ventral lateral preoptic (VLPO) area.25 VLPO neurons are sleep active25 and may also promote sleep because cytotoxic lesions of this area cause long-lasting insomnia.26 Because VLPO neurons express the inhibitory neurotransmitters γ-butyric acid and galanin,27 we had hypothesized that VLPO neurons could promote sleep via inhibition of TMN neurons. However, the results of the present study and the data from the HDC-null mice suggest that this pathway is not critical for the homeostatic regulation of sleep. However, this pathway may still critically inhibit wakefulness under more specific conditions that we did not examine, eg, in response to novelty. They may also sustain wakefulness during cataplectic attacks in narcoleptic humans and canines, since putative histamine neurons are active during the cataplexy in narcoleptic dogs.28
In addition to histaminergic neurons in the TMN, there are several other neuronal groups implicated in arousal in the brain. They include hypocretin neurons in the lateral hypothalamus, cholinergic neurons in the basal forebrain and mesopontine tegmentum, noradrenergic neurons in the locus coeruleus, and serotonergic neurons in the dorsal raphe.29 VLPO neurons project to all of these brain areas,27 raising the possibility that spontaneous sleep requires the simultaneous inhibition of multiple arousal systems. In fact, it has been suggested that these multiple arousal systems are redundant, so that no one system is absolutely necessary for the occurrence of waking.29 Therefore, loss of TMN neurons could be largely compensated for by activity of other arousal systems. An alternate view that is also consistent with the current results is that various arousal systems promote arousal during different times or situations and that loss of particular arousal systems may cause selective deficits in wakefulness instead of global changes in levels of wakefulness. Future studies using multiple lesions of the TMN and other brain areas implicated in arousal are needed to clarify the roles of different arousal systems in waking and sleep.
Acknowledgments
Supported by NIH grants NS30140, AG09975, AG15853, MH55772 and Medical Research Service of the Department of Veterans Affairs.
Footnotes
Disclosure Statement This is not an industry supported study. Drs. Gerashchenko, Chou, Blanco-Centurion, Saper, and Shiromani have indicated no financial conflicts of interest.
References
- 1.Panula P, Pirvola U, Auvinen S, Airaksinen MS. Histamine-immunoreactive nerve fibers in the rat brain. Neuroscience. 1989;28:585–610. doi: 10.1016/0306-4522(89)90007-9. [DOI] [PubMed] [Google Scholar]
- 2.Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol. 2001;63:637–72. doi: 10.1016/s0301-0082(00)00039-3. [DOI] [PubMed] [Google Scholar]
- 3.Haas H, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci. 2003;4:121–30. doi: 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- 4.Steininger TL, Alam MN, Gong H, Szymusiak R, McGinty D. Sleep-waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res. 1999;840:138–47. doi: 10.1016/s0006-8993(99)01648-0. [DOI] [PubMed] [Google Scholar]
- 5.Vanni-Mercier G, Gigout S, Debilly G, Lin JS. Waking selective neurons in the posterior hypothalamus and their response to histamine H3-receptor ligands: an electrophysiological study in freely moving cats. Behav Brain Res. 2003;144:227–41. doi: 10.1016/s0166-4328(03)00091-3. [DOI] [PubMed] [Google Scholar]
- 6.Strecker RE, Nalwalk J, Dauphin LJ, et al. Extracellular histamine levels in the feline preoptic/anterior hypothalamic area during natural sleep-wakefulness and prolonged wakefulness: an in vivo microdialysis study. Neuroscience. 2002;113:663–70. doi: 10.1016/s0306-4522(02)00158-6. [DOI] [PubMed] [Google Scholar]
- 7.Von Economo C. Sleep as a problem of localization. J Nerv Ment Dis. 1930;71:249–59. [Google Scholar]
- 8.Ranson SW. Somnolence caused by hypothalamic lesions in the monkey. Arch Neurol Psychiat. 1939;41:1–23. [Google Scholar]
- 9.Nauta WJH. Hypothalamic regulation of sleep in rats. An experimental study J Neurophysiol. 1946;9:285–316. doi: 10.1152/jn.1946.9.4.285. [DOI] [PubMed] [Google Scholar]
- 10.Denoyer M, Sallanon M, Buda C, Kitahama K, Jouvet M. Neurotoxic lesion of the mesencephalic reticular formation and/or the posterior hypothalamus does not alter waking in the cat. Brain Res. 1991;539:287–303. doi: 10.1016/0006-8993(91)91633-c. [DOI] [PubMed] [Google Scholar]
- 11.Yanai K, Zhao XL, Watanabe T. Excitotoxic lesions of histaminergic neurons by excitatory amino acid agonists in the rat brain. Neurosci Lett. 1997;232:159–62. doi: 10.1016/s0304-3940(97)00609-5. [DOI] [PubMed] [Google Scholar]
- 12.Ohtsu H, Tanaka S, Terui T, et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett. 2001;502:53–6. doi: 10.1016/s0014-5793(01)02663-1. [DOI] [PubMed] [Google Scholar]
- 13.Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS. Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J Neurosci. 2002;22:7695–711. doi: 10.1523/JNEUROSCI.22-17-07695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang ZL, Qu WM, Li WD, et al. Arousal effect of orexin A depends on activation of the histaminergic system. Proc Natl Acad Sci U S A. 2001;98:9965–70. doi: 10.1073/pnas.181330998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Airaksinen MS, Alanen S, Szabat E, Visser TJ, Panula P. Multiple neurotransmitters in the tuberomammillary nucleus: comparison of rat, mouse, and guinea pig. J Comp Neurol. 1992;323:103–16. doi: 10.1002/cne.903230109. [DOI] [PubMed] [Google Scholar]
- 16.Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001;21:9273–9. doi: 10.1523/JNEUROSCI.21-23-09273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerashchenko D, Kohls MD, Greco M, et al. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J Neurosci. 2001;21:7273–83. doi: 10.1523/JNEUROSCI.21-18-07273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic Press, 1986.
- 19.Chou TC, Bjorkum AA, Gaus SE, Lu J, Scammell TE, Saper CB. Afferents to the ventrolateral preoptic nucleus. J Neurosci. 2002;22:977–90. doi: 10.1523/JNEUROSCI.22-03-00977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Senba E, Daddona PE, Watanabe T, Wu JY, Nagy JI. Coexistence of adenosine deaminase, histidine decarboxylase, and glutamate decarboxylase in hypothalamic neurons of the rat. J Neurosci. 1985;5:3393–402. doi: 10.1523/JNEUROSCI.05-12-03393.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marcus JN, Aschkenasi CJ, Lee CE, et al. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- 22.Gerashchenko D, Blanco-Centurion C, Greco MA, Shiromani PJ. Effects of lateral hypothalamic lesion with the neurotoxin hypocretin-2-saporin on sleep in Long-Evans rats. Neuroscience. 2003;116:223–35. doi: 10.1016/s0306-4522(02)00575-4. [DOI] [PubMed] [Google Scholar]
- 23.Lamberty Y, Margineanu DG, Dassesse D, Klitgaard H. H3 agonist immepip markedly reduces cortical histamine release, but only weakly promotes sleep in the rat. Pharmacol Res. 2003;48:193–8. doi: 10.1016/s1043-6618(03)00094-x. [DOI] [PubMed] [Google Scholar]
- 24.Lin JS, Sakai K, Vanni-Mercier G, Jouvet M. A critical role of the posterior hypothalamus in the mechanisms of wakefulness determined by microinjection of muscimol in freely moving cats. Brain Res. 1989;479:225–40. doi: 10.1016/0006-8993(89)91623-5. [DOI] [PubMed] [Google Scholar]
- 25.Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–9. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- 26.Lu J, Greco MA, Shiromani P, Saper CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000;20:3830–42. doi: 10.1523/JNEUROSCI.20-10-03830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–21. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.John J, Wu MF, Boehmer LN, Siegel JM. Cataplexy-active neurons in the hypothalamus; implications for the role of histamine in sleep and waking behavior. Neuron. 2004;42(4):619–34. doi: 10.1016/s0896-6273(04)00247-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones BE. Arousal systems. Front Biosci. 2003;8:S438–51. doi: 10.2741/1074. [DOI] [PubMed] [Google Scholar]