

Abstract
Background
We investigated whether decreased myofilament calcium contractile activation may, in part, contribute to heart failure.
Methods and Results
Calcium concentration required for 50% activation and Hill coefficient for fibers from nonfailing and failing human hearts at pH 7.1 were not different. Maximum calcium-activated force (Fmax) was also not different. However, at pH 6.8 and 6.9, differences were seen in myofilament calcium activation between nonfailing and failing hearts. At lower pH, failing myocardium was shifted left on the calcium axis compared with nonfailing myocardium, which suggested an increase in myofilament calcium responsiveness. Increased inorganic phosphate concentration decreased maximal force development by 56% in nonfailing and 36% in failing myocardium and shifted the calcium-force relationship by 2.01±0.22 versus 0.86±0.13 μmol/L, respectively (P<0.05). Addition of cAMP resulted in a 0.56 μmol/L shift toward higher intracellular calcium concentrations in nonfailing myocardium and a 1.04 μmol/L shift in failing myocardium. Protein kinase A in the presence of cAMP resulted in a further rightward shift in nonfailing human myocardium but did not further shift the calcium-force relationship in fibers from failing hearts. cGMP also resulted in a greater decrease in myofilament calcium sensitivity in fibers from failing hearts.
Conclusions
We propose that changes at the level of the thin myofilaments result in differential responses to changes in the intracellular milieu in nonfailing versus failing myocardium.
Keywords: myocardium, heart failure, calcium, cGMP, myocytes
One widely held hypothesis on the cause of contractile dysfunction in heart failure is that it is triggered by abnormal excitation-contraction coupling due to alterations in intracellular calcium availability and mobilization.1–5 Myocardial contractility impairment at the level of the myocyte in the failed human heart remains controversial.6 A more likely hypothesis with regard to the negative force-interval relationship seen at higher frequencies of contraction is a change in coupling of the sarcoplasmic reticulum with transsarcolemmal calcium influx.3
Another hypothesis to explain depressed contractility seen in heart failure is a change in myofilament calcium responsiveness.3,7 Reduced calcium sensitivity or decreased cooperation between the thick and thin myofilaments could result in reduced contractile activation and force development. In human heart failure, myofilament calcium responsiveness has been reported to be both changed8,9 and unchanged.10 Nevertheless, in human and experimental heart failure, several changes have been reported to occur. These include an isoform shift in troponin T (TnT) and a decrease in myosin light-chain kinase 2.6,11–13 These changes are believed to be partly responsible for a decrease in myofibrillar ATPase activity,14,15 a decreased cross-bridge cycling rate,16 and an altered responsiveness to agents that act on the myofilaments.6,8,17
Questions on the remodeling process are centered on the functional significance of a particular isoform population and altered distribution. Although changes in distribution of myosin isoforms occur with changes in cardiac workload, the myosin isoenzyme population does not always correlate with function.18 The question arises as to whether thin-filament functional properties are altered during long-term changes in myocardial activity and hemodynamic load.
We have addressed the question of whether failure to demonstrate differences in contractile activation in failing human myocardium reflects experimental conditions and substrate concentrations under which experiments were performed. We compared and examined effects of cAMP and protein kinase A (PKA), cGMP, acidosis, and inorganic phosphate in nonfailing and failing human myocardial fibers.
Methods
Human Hearts
Human tissues were obtained from 21 hearts (10 idiopathic dilated and 11 ischemic cardiomyopathic). Nonfailing human myocardium was obtained from 20 brain-dead human donors. No clinical or echocardiographic evidence indicated left ventricular dysfunction in tissue from any subjects.
Skinned-Fiber Preparations
Muscle strips of uniform size were dissected. A modified Krebs-Henseleit solution was used to bathe the strips. Muscles were stretched until no further increase occurred in active twitch force. Preparations were electrically paced. Electrical stimulus was turned off, and bath temperature was lowered to 22°C.
Fibers were exposed to a skinning solution that contained 250 μg/mL saponin at pH 7.1 as previously described.8 Total salt concentrations and absolute stability constants used to calculate compositions of the solutions were as reported by Fabiato.19
Interventions were performed on the skinned fiber preparations throughout a series of calcium activations: (1) changes in pH (ie, 7.1, 6.9, and 6.8); (2) addition of 15 mmol/L inorganic phosphate; (3) addition of 10 μmol/L dibutyryl cAMP (D-cAMP), with or without PKA; and (4) addition of 10 μmol/L 8-bromo-cGMP. To examine the effects of exogenous cAMP-dependent PKA, we added 1 mg/mL PKA in the presence of D-cAMP to the activating solutions at each pCa. Force versus [Ca2+] was fitted to the modified Hill equation.8
Materials
All chemicals were obtained from Sigma Chemical Co and were of the highest analytical grade.
Statistics
Data shown are mean±SEM. Calcium-force relationships were fitted individually to the Hill equation, and parameters were then pooled. Hill parameters were tested for statistical significance with ANOVA. P<0.05 was required for statistical significance.
Results
Effects of pH on Ventricular Myocardium From Nonfailing and Failing Human Hearts
Figure 1 shows effects of decreases in pH (7.1, 6.9, and 6.8) on calcium-force relationships in nonfailing and failing myocardium. At pH 7.1, no difference was seen in the calcium-force relationship between nonfailing and failing myocardium (Table 1 and Figure 1A). Maximal calcium-activated forces for nonfailing and failing groups were not significantly different at pH 7.1 (1.1±0.32 versus 0.98±0.33 g/mm2).
Figure 1.
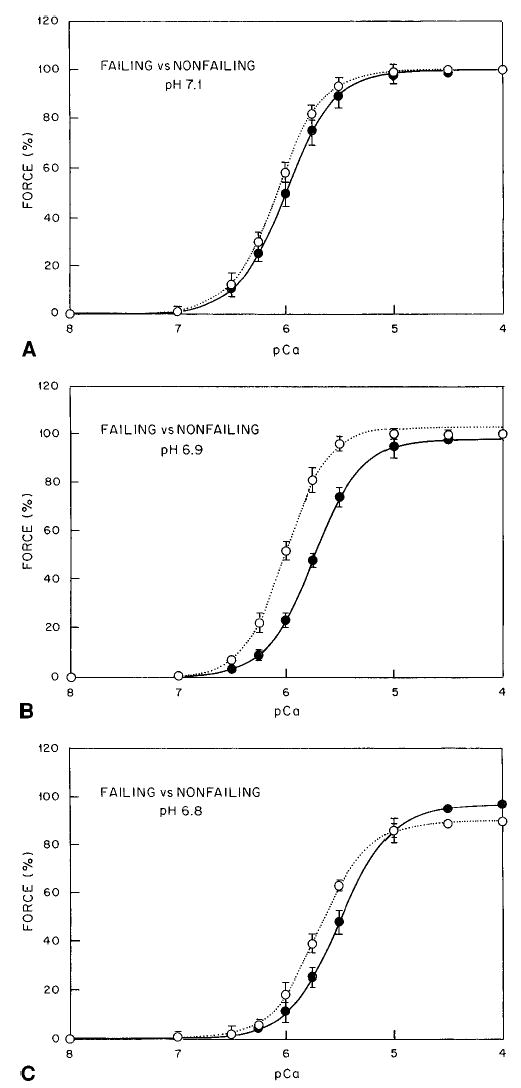
Changes in pH in nonfailing and failing myocardium. A, pH 7.1. B, pH 6.9. C, pH 6.8. Solid line and • indicates non-failing myocardium; dotted line and ○, failing myocardium.
TABLE 1.
Effect of pH on the Calcium-Force Relationship
| pH 7.1
|
pH 6.9
|
pH 6.8
|
||||
|---|---|---|---|---|---|---|
| Nonfailing (n=20) | Failing (n=21) | Nonfailing (n=13) | Failing (n=18) | Nonfailing (n=12) | Failing (n=12) | |
| [Ca2+]50%, μmol/L | 1.01±0.12 | 0.85±0.11 | 1.80±0.08† | 1.00±0.09* | 3.23±0.22† | 2.07±0.18*† |
| n | 1.89±0.14 | 2.02±0.16 | 1.97±0.11 | 2.25±0.11 | 1.79±0.21 | 1.93±0.23 |
| Fmax, % | 100 | 100 | 98±5 | 103±8 | 97±4 | 90±3† |
Numbers in parentheses indicate number of muscle preparations per group. n indicates Hill coefficients.
P<0.05 vs nonfailing human myocardium
P<0.05 vs pH 7.1.
As expected, a rightward shift occurred on the calcium axis for nonfailing and failing myocardium with decreases in pH (Figures 1B and 1C and 2A and 2B). In nonfailing fibers, a decrease in pH from 7.1 to 6.9 shifted the calcium-force relationship toward higher [Ca2+] by 0.79±0.32 μmol/L (Figure 2A). Further decreases in pH from 7.1 to 6.8 shifted the calcium-force relationship toward even higher [Ca2+], by 2.22±0.30 μmol/L. In fibers from failing hearts, a decrease in pH from 7.1 to 6.9 did not significantly shift the calcium-force relationship toward higher [Ca2+] (change of 0.15 μmol/L; Figure 2B), which reveals a difference in myofilament calcium activation at pH 6.9 between nonfailing and failing myocardium (Figure 1B and 1C). Further decreases in pH to 6.8 shifted the calcium-force relationship toward higher [Ca2+] (by 1.22±0.42 μmol/L) compared with the calcium concentration required for 50% activation ([Ca2+]50%) at pH 7.1 (Figure 1C). Maximal calcium-activated force was not different at pH 7.1 or 6.9 in nonfailing and failing myocardium. However, at pH 6.8, a lower maximal calcium-activated force occurred in fibers from failing hearts at the same pH (Figure 2B).
Figure 2.
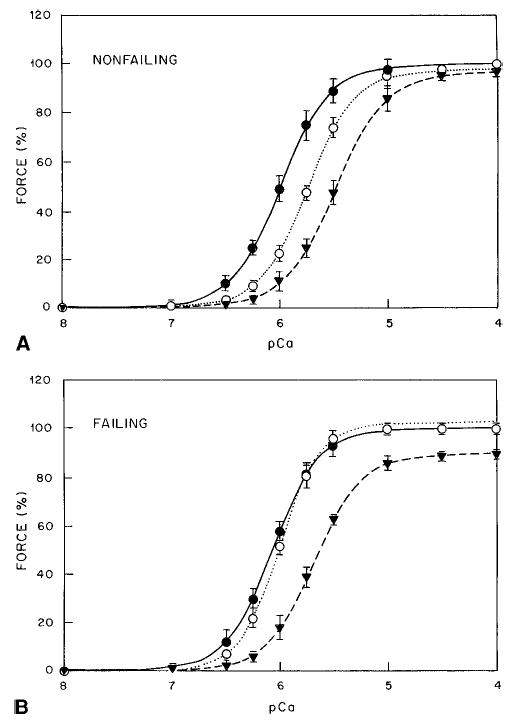
Calcium-force relationship in nonfailing (A) and failing (B) myocardium to changes in pH. Solid line and • indicates pH 7.1; dotted line and ○, pH 6.9; dashed line and ▾, pH 6.8.
Ratio of Δp [Ca50%]/ΔpH was 5.15 (pH 7.1 to 6.9) and 7.20 (pH 7.1 to 6.8) for fibers from nonfailing hearts and 2.30 (pH 7.1 to 6.9) and 4.10 (pH 7.1 to 6.8) for fibers from failing hearts. Results show that fibers from failing hearts are more resistant to changes in pH (ie, acidosis) compared with nonfailing fibers.
Effect of Increases in Inorganic Phosphate Concentration in Nonfailing and Failing Human Myocardium
Addition of inorganic phosphate shifted nonfailing myocardium to higher [Ca2+], with a larger reduction in maximal calcium-activated force (Table 2). Effects of 15 mmol/L phosphate on the calcium-force relationships are depicted in Figure 3, which shows an increase in [Ca2+]50% of 2.01±0.22 μmol/L in nonfailing and of 0.86±0.13 μmol/L in failing myocardium (P<0.05 compared with nonfailing myocardium). With 15 mmol/L inorganic phosphate, maximal Ca2+-activated force (Fmax) was decreased by 56±4% versus 34±2% for nonfailing versus failing myocardium (P<0.05 compared with nonfailing myocardium).
TABLE 2.
Effect of Inorganic Phosphate on the Calcium-Force Relationship at pH 7.1
| Control
|
Inorganic Phosphate (15 mmol/L)
|
|||
|---|---|---|---|---|
| Nonfailing (n=4) | Failing (n=5) | Nonfailing (n=4) | Failing (n=5) | |
| [Ca2+]50%, μmol/L | 1.01±0.09 | 0.95±0.08 | 3.02±0.22† | 1.81±0.13*† |
| n | 2.14±0.22 | 2.32±0.27 | 2.10±0.20 | 2.40±0.31 |
| Fmax, % | 100 | 100 | 44±4† | 64±2*† |
Numbers in parentheses indicate number of muscle preparations per group. n indicates Hill coefficient.
P <0.05 vs nonfailing mycardium
P<0.05 vs control.
Figure 3.
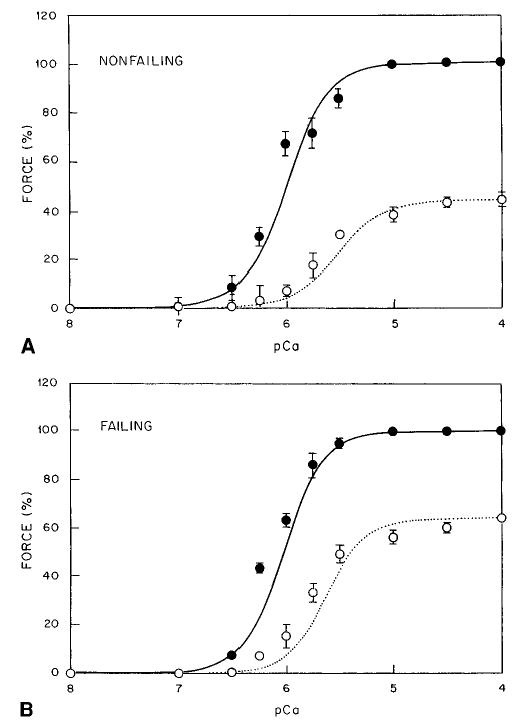
Inorganic phosphate in nonfailing (A) and failing (B) myocardium. Solid line and • indicates control; dotted line and ○, 15 mmol/L inorganic phosphate.
Effect of cAMP and PKA in Nonfailing and Failing Human Myocardium
As shown in Table 3 and Figure 4, in the presence of 10 μmol/L cAMP, force was shifted toward higher [Ca2+], from 1.03±0.09 to 1.59±0.11 μmol (change, 0.56 μmol) compared with 0.91±0.08 to 1.95±0.06 μmol (change, 1.04 μmol) for nonfailing and failing myocardium, respectively. Because the results presented above can be due to different amounts of endogenous protein kinase A (PKA) in nonfailing and failing myocardial skinned-fiber preparations, we added 1 mg/mL of PKA in the presence of cAMP (Table 3). As shown in Figure 4A, in the presence of 10 μmol/L cAMP, addition of PKA resulted in a further rightward shift to 1.93±0.13 μmol in nonfailing myocardium when compared with [Ca2+]50% in the presence of cAMP alone (change, 0.34 μmol). In contrast, addition of PKA to failing myocardium did not further shift the calcium-force relationship (Figure 4B). As shown in Table 3, addition of cAMP or PKA did not affect Fmax or the Hill coefficient.
TABLE 3.
Effect of D-cAMP and PKA on the Calcium-Force Relationship
| [Ca2+]50%, μmol/L | n | Fmax, % | |
|---|---|---|---|
| Nonfailing (n=4) | 1.03±0.09 | 2.23±0.21 | 100 |
| +10 μmol/L D-cAMP | 1.59±0.11* | 2.40±0.38 | 100 |
| +10 μmol/L D-cAMP | 1.93±0.13* | 2.23±0.29 | 100 |
| +PKA | |||
| Failing (n=6) | 0.91±0.08 | 2.44±0.36 | 100 |
| +10 μmol/L D-cAMP | 1.95±0.06*† | 2.28±0.26 | 100 |
| +10 μmol/L D-cAMP | 1.99±0.08* | 2.21±0.32 | 100 |
| +PKA |
Numbers in parentheses indicate number of muscle preparations per group. n indicates Hill coefficient.
P<0.05 vs predrug condition
P<0.05 vs nonfailing conditions.
Figure 4.
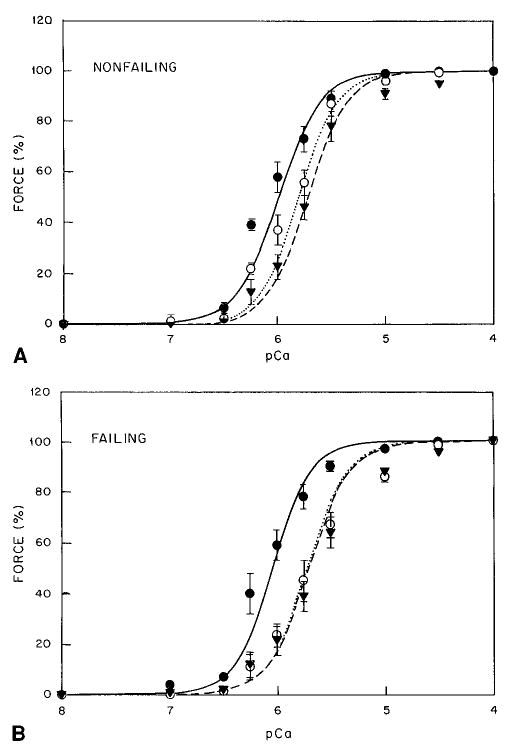
Effect of cAMP and PKA on nonfailing (A) and failing (B) myocardium. Solid line and • indicates control; dotted line and ○, with 10 μmol/L D-cAMP; dashed line and ▾, with 10 μmol/L D-cAMP and 1 mg/mL PKA.
Effect of cGMP in Nonfailing and Failing Human Myocardium
Studies have suggested that myocardial cGMP may play a role in regulation of myocardial contraction and relaxation.20 cGMP did not change myofilament cooperativity of Hill coefficient or Fmax (Figure 5). However, it did result in a rightward shift to higher [Ca2+]i (change of 0.2 μmol/L) in nonfailing myocardium (Table 4). In failing myocardium, 10 μmol/L cGMP resulted in a greater decrease in myofilament calcium sensitivity and a shift to higher [Ca2+]i (change of 0.45 μmol/L). The greater shift on the calcium axis seen in failing myocardium was not significantly different from the change in [Ca2+]50% in nonfailing myocardium.
Figure 5.
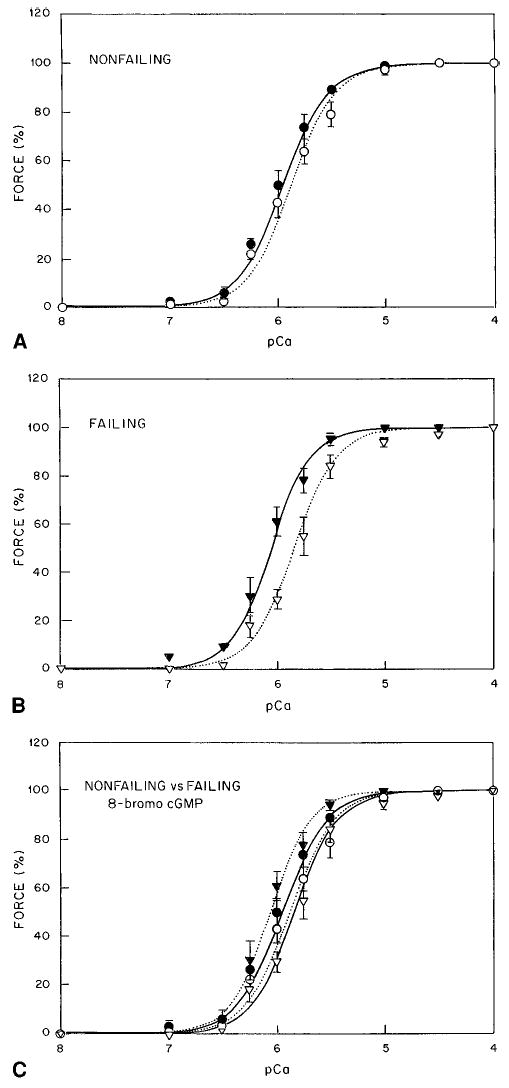
Effect of cGMP on nonfailing (A) and failing (B) myocardium. C, Comparison of effect of cGMP in failing and nonfailing myocardium. Solid line and •, control in nonfailing myocardium; dotted line and ○, 10 μmol/L 8-bromo-cGMP in nonfailing myocardium; solid line and ▾, control in failing myocardium; and dotted line and ▿, 10 μmol/L 8-bromo-cGMP in failing myocardium.
TABLE 4.
Effect of 8-Br-cGMP on the Calcium-Force Relationship
| [Ca2+]50%, mmol/L | n | Fmax, % | |
|---|---|---|---|
| Nonfailing (n=4) | 1.12±0.03 | 2.02±0.28 | 100 |
| +10 mmol/L 8-Br-cGMP | 1.32±0.05* | 2.11±0.32 | 100 |
| Failing (n=4) | 1.00±0.03 | 2.32±0.31 | 100 |
| +10 mmol/L 8-Br-cGMP | 1.45±0.02* | 2.18±0.20 | 100 |
Numbers in parentheses indicate number of muscle preparations per group. n indicates Hill coefficient.
P<0.05 vs predrug condition.
Discussion
Mechanisms of depressed contractile function in heart failure are presently unknown, but they probably involve alterations in calcium handling and myofilament function. However, it is unknown how or to what degree these alterations in cellular processes contribute to the decline of in vivo cardiac pump function seen clinically.
Experimental animal data have consistently demonstrated depressed myocyte function in failing hearts. The shift in the force-interval relationship seen at higher stimulation rates has been suggested to be associated with a change in myofilament calcium responsiveness due to the accumulation of inhibiting products such as hydrogen ions or inorganic phosphate or altered coupling between transsarcolemmal calcium influx and sarcoplasmic reticulum calcium release.3,7
Heart failure is associated with changes in the distribution and content of contractile proteins.11–13,21,22 Investigators also have found decreased myocardial myofibrillar ATPase activity in failing human heart.6,14,15 In failing human hearts, β-myosin heavy chain and levels of β-myosin heavy-chain mRNA are unchanged,23 a finding consistent with the present results with similar Fmax. Hence, alterations in other contractile proteins probably play a role in determination of the contractile properties of failing human myocardium. Several observations support this hypothesis. Recent reports have shown increased expression of a fetal cardiac isoform of TnT in human heart failure.11,13 Furthermore, investigators have shown that shifts in TnT isoform expression correlate with changes in the calcium responsiveness of the myofilaments.18,24
Margossian et al12 reported a marked decrease in myosin light-chain content in human heart failure, whereas others have demonstrated an increase in the atrial form of myosin light chain-1. Morano et al25 have shown that alterations in myosin light chain–actin interaction can have a profound effect on tension development and calcium responsiveness. Thus, changes in myosin light chain in heart failure may be of functional consequence for contractile activation.
Changes in myofilament calcium sensitivity that we observed in response to pH, inorganic phosphate, cAMP, and cGMP are probably achieved through different mechanisms. Our findings at pH 7.1 are also similar to most models of hypertrophy studied, which demonstrate similar calcium-force relationships.26,27 Our experiments showed that reduction of pH from 7.1 to 6.8 decreased sensitivity to [Ca2+]i without affecting Fmax. However, at a lower pH, 6.8, fibers from failing hearts demonstrated a significant decrease. At more acidic pH, we found, similar to results in skinned ventricular fibers from cardiomyopathic hamsters, that the calcium-force relationship for failing myocardium was shifted to the left on the calcium axis, which indicated an increased sensitivity of the myofilaments to calcium.27
Hydrogen-ion concentration decreases affinity of troponin C (TnC) for calcium. Therefore, the decrease in Fmax seen in failing myocardium at pH 6.8 and smaller shifts in myofilament calcium sensitivity suggest adaptive changes at the level of TnC. We recently reported 2 missense point mutations in TnC in failing myocardium that result in a decrease in myofilament calcium sensitivity.28 This suggests that differences in calcium activation may reside at the level of TnC.
Inorganic phosphate greatly inhibits the Fmax in skinned fibers.29,30 Kentish31 reported both depressant effects and desensitization of the myofilaments in the presence of inorganic phosphate. Our results correlate well with the decrease in Fmax and decreased sensitivity of the myofilaments to calcium in response to 15 mmol/L phosphate. Fmax can decrease as a result of uncoupling of cross-bridges.32
Inorganic phosphate increases the population of weak cross-bridges and results in a decrease in Fmax and a rightward shift on the calcium axis. In failing hearts, greater inhibition and reduction in force would be expected to be present. However, in myocardium from failing hearts with reduced myofibrillar ATPase activity, inhibitory effects might be overridden under conditions of prolonged cross-bridge attachment. Therefore, under these conditions, the effect of inorganic phosphate should be reduced.
Changes in cross-bridge cycling rate have been shown to shift the calcium-force relationship.33,34 We have previously shown that myocardium from failing hearts has a slower cross-bridge cycling rate.16 Slowing of the cross-bridge cycling rate results in a leftward shift in the calcium-axis due to longer TnC calcium interactions. This might explain why inorganic phosphate had a lesser effect on the Fmax and a lesser shift to higher calcium concentrations in failing myocardium. Longer TnC-calcium interactions in failing myocardium should also result in a lesser shift to higher [Ca2+]i with lowering of intracellular pH, which is supported by our experimental observations.
Whether contractile protein phosphorylation is altered in vivo in heart failure, as suggested for other regulatory proteins (eg, phospholamban), is unknown.35 To account for varying levels of contractile protein phosphorylation, we applied protein kinase to maximally phosphorylate the contractile proteins. However, it is possible that maximal phosphorylation might mask (by saturation) potential intrinsic alterations in contractile function induced by heart failure. We attempted to resolve this issue by studying both the native state and saturated levels.
Presence of cAMP caused a decrease in sensitivity of the myofilaments in both nonfailing and failing fibers. One explanation is that cAMP, in the presence of native PKA, induced phosphorylation of troponin I (TnI). In the presence of cAMP, muscles from failing hearts exhibited a greater shift to higher [Ca2+]i than that seen in nonfailing myocardium. This indicates either changes in the composition or activation of TnI or differing amounts of PKA. However, no isoforms of TnI have been identified in failing human myocardium.36,37 PKA content has not been reported to be different between nonfailing and failing myocardium.38 A more likely explanation is that the level of phosphorylation in failing fibers is lower than in nonfailing myocardium, as is similarly seen for phospholamban Ser-1635 and Threo-17. Failing human myocardium has been reported to have lower concentrations of intracellular cAMP.39 In support of this concept, addition of cAMP-dependent PKA in the presence of cAMP did not cause a significant shift to the right on the calcium axis in myocardium from failing hearts. Shifts seen in nonfailing myocardium in the presence of both cAMP and PKA were similar to the shift seen in failing fibers with cAMP alone.
Although experimental conditions were the same between nonfailing and failing tissues, differences in the activation of TnI remain a possible explanation. If different phosphorylation levels existed, addition of cAMP-dependent PKA in the nonfailing muscles should have shifted the calcium-force relationship to the right by as much as in the failing fibers unless changes occurred in the structure or function of TnI. This is concurrent with the present experimental results.
TnT populations have been shown to affect myofilament calcium responsiveness. However, PKA has a higher affinity for phosphorylation of TnI compared with TnT. Current concepts indicate that TnI promotes a “closed state” in which cross-bridges interact with the thin filament but in a weak non–force-generating reaction. Phosphorylation at the PKA sites of TnI appears to reduce the affinity of cross-bridges for the thin filament. Lesser phosphorylation may reflect differences in the amount of light chain-2 that result in stiffer cross-bridges or a stronger calcium-regulated binding state in failing myocardium. This explains the reduced myofilament responsiveness to changes in pH and inorganic phosphate.
Protein kinase C is the only kinase known to phosphorylate both cardiac TnI and TnT in vitro.40 However, we previously reported that protein kinase C activation resulted in a rightward shift on the calcium axis and a decrease in Fmax.17 Decreased Fmax and change in the Hill coefficient suggests phosphorylation of TnT.17 Failing human tissues have been shown to express 2 TnT isoforms and were less affected by protein kinase C activation. Both TnT and TnI have ≥2 phosphorylation sites.40 Therefore, our present data suggest that phosphorylation of the 2 TnT isoforms cannot explain the observed changes in myofilament calcium-activation seen with cAMP and PKA. However, differences at phosphorylation sites of TnI could explain our experimental observations.
cGMP stimulates cAMP-dependent phosphodiesterase, which results in an increase in cAMP hydrolysis. cGMP activation of cGMP-protein kinase results in phosphorylation of TnI and should result in a decrease in myofilament calcium responsiveness. If, as proposed, failing myocardium has significantly less cAMP, then a lesser inhibitory effect would have been expected for the cGMP-induced rightward shift on the calcium axis. Our data support this hypothesis in that cGMP shifted the calcium-force relationship by 0.2 μmol/L in nonfailing myocardium and by 0.45 μmol/L in failing myocardium.
In conclusion, thin myofilament regulation clearly can affect force development. Although differences in experimental findings have been reported by investigators who used agents targeted at myofilament calcium sensitivity,8,41 we propose that adaptational changes may not always be detected by standard steady-state calcium-force relationship determinations. Differences in isotype, structural interaction, phosphorylation level, or intracellular milieu may alter apparent myofibrillar calcium responsiveness in human myocardium.
Acknowledgments
The present study was supported in part by National Institute of Health grants (2RO1-HL-49574 and R44-HL-52249 to J.K.G. and R.J.H. and KO8-HL-03561 and R29-HL-57623 to R.J.H.) and by grants from the German Research Foundation (Schm 1116/1-1 to U.S. and Schw 607/3-1 to R.H.G.S.). We thank NDRI and Gwathmey Inc for technical assistance.
References
- 1.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt U, Hajjar RJ, Helm PA, Kim CS, Doye AA, Gwathmey JK. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol. 1998;30:1929–1937. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- 3.Gwathmey JK, Slawsky MT, Hajjar RJ, Briggs GM, Morgan JP. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J Clin Invest. 1990;85:1599–1613. doi: 10.1172/JCI114611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulieri LA, Hasenfuss G, Leavitt B, Allen PD, Aplert NR. Altered myocardial force-frequency relation in human heart failure. Circulation. 1992;86:2017–2018. doi: 10.1161/01.cir.85.5.1743. [DOI] [PubMed] [Google Scholar]
- 5.Pieske B, Kretschmann B, Meyer M, Holubarsch C, Weirich J, Posival H, Minami K, Just H, Hasenfuss G. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation. 1995;92:1169–1178. doi: 10.1161/01.cir.92.5.1169. [DOI] [PubMed] [Google Scholar]
- 6.Solaro RJ, Gao L, Olson CC, Gwathmey JK. Control of myofilament activation in heart failure. Circulation. 1993;87(suppl 7):VII-38–VII-43. [Google Scholar]
- 7.Gwathmey JK, Ingwall JS. Basic pathophysiology of congestive heart failure. Cardiol Rev. 1995;3:282–291. [Google Scholar]
- 8.Hajjar RJ, Gwathmey JK, Briggs GM, Morgan JP. Differential effect of DPI 201–106 on the sensitivity of the myofilaments to Ca2+ in intact and skinned trabeculae from control and myopathic human hearts. J Clin Invest. 1988;82:1578–1584. doi: 10.1172/JCI113769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D’Agnolo A, Luciani GB, Mazzucco A, Gallucci V, Salviati G. Contractile properties and Ca2+ release activity of the sarcoplasmic reticulum in dilated cardiomyopathy. Circulation. 1992;85:518–525. doi: 10.1161/01.cir.85.2.518. [DOI] [PubMed] [Google Scholar]
- 10.Schwinger RHG, Bohm M, Koch A, Schmidt U, Morano I, Eissner HJ, Uberfuhr P, Reichart B, Erdmann E. The failing human heart is unable to use the Frank-Starling mechanism. Circ Res. 1994;74:959–969. doi: 10.1161/01.res.74.5.959. [DOI] [PubMed] [Google Scholar]
- 11.Anderson PAW, Greig A, Mark TM, Malouf NN, Oakeley AE, Ungerleider RM, Allen PD, Kay BK. Molecular basis of human cardiac troponin T isoforms expressed in the developing, adult, and failing heart. Circ Res. 1995;76:681–686. doi: 10.1161/01.res.76.4.681. [DOI] [PubMed] [Google Scholar]
- 12.Margossian SS, White HD, Caulfield JB, Norton P, Taylor S, Slayter HS. Light chain 2 profile and activity of human ventricular myosin during dilated cardiomyopathy: identification of a causal agent for impaired myocardial function. Circulation. 1992;85:1720–1733. doi: 10.1161/01.cir.85.5.1720. [DOI] [PubMed] [Google Scholar]
- 13.Anderson PAW, Malouf NN, Oakeley AE, Pagani ED, Allen PD. Troponin T isoform expression in humans. Circ Res. 1991;69:1226–1233. doi: 10.1161/01.res.69.5.1226. [DOI] [PubMed] [Google Scholar]
- 14.Alpert NR, Gordon MS. Myofibrillar adenosine triphosphatase activity in congestive heart failure. Am J Physiol. 1962;202:940–946. doi: 10.1152/ajplegacy.1962.202.5.940. [DOI] [PubMed] [Google Scholar]
- 15.Pagani ED, Alousi AA, Grant AM, Older TM, Dziuban SW, Allen PD. Changes in myofibrillar content and Mg-ATPase activity in ventricular tissue from patients with heart failure caused by coronary artery disease, cardiomyopathy, or mitral valve insufficiency. Circ Res. 1988;63:380–385. doi: 10.1161/01.res.63.2.380. [DOI] [PubMed] [Google Scholar]
- 16.Hajjar RJ, Gwathmey JK. Cross-bridge dynamics in human ventricular myocardium: regulatory properties of contractivity in the failing heart. Circulation. 1992;86:1819–1826. doi: 10.1161/01.cir.86.6.1819. [DOI] [PubMed] [Google Scholar]
- 17.Gwathmey JK, Hajjar RJ. Effect of protein kinase C activation on sarcoplasmic reticulum function and apparent myofibrillar Ca2+ sensitivity in intact and skinned muscles from normal and diseased human myocardium. Circ Res. 1990;67:744–752. doi: 10.1161/01.res.67.3.744. [DOI] [PubMed] [Google Scholar]
- 18.Solaro RJ, el-Saleh SC, Kentish J, Meyer A, Martin A. Transitions in isoform populations of thick and thin filament proteins and calcium activation of force and ATP hydrolysis by cardiac myofilaments. Prog Clin Biol Res. 1989;315:487–502. [PubMed] [Google Scholar]
- 19.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–391. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 20.George WJ, Busuttil RW, Paddock RJ, White LA, Ignarro LJ. Opposing regulatory influences of cyclic guanosine monophosphate and cyclic adenosine monophosphate in the control of cardiac muscle contraction., In: Roy P.-E., Harris P., eds. Recent Advances in Studies on Cardiac Structure and Metabolism Baltimore, Md: University Park Press; 1975: 243–250. [PubMed]
- 21.Solaro RJ. Myosin and why hearts fail. Circulation. 1992;85:1945–1947. doi: 10.1161/01.cir.85.5.1945. [DOI] [PubMed] [Google Scholar]
- 22.Solaro RJ. Troponin I, stunning, hypertrophy, and failure of the heart. Circ Res. 1999;84:122–124. doi: 10.1161/01.res.84.1.122. [DOI] [PubMed] [Google Scholar]
- 23.Boheler KR, Carrier L, de la Bastie D, Allen PD, Komajda M, Mecardier JJ, Schwartz K. Skeletal actin mRNA increases in the human heart during ontogenic development and is the major isoform of control and failing adult hearts. J Clin Invest. 1991;88:323–330. doi: 10.1172/JCI115295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nassar R, Malouf NN, Kelly MB, Oakeley AE, Anderson PAW. Force-pCa relation and troponin T isoforms of rabbit myocardium. Circ Res. 1991;69:1470–1475. doi: 10.1161/01.res.69.6.1470. [DOI] [PubMed] [Google Scholar]
- 25.Morano I, Ritter O, Bonz A, Timek T, Vahl CF, Michel G. Myosin light chain-actin interaction regulates cardiac contractility. Circ Res. 1995;76:720–725. doi: 10.1161/01.res.76.5.720. [DOI] [PubMed] [Google Scholar]
- 26.Veksler V, Ventura-Clapier R. In situ study of myofibrils, mitochondria and bound creatine kinases in experimental cardiomyopathies. Mol Cell Biochem. 1994;133–134:287–298. doi: 10.1007/978-1-4615-2612-4_19. [DOI] [PubMed] [Google Scholar]
- 27.Veksler VI, Ventura-Clapier R, Lechene P, Vassort G. Functional state of myofibrils, mitochondria, and bound creatine kinase in skinned ventricular fibers of cardiomyopathic hamsters. J Mol Cell Cardiol. 1988;20:329–342. doi: 10.1016/s0022-2828(88)80067-1. [DOI] [PubMed] [Google Scholar]
- 28.Liao R, Gwathmey JK, Wang CK. Functional significance of two missense mutations in troponin C found in human myocardium with idiopathic dilated cardiomyopathy. Circulation. 1998;98:I-625. Abstract. [Google Scholar]
- 29.Solaro RJ, Holroyde MJ, Herzig JW, Peterson J. Cardiac relaxation and myofibrillar interactions with phosphate and vanadate. Eur Heart J. 1980;1(suppl A):21–27. doi: 10.1093/eurheartj/1.suppl_1.21. [DOI] [PubMed] [Google Scholar]
- 30.Herzig J, Peterson JW, Ruegg JC, Solaro RJ. Vanadate and phosphate ions reduce tension and increase cross-bridge kinetics in chemically skinned heart muscle. Biochim Biophys Acta. 1981;672:191–196. doi: 10.1016/0304-4165(81)90392-5. [DOI] [PubMed] [Google Scholar]
- 31.Kentish JC. The effects of inorganic phosphate and creatine phosphate on force production in skinned muscles from rat ventricle. J Physiol (Lond) 1986;370:584–604. doi: 10.1113/jphysiol.1986.sp015952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gwathmey JK, Hajjar RJ, Solaro RJ. Contractile deactivation and uncoupling of cross-bridges: effects of 2,3-butanedione monoxime on mammalian myocardium. Circ Res. 1991;69:1280–1292. doi: 10.1161/01.res.69.5.1280. [DOI] [PubMed] [Google Scholar]
- 33.Brandt PW, Cox RN, Kawai M, Robinson T. Regulation of tension in skinned muscle fibers: effect of cross-bridge kinetics on apparent Ca2+ sensitivity. J Gen Physiol. 1982;79:997–1016. doi: 10.1085/jgp.79.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morano I, Bletz C, Wojciechowski, Ruegg JG. Modulation of cross-bridge kinetics by myosin isoenzymes in skinned human heart fibers. Circ Res. 1991;68:614–618. doi: 10.1161/01.res.68.2.614. [DOI] [PubMed] [Google Scholar]
- 35.Schwinger RH, Munch G, Bolck B, Karczewski P, Krause EG, Erdmann E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serine-16 phospholamban phosphorylation. J Mol Cell Cardiol. 1999;31:479–491. doi: 10.1006/jmcc.1998.0897. [DOI] [PubMed] [Google Scholar]
- 36.Hunkeler NM, Kullman J, Murphy AM. Troponin I isoform expression in human heart. Circ Res. 1991;69:1409–1414. doi: 10.1161/01.res.69.5.1409. [DOI] [PubMed] [Google Scholar]
- 37.Sasse S, Brand NJ, Kyprianou P, Dhoot GK, Wade R, Arai M, Periasamy M, Yacoub MH, Barton PJR. Troponin I gene expression during human cardiac development and end-stage heart failure. Circ Res. 1993;72:932–938. doi: 10.1161/01.res.72.5.932. [DOI] [PubMed] [Google Scholar]
- 38.Bohm M, Reiger B, Schwinger RH, Erdmann E. cAMP concentrations, cAMP dependent protein kinase activity, and phospholamban in non-failing and failing myocardium. Cardiovasc Res. 1994;28:1713–1719. doi: 10.1093/cvr/28.11.1713. [DOI] [PubMed] [Google Scholar]
- 39.Feldman MD, Copelas L, Gwathmey JK, Phillips PJ, Schoen F, Grossman W, Morgan JP. Deficient production of cyclic AMP: pharmacologic evidence of an important cause of contractile dysfunction in patients with end-stage heart failure. Circulation. 1987;75:331–339. doi: 10.1161/01.cir.75.2.331. [DOI] [PubMed] [Google Scholar]
- 40.Katoh N, Wise BC, Kuo JF. Phosphorylation of cardiac troponin inhibitory subunit (troponin I) and tropomyosin-binding subunit (troponin T) by cardiac phospholipid-sensitive Ca2+-dependent protein kinase. Biochemistry. 1983;209:189–195. doi: 10.1042/bj2090189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bohm M, Diet F, Kemkes B, Wankerl M, Erdmann E. Inotropic response to DPI 201–106 in the failing human heart. Br J Pharmacol. 1989;98:275–283. doi: 10.1111/j.1476-5381.1989.tb16892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]