

Abstract
Background
Failing human myocardium is characterized by abnormal relaxation, a deficient sarcoplasmic reticulum (SR) Ca2+ uptake, and a negative frequency response, which have all been related to a deficiency in the SR Ca2+ ATPase (SERCA2a) pump.
Methods and Results
To test the hypothesis that an increase in SERCA2a could improve contractile function in cardiomyocytes, we overexpressed SERCA2a in human ventricular myocytes from 10 patients with end-stage heart failure and examined intracellular Ca2+ handling and contractile function. Overexpression of SERCA2a resulted in an increase in both protein expression and pump activity and induced a faster contraction velocity (26.7±6.7% versus 16.6±2.7% shortening per second, P<0.005) and enhanced relaxation velocity (32.0±10.1% versus 15.1±2.4%, P<0.005). Diastolic Ca2+ was decreased in failing cardiomyocytes overexpressing SERCA2a (270±26 versus 347±30 nmol/L, P<0.005), whereas systolic Ca2+ was increased (601±38 versus 508±25 nmol/L, P<0.05). In addition, the frequency response was normalized in cardiomyocytes overexpressing SERCA2a.
Conclusions
These results support the premise that gene-based therapies and targeting of specific pathways in human heart failure may offer a new modality for the treatment of this disease.
Keywords: contractility, myocytes, gene therapy, sarcoplasmic reticulum
Contraction and relaxation in cardiac myocytes are tightly regulated by intrinsic mechanisms that govern the sequential rise and fall of cytosolic Ca2+. During depolarization, Ca2+ entry through the L-type Ca2+ channels triggers the release of Ca2+ from the sarcoplasmic reticulum (SR) through ryanodine receptors, resulting in activation of the contractile proteins. In human cardiomyocytes, the removal of Ca2+ from the cytoplasm is governed mainly by the SR Ca2+ ATPase (SERCA2a) pump and to a lesser extent the Na/Ca exchanger.1 Cardiomyocytes isolated from failing human hearts are characterized by contractile dysfunction, including prolonged relaxation, reduced systolic force, and elevated diastolic force.2–5 These contractile abnormalities are paralleled by abnormal Ca2+ homeostasis, such as reduced SR Ca2+ release, elevated diastolic Ca2+, and reduced rate of Ca2+ removal.6 In addition, failing human myocardium is characterized by a frequency-dependent decrease in systolic force and Ca2+, as opposed to normal myocardium, in which an increase in pacing rate results in potentiation of contractility and an increase in SR Ca2+ release.3,6–8 In the failing heart, the decrease in SR Ca2+ load has been linked to a decrease in SERCA2a function.9 However, there has been long-standing controversy as to whether the protein level of SERCA2a is decreased in failing human hearts.1,6,9–12 In addition, because human cardiomyocytes rely on the SERCA2a pump to a lesser degree than do rodents (≈60% versus ≈90%), the direct implication of a reduction in SERCA2a activity in human failing hearts for overall contractile function has not been clear. In this study, we overexpressed SERCA2a by adenoviral gene transfer in viable human cardiomyocytes from failing and nonfailing human hearts.
Methods
Failing human ventricular myocardial tissue was obtained from 10 explanted hearts (5 ischemic and 5 dilated cardiomyopathy) and nonfailing tissue from 3 donor hearts. Myocytes were isolated from 1 g of endocardial tissue removed from the free wall of the left ventricle by enzymatic digestion as previously described.5 The proportions of rod-shaped viable cells at the time of isolation were 28±5% (n = 10) for failing and 35±8% (n = 3) for nonfailing cardiomyocytes (P>0.1), and at 24 hours after infection, they were 19±6% (n = 10) and 24 ± 7% (n = 3) (P>0.1). After isolation, the cells were resuspended in F10 medium with 0.164 U/100 mL insulin, 50 U/mL penicillin, and 50 U/mL streptomycin, equilibrated to pH 7.4 and infected with the adenoviruses at a multiplicity of infection (MOI) of 100. Two first-generation type 5 recombinant adenoviruses were used in the study: Ad.GFP, which carries the green fluorescent protein under the control of the cytomegalovirus promoter, and Ad.SERCA2a, which carries both the SERCA2a and GFP genes, each under the control of separate cytomegalovirus promoters. After 24 hours, myocytes were placed in a flow chamber on the stage of an inverted microscope, superfused with oxygenated Krebs-Henseleit solution, and electrically stimulated with biphasic pulse (0.2 Hz, 50% above threshold).5 Contraction amplitude and rates of contraction and relaxation were recorded online with a video edge-detection system and data acquisition software (Ion Optix). The fluorescent Ca2+ indicator fura 2 (Molecular Probes) was used to measure intracellular Ca2+ with a dual-excitation spectrofluorometer (IonOptix) as described previously.13 We isolated SR membranes from ventricular myocytes, and SERCA2a activity assays were carried out on the basis of a pyruvate/NADH coupled reaction at [Ca2+] of 10 μmol/L as previously described.13 SDS-PAGE was performed on the isolated membranes under reducing conditions on a 7.5% separation gel with a 4% stacking gel and immunoblotted with 1:2500 diluted monoclonal anti-SERCA2 antibody (Affinity BioReagents). The blot was then incubated in a chemiluminescence system and exposed to an X-OMAT AR x-ray film (Fuji Films) for 1 minute. Data were presented as mean ± SD and were analyzed with a 1-way ANOVA, with statistical differences identified at P<0.05.
Results
The coexpression of GFP allowed us to identify the cells that were infected and expressing the transgene (Figure 1a). Figure 1b shows tracings from representative cardiomyocytes isolated from failing hearts, which are characterized by decreased shortening and prolonged relaxation compared with the donor nonfailing cardiomyocytes. Overexpression of SERCA2a in failing cardiomyocytes induced a faster contraction velocity (26.7±6.7% versus 16.6±2.7% shortening per second, P<0.005) and enhanced relaxation velocity (32.0±10.1% versus 15.1±2.4%, P<0.005). Diastolic Ca2+ was decreased in failing cardiomyocytes overexpressing SERCA2a (270±26 versus 347±30 nmol/L, P<0.005), whereas systolic Ca2+ was increased (601±38 versus 508±25 nmol/L, P<0.05). Because a negative frequency response is a distinctive characteristic of failing myocardium, we tested whether an increase in SERCA2a expression restores the frequency response to normal. As shown in Figure 1c, increasing the frequency of stimulation in nonfailing cardiomyocytes induced an increase in contraction and intracellular Ca2+ with little change in diastolic Ca2+. In failing cardiomyocytes, increasing the frequency of stimulation induced a decrease in contraction and a large increase in diastolic cell length and diastolic Ca2+. Overexpression of SERCA2a in failing cardiomyocytes restored the frequency response, with increasing contraction at increasing frequencies. However, at high stimulation frequencies (2 Hz), both diastolic Ca2+ and cell length increased, but to a lesser degree than in failing cardiomyocytes infected with Ad.GFP. To verify that overexpression of SERCA2a in the human cardiomyocytes resulted in enhanced SERCA2a expression and in increased SR pump activity, we also examined immunoblots from infected cardiomyocytes and measured enzymatic activity of the SERCA2a. As shown in Figure 2, infection of cardiomyocytes with Ad.SERCA2a resulted in increased expression of SERCA2a protein and enhanced Ca2+ ATPase activity (43.2±3.8 versus 72.6±5.1 nmol · mg−1 · min−1, n = 6, P<0.01).
Figure 1.
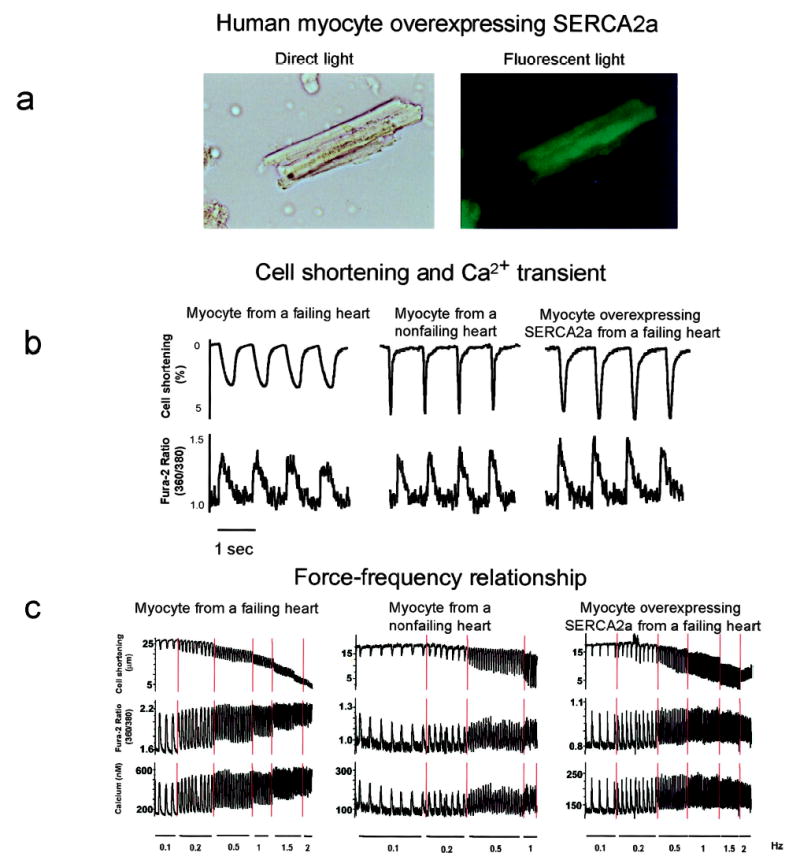
a, After isolation, failing human cardiomyocytes were infected with Ad.SERCA2a. Twenty-four hours after infection, a cardiomyocyte is visualized with white light and at 510 nm with single excitation peak at 490 nm of blue light. Coexpression of GFP demonstrates visually that SERCA2a is being expressed in cell. b, Recordings from cardiomyocytes isolated from donor nonfailing heart and from failing heart infected with either Ad.GFP or Ad.SERCA2a, stimulated at 1 Hz at 37°C. Failing cell had a characteristic decrease in contraction and prolonged relaxation along with a prolonged Ca2+ transient. Overexpression of SERCA2a in failing cardiomyocyte normalized these parameters. c, Recordings from same cardiomyocytes as in b stimulated at increasing frequencies. Failing cardiomyocyte demonstrated a decrease in contraction amplitude and an increase in diastolic tone and Ca2+. Overexpression of SERCA2a restored frequency-dependent increase in contraction amplitude and mitigated increase in diastolic Ca2+ and length.
Figure 2.
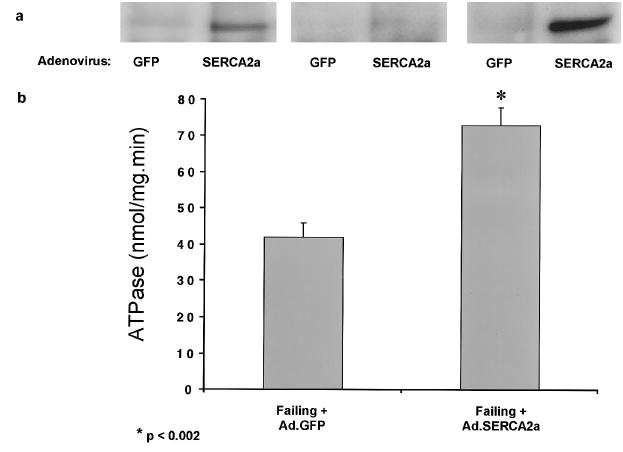
a, Immunoblots of SERCA2a from crude membranes of failing cardiomyocytes infected for 24 hours with Ad.SERCA2a or Ad.GFP. All representative lanes are from failing hearts. Each pair of immunoreactive densities represents paired immunoblots from 1 preparation of failing cardiomyocytes that were infected with either Ad.GFP or Ad.SERCA2a. b, Paired measurements of ATPase activity performed at [Ca2+] of 10 μmol/L in membrane preparations from failing cardiomyocytes infected for 24 hours with Ad.SERCA2a or Ad.GFP.
Discussion
Cardiac myocytes from failing human hearts of any etiology show a significant impairment of velocities of contraction and relaxation under low stimulation rate, and an alteration of contraction amplitude occurs at higher frequencies of stimulation than with nonfailing myocytes,13–15 contributing to the systolic and diastolic dysfunction in failing hearts. Abnormalities of the Ca2+ uptake by SERCA2a have been shown both in animal models of heart failure and in humans to account for the described functional abnormalities. Restoring the protein levels and function therefore represents a strategy to revoke the defects. Overexpressing SERCA2a was shown to reverse the contractile abnormalities of failing hearts, as we and others have validated in animal models.13–15 Transgenic mice overexpressing SERCA2a were characterized by higher myocardial contractility, including increased rates of pressure development for contraction and relaxation.16,17 Furthermore, in animal models in vivo, overexpressing SERCA2a improved contractile parameters,18 indicating that enhancing contractility at the cellular level does translate into improved ventricular performance. However, species-dependent differences need to be evaluated in the interpretation of a model and its translation to pathophysiology in humans. In particular, species-dependent differences are known to exist at the level of the SR in terms of Ca2+ removal during relaxation. Therefore, it was not clear whether gene transfer of SERCA2a could be used to restore contractility in human cardiomyocytes isolated from failing hearts. The difficulties in manipulating human myocytes, especially from diseased hearts, have thus far limited studies on the feasibility and efficacy of gene therapy in humans, with a growing experience in animal models. In this study, we were able to overexpress SERCA2a in human cardiac myocytes, and we showed that this translated into a normalization of the major characteristic abnormalities of contraction and calcium handling at the cellular level with an enhanced contraction amplitude and velocities of contraction and relaxation, an increase in peak Ca2+, and abbreviation of the calcium transient.
Heart failure continues to be a growing health problem in the United States, especially as the population ages. Up to now, treatment regimens can slow the progress of the disease without clearly reversing it.19 Gene-based therapies and targeting specific pathways in human heart failure may offer a new modality for the treatment of this disease. Overexpression of SERCA2a increases contractility in the short term, but whether targeted gene transfer has long-term benefits, as opposed to the long list of failed inotropic agents, is not yet known. Further experimental work will be necessary to validate the premise that improving contractile parameters at the cellular level can affect overall ventricular performance and survival in heart failure.
Acknowledgments
This work was supported in part by NIH grants HL-50361 and HL-57623 (Dr Hajjar), HL-54202 and HL-59521 (Dr Rosenzweig), and HL-49574 and HL-60323 (Dr Gwathmey); a Doris Duke Charitable Foundation Clinician Scientist Award and American Federation of Aging Research Grant (Dr Hajjar); and British Heart Foundation Grants 97064 and 98043 (Dr Harding). Dr Rosenzweig is an Established Investigator of the American Heart Association. The authors would like to thank the cardiac surgeons at Massachusetts General Hospital for assistance in tissue acquisition and the National Disease Research Interchange (NDRI) for technical assistance.
References
- 1.Arai M, Matsui H, Periasamy M. Sarcoplasmic reticulum gene expression in cardiac hypertrophy and heart failure. Circ Res. 1994;74:555–564. doi: 10.1161/01.res.74.4.555. [DOI] [PubMed] [Google Scholar]
- 2.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 3.Gwathmey JK, Slawsky MT, Hajjar RJ, Briggs GM, Morgan JP. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J Clin Invest. 1990;85:1599–1613. doi: 10.1172/JCI114611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harding SE, Jones SM, O’Gara P, del Monte F, Vescovo G, Poole-Wilson PA. Isolated ventricular myocytes from failing and non-failing human heart: the relation of age and clinical status of patients to isoproterenol response. J Mol Cell Cardiol. 1992;24:549–564. doi: 10.1016/0022-2828(92)91843-t. [DOI] [PubMed] [Google Scholar]
- 5.del Monte F, O’Gara P, Poole-Wilson PA, Yacoub M, Harding SE. Cell geometry and contractile abnormalities of myocytes from failing human left ventricle. Cardiovasc Res. 1995;30:281–290. [PubMed] [Google Scholar]
- 6.Schmidt U, Hajjar RJ, Helm PA, Kim CS, Doye AA, Gwathmey JK. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol. 1998;30:1929–1937. doi: 10.1006/jmcc.1998.0748. [DOI] [PubMed] [Google Scholar]
- 7.Mulieri LA, Hasenfuss G, Leavitt B, Allen PD, Alpert NR. Altered myocardial force-frequency relation in human heart failure. Circulation. 1992;85:1743–1750. doi: 10.1161/01.cir.85.5.1743. [DOI] [PubMed] [Google Scholar]
- 8.Davies CH, Davia K, Bennett JG, Pepper JR, Poole-Wilson PA, Harding SE. Reduced contraction and altered frequency response of isolated ventricular myocytes from patients with heart failure. Circulation. 1995;92:2540–2549. doi: 10.1161/01.cir.92.9.2540. [DOI] [PubMed] [Google Scholar]
- 9.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. doi: 10.1161/01.res.75.3.434. [DOI] [PubMed] [Google Scholar]
- 10.Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H, Hasenfuss G. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92:778–784. doi: 10.1161/01.cir.92.4.778. [DOI] [PubMed] [Google Scholar]
- 11.Mercadier JJ, Lompre AM, Duc P, Boheler KR, Fraysse JB, Wisnewsky C, Allen PD, Komajda M, Schwartz K. Altered sarcoplasmic reticulum Ca2+-ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Invest. 1990;85:305–309. doi: 10.1172/JCI114429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwinger RH, Bohm M, Schmidt U, Karczewski P, Bavendiek U, Flesch M, Krause EG, Erdmann E. Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+ uptake and Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–3228. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- 13.Hajjar RJ, Kang JX, Gwathmey JK, Rosenzweig A. Physiological effects of adenoviral gene transfer of sarcoplasmic reticulum calcium ATPase in isolated rat myocytes. Circulation. 1997;95:423–429. doi: 10.1161/01.cir.95.2.423. [DOI] [PubMed] [Google Scholar]
- 14.Hajjar RJ, Schmidt U, Kang JX, Matsui T, Rosenzweig A. Adenoviral gene transfer of phospholamban in isolated rat cardiomyocytes: rescue effects by concomitant gene transfer of sarcoplasmic reticulum Ca2+ ATPase. Circ Res. 1997;81:145–153. doi: 10.1161/01.res.81.2.145. [DOI] [PubMed] [Google Scholar]
- 15.Giordano FJ, He H, McDonough P, Meyer M, Sayen MR, Dillmann WH. Adenovirus-mediated gene transfer reconstitutes depressed sarcoplasmic reticulum Ca2+-ATPase levels and shortens prolonged cardiac myocyte Ca2+ transients. Circulation. 1997;96:400–403. doi: 10.1161/01.cir.96.2.400. [DOI] [PubMed] [Google Scholar]
- 16.Baker DL, Hashimoto K, Grupp IL, Ji Y, Reed T, Loukianov E, Grupp G, Bhagwhat A, Hoit B, Walsh R, Marban E, Periasamy M. Targeted overexpression of the sarcoplasmic reticulum Ca2+-ATPase increases cardiac contractility in transgenic mouse hearts. Circ Res. 1998;83:1205–1214. doi: 10.1161/01.res.83.12.1205. [DOI] [PubMed] [Google Scholar]
- 17.He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, Bluhm WF, Meyer M, Sayen MR, Swanson E, Dillmann WH. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest. 1997;100:380–389. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyamoto MI, Guerrero JL, Schmidt U, Gwathmey JK, Dec GW, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves LV function in aortic-banded rats in transition to heart failure. Circulation. 1998;98:736. doi: 10.1073/pnas.97.2.793. Abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevenson LW. Inotropic therapy for heart failure. N Engl J Med. 1998;339:1848–1850. doi: 10.1056/NEJM199812173392511. [DOI] [PubMed] [Google Scholar]