

Abstract
Inflammation and oxidative stress are important factors in the pathogenesis of diabetes and contribute to the pathogenesis of diabetic complications. Periodontitis is an inflammatory disease that is characterized by increased oxidative stress, and the risk for periodontitis is increased significantly in diabetic subjects. In this study, we examined the superoxide (O2−)-generating reduced nicotinamide adenine dinucleotide phosphate-oxidase complex and protein kinase C (PKC) activity in neutrophils. Fifty diabetic patients were grouped according to glycemic control and the severity of periodontitis. Neutrophils from diabetic patients with moderate [amount of glycated hemoglobin (HbA1c) between 7.0% and 8.0%] or poor (HbA1c >8.0%) glycemic control released significantly more O2− than neutrophils from diabetic patients with good glycemic control (HbA1c <7.0%) and neutrophils from nondiabetic, healthy individuals upon stimulation with 4β-phorbol 12-myristate 13-acetate or N-formyl-Met-Leu-Phe. Depending on glycemic status, neutrophils from these patients also exhibited increased activity of the soluble- and membrane-bound forms of PKC, elevated amounts of diglyceride, and enhanced phosphorylation of p47-phox during cell stimulation. In addition, we report a significant correlation between glycemic control (HbA1c levels) and the severity of periodontitis in diabetic patients, suggesting that enhanced oxidative stress and increased inflammation exacerbate both diseases. Thus, hyperglycemia can lead to a novel form of neutrophil priming, where elevated PKC activity results in increased phosphorylation of p47-phox and O2− release.
Keywords: signal transduction, diabetes
INTRODUCTION
Diabetes mellitus (DM) encompasses a heterogeneous group of disorders with the common characteristic of altered glucose tolerance and impaired lipid and carbohydrate metabolism. It is estimated that 6.5% of the population in the United States has diabetes, with only half of the affected individuals diagnosed. The incidence of this disease continues to increase at an alarming rate [1]. The substantial increase in obesity, which is the main risk factor for type 2 diabetes found in National Health and Nutrition Examination Survey III, indicates that diabetes will continue to be a major problem in the U.S. population [2].
Stimulated neutrophils produce large quantities of superoxide (O2−) and hydrogen peroxide, which are major components of the oxygen-dependent, antimicrobial arsenal of these cells [3]. The reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, which catalyzes the generation of O2− and peroxide, is dormant/dissociated in unstimulated cells and consists of membrane-bound (cytochrome b558), cytoskeletal-associated (p67-phox), and cytoplasmic proteins (p47-phox, Rac). Upon stimulation, the oxidase is assembled/activated on the inner surface of the plasmalemma and phagosomal membrane [3, 4]. p47-phox is an adaptor protein that facilitates assembly of the oxidase after undergoing phosphorylation on multiple serine residues in its C-terminal region [5]. Protein kinase C (PKC) catalyzes the phosphorylation of p47-phox after activation by its natural activator sn-1,2-diacylglycerol (DAG) or the tumor promoter phorbol 12-myristate 13-acetate (PMA) [6, 7]. PMA binds to the DAG site on PKC [7]. Phagocytic leukocytes from diabetic patients release enhanced amounts of O2−/peroxide when compared with normal cells [8–11]. The biochemical mechanism(s) responsible for this is not known.
Hyperglycemia affects cells through a variety of mechanisms [12]. For example, increased concentrations of glucose promote de novo synthesis of DAG in cells through the glycolytic/glycerol-3-phosphate acyltransferase pathway. The resulting increases in DAG trigger activation of PKC with physiological consequences (for review, see ref. [13]). In addition, numerous proteins undergo nonenzymatic glycosylation in a hyperglycemic environment to form advanced glycation end products (AGE), which accumulate in diabetic plasma and tissues (e.g., ref. [14]). A well-characterized receptor for AGE is present on monocytes, macrophages, and neutrophils [15, 16]. AGE alone triggers minimal O2−/peroxide production by neutrophils but markedly increases the production of these reactive oxygen species upon subsequent stimulation of the cells with the chemoattractant N-formyl-Met-Leu-Phe (fMLP) [16–18]. AGE have been implicated in susceptibility oral infections, exaggerated inflammatory responses, and destruction of alveolar bone, which accompanies diabetes [19, 20].
Chronic periodontitis is the most common type of periodontal disease and results from extension of the inflammatory process initiated by bacteria in the gingiva to the supporting periodontal tissues. A reciprocal relationship exists between diabetes and periodontal disease [21–24]. Periodontal infections, like other infections, have a significant impact on diabetic control [21, 22]. Conversely, diabetes is a significant risk factor for the development of periodontal disease and aggravates the severity of periodontal infections [23]. Factors that increase the severity of inflammatory diseases, such as altered neutrophil function, have been associated with the pathogenesis of diabetes [24]. The comorbidity of these two inflammatory diseases suggests that there are common elements of pathogenesis related to risk for both conditions.
In this paper, we report a novel mechanism of neutrophil priming, where elevated PKC activity results in increased phosphorylation of p47-phox and O2− release. Moderate or poor glycemic control in diabetic patients raises PKC protein levels and activity causing neutrophil priming. The increased inflammation and oxidative stress are consistent with increased risk for inflammatory complications of diabetes, including periodontitis.
MATERIALS AND METHODS
Selection of subjects
The Institutional Review Board at Boston University Medical Center (MA) approved this study, and informed consent was obtained from all subjects prior to evaluation. In total, 50 diabetic patients (30 males and 20 females; mean age 50.9 ± 1.3 years), 45 nondiabetic, healthy subjects (30 males and 15 females; mean age 48.3 ± 1.9 years), and six patients with chronic periodontitis (46.7 ± 3.4 years) were enrolled. Diabetic subjects were recruited from the Diabetic Clinic at Boston University Medical Center, and diagnosis of diabetes was based on the criteria of the American Diabetes Association [25]. Briefly, for a subject to be diagnosed as “diabetic,” one of the following criteria was met: symptoms of diabetes (polyuria, polydipsia, unexplained weight loss) plus a casual (any time of day without regard to time since last meal) plasma-glucose concentration of ≥200 mg/dl (11.1 mmol/l); fasting (no caloric intake for at least 8 h) plasma-glucose levels ≥120 mg/dl (6.7 mmol/l); and plasma-glucose levels ≥200 mg/dl (11.1 mmol/l) during an oral glucose-tolerance test. Nondiabetic, healthy and chronic periodontitis subjects were recruited from the patient pool of the Clinical Research Center at Boston University School of Dental Medicine, a unit of the General Clinical Research Center at Boston University Medical Center. Diabetic subjects signed a release form for their medical records prior to baseline.
Patients were grouped by glycemic control, as defined by the American Diabetes Association, based on the amount of glycated hemoglobin (HbA1c) in erythrocytes, and HbA1c <7.0%: good control; HbA1c 7.0–8.0%: moderate control; and HbA1c >8.0%: poor control requiring treatment [25, 26].
At baseline, the medical and dental status of subjects was reviewed and recorded. All diabetic subjects were at least 21 years of age, able to give written, informed consent, did not have any other systemic disease in addition to diabetes, and had a minimum of 16 of their natural teeth excluding their third molars. Nondiabetic, healthy subjects, meeting the same inclusion criteria, were included as controls. Individuals with active infectious diseases (e.g., hepatitis, AIDS, tuberculosis), chronically treated with medications known to affect periodontal tissues (phenytoin, cyclosporin-A, or calcium channel blockers) and females who were lactating or pregnant, were excluded.
Periodontal status was evaluated based on radiographic findings (bone level as a percentage of total tooth length) and clinical attachment loss. The severity of periodontitis was assessed by measuring pocket depth; pockets ≥5 mm but less than 7 mm on two nonadjacent teeth were defined as moderate, and pockets ≥7 mm on two nonadjacent teeth were defined as severe [27]. The demographics and the characteristics of the DM patients and healthy controls are described in Table 1.
TABLE 1.
Demographic Data of Study Subjects
| Diabetes mellitus (DM) |
|||||||
|---|---|---|---|---|---|---|---|
| Periodontitis |
|||||||
| Healthy controls (HC) | Total | No (DM-NP) | Moderate (DM-MP) | Severe (DM-SP) | Chronic periodontitis (CP) | ||
| N | 45 | 50 | 16 | 20 | 14 | 6 | |
| Type of diabetes | Type I | – | 12 | 3 | 5 | 4 | – |
| Type II | 38 | 13 | 15 | 10 | |||
| Duration of diabetes (year; mean±sem) | – | 10.70 ± 1.43 | 7.59 ± 0.31 | 8.85 ± 0.25 | 10.24 ± 0.50 | – | |
| Age (mean±sem) | 48.3 ± 1.9 | 50.9 ± 1.3 | 48.6 ± 3.7 | 50.7 ± 3.9 | 56.0 ± 2.9 | 46.7 ± 3.4 | |
| Gender | Male | 30 | 30 | 10 | 12 | 8 | 5 |
| Female | 15 | 20 | 6 | 8 | 6 | 1 | |
| Race | Asian | 9 | 3 | 3 | 0 | 0 | 0 |
| Caucasian | 15 | 20 | 6 | 8 | 6 | 1 | |
| African-American | 14 | 26 | 6 | 12 | 8 | 1 | |
| Hispanic | 5 | 8 | 3 | 3 | 2 | 1 | |
| Smoking status | Smoker | 16 | 17 | 6 | 9 | 6 | 0 |
| Nonsmoker | 29 | 33 | 10 | 11 | 8 | 6 | |
Isolation of human peripheral blood neutrophils
Peripheral venous blood (45 ml) was obtained from each subject in heparinized (10 U/ml) glass tubes, and neutrophils were isolated using a discontinuous gradient system [28]. Peripheral blood was layered on a mixture of MonoPoly (Flow Laboratories, McLean, VA), Histopaque 1119, and Histopaque 1077 (Sigma Chemical Co., St. Louis, MO), and the tubes were centrifuged at 500 g for 30 min. The neutrophil-rich layer was collected, and contaminating erythrocytes were lysed with a hypotonic NH4Cl buffer (155 mM NH4Cl, 10 mM KHCO3, 120 μM EDTA, pH 7.4). Cells were washed twice with phosphate-buffered saline (PBS; 13.3 mM Na2HPO4, 6.7 mM KH2PO4, pH 7.2, 0.15 M NaCl), resuspended in 50 ml PBS, and counted with a hemocytometer. Cell preparations were routinely 99% neutrophils with ≥95% viability, as determined by trypan blue exclusion.
O2− release
O2− release was monitored spectrophotometrically at 37°C by measuring O2− dismutase-inhibitable reduction of ferricytochrome c at 550 nm [29]. Assays were carried out in 96-well microtiter plates with flat-bottomed wells (Linbro type, Flow Laboratories). Five separate samples were used for each condition. Control wells contained all of the components of the assay mixture plus O2− dismutase (20 U/ml) to correct for ferricytochrome c reduction by agents other than O2−. Cells (1.5 × 105) were suspended in PBS (200 μl/well) and stimulated by the addition of PMA (300 nM) or fMLP (1.0 μM) and the absorbance (optical density) at 550 nm, recorded in a Vmax kinetic microplate reader (Molecular Devices, Sunnyvale, CA). O2− release was measured under conditions of linearity with respect to time and cell number and is expressed as nmole O2−/min/105 PMNs.
Subfractionation of neutrophils
Subfractionation of neutrophils was performed at 4°C as described by Wolfson and colleagues [30] with minor modifications. Neutrophils (10 × 106 cells/ml) were suspended in Hanks’ balanced salt solution (HBSS; 5.37 mM KCl, 0.44 mM KH2PO4, 136.89 mM NaCl, 0.34 mM Na2HPO4, 4.17 mM NaHCO3) without magnesium and calcium, preincubated for 10 min at 37°C, and then treated with PMA [300 nM final concentration in 0.10% dimethyl sulfoxide (DMSO)] or 0.10% DMSO alone for 10 min. Assays were terminated by immersing the reaction tubes in an ice bath for 5 min. Cells were then washed twice with HBSS without magnesium and calcium, suspended at 5 × 107 cells/ml in extraction buffer [20 mM Tris·HCl, pH 7.5, 10 mM EGTA, 2.0 mM EDTA, 1.0 mM phenylmethylsulfonyl fluoride, 50 mM 2-mercaptoethanol (2-ME), 20 U leupeptin/ml, and 0.0122 U aprotinin/ml], and sonicated at 15 W for 15 s. An aliquot of the lysate was saved, and the remaining sonicate was centrifuged at 100,000 g for 60 min. The supernatant fluid was collected and is referred to as the cytosol-rich or soluble fraction. The pellet was resuspended to the original volume in extraction buffer, sonicated again at 15 W for 15 S, and incubated for 1 h at 4°C in the presence of 0.30% (final concentration) Triton X-100. The resulting suspension is referred to as the membrane-rich fraction.
PKC assay
Calcium-dependent PKC activity was assayed as described previously [31, 32] with minor modifications. This enzyme was assayed by measuring the transfer of 32P from [γ-32P]adenosine 5′-triphosphate (ATP) into histone III-S at 30°C. The standard assay mixture contained 25 mM Tris·HCl, pH 7.4, 10 mM MgCl2, 0.50 mM CaCl2, 0.20 mg/ml histone type III-S, 20 μg/ml diolein (DAG), and 50 μg/ml phosphatidylserine (PS). The 100,000 g soluble and membrane fractions were used as the source of PKC, and the reactions were initiated by the addition of 50 μM [γ-32P]ATP (1.0 μCi). Reactions were terminated by the addition trichloroacetic acid (TCA) to a final volume of 25%. The resulting precipitates were collected by vacuum filtration on polycarbonate filters (type HA, 0.45 μm; Costar, Cambridge, MA) and washed three times with 5.0% TCA. After drying, the amount of radioactivity on the filters was quantified by β-liquid scintillation counting (LKB, Bromma, Sweden). Assays were performed in the presence of Ca2+ alone, DAG/PS alone, and Ca2+/DAG/PS, and the values for Ca2+ and DAG/PS alone were subtracted from the value for Ca2+/DAG/PS. Activity measured under these conditions was linear from 1.0 to 7.0 min and was directly proportional to the quantity of the “cell fractions” present in the assay between 0.75 × 105 and 2.25 × 105 cell equivalents. Activity is expressed as picomoles of 32P incorporated per minute per 107 cells.
Western blot
Cell lysates (60 μg/ml) were placed in sodium dodecyl sulfate (SDS) sample buffer [62.5 mM Tris-HCl, pH 6.8, 2% w/v SDS, 10% glycerol, 50 mM dithiothreitol, 0.01% w/v bromophenol blue] and boiled for 5 min. Samples and standards (Bio-Rad, Hercules, CA) were loaded on 10% SDS-polyacrylamide gel electrophoresis (PAGE) acrylamide gels. Proteins were transferred from the gel to a polyvinylidene difluoride membrane, and the membrane was incubated with p47-phox antibody (1:200) overnight at 4°C in 20 mM Tris–HCl (pH 7.5) containing 250 mM NaCl, 0.10% (v/v) Tween 20, and 5.0% (w/v) bovine serum albumin. The membrane was washed three times (10 min/wash) with 20 mM Tris–HC1, pH 7.4, containing 150 mM NaCl and 0.10% (v/v) Tween 20 (TBST) and then incubated with the second antibody (donkey anti-goat immunoglobulin G-horseradish peroxidase conjugate; 1:3000) in TBST for 1 h at room temperature. Membranes were again washed three times in TBST. Bands were visualized after incubation of membranes for 5 min at room temperature in a luminol-enhanced chemiluminescence detection system (Pierce, Rockford, IL) followed by autoradiography.
To assess the relevant isoforms of PKC being activated, neutrophils were incubated with GF109203X {2-[1-(3-dimethylamino-propyl)indol-3-yl]-3-(1H-indol-3-yl)maleimide; also known as bisindolylmaleimide I or Gö 6850}, a potent and selective inhibitor of PKC-α, -β1, and -β2 at 5 μM for 15 min, or staurosporine, a nonspecific and potent inhibitor of kinases including PKC, PKA, and PKG at 0.1 μM for 10 min [33–35]. O2− generation postincubation was assayed as described above.
Measurement of diglyceride
Diglyceride was extracted from neutrophils by the method of Bligh and Dyer [36] and converted stoichiometrically to [32P]phosphatidate ([32P]PA) by incubation with [γ-32P]ATP and Escherichia coli diglyceride kinase [37]. The resulting [32P]PA was isolated by thin-layer chromatography, visualized by autoradiography, scraped from the plate, and quantified by scintillation counting [37].
Immunoblotting/detection of phosphorylated p47-phox in neutrophils
Phosphorylation of p47-phox in stimulated neutrophils was monitored by Western blotting with a phosphospecific antibody [pAb; pPKC (S) Ab] obtained from Cell Signaling Technology (Beverly, MA) [38]. The preferred sequence recognized by this pAb is phospho-Ser flanked by Lys or Arg at the −2 and +2 positions and with a hydrophobic residue at the +1 position. The specificity of this pAb is not rigid, and motifs similar to the preferred sequence are also recognized by this pAb. A mouse monoclonal antibody (mAb) raised against nonphosphorylated, recombinant p47-phox was a gift from Dr. Paul Heyworth (Scripps Research Center, La Jolla, CA). Neutrophils (1.9 × 106/ml) were stimulated in disposable, 1-cm plastic cuvettes at 37°C. The standard assay mixture consisted of a modified Dulbecco’s PBS medium (135 mM NaCl, 2.7 mM KCl, 16.2 mM Na2PO4, 1.47 mM KH2PO4, 0.90 mM CaCl2, and 0.50 mM MgCl2, pH 7.35) containing 7.5 mM D-glucose. Cells were incubated in this medium for 10.0 min prior to stimulation with 300 nM PMA. At the appropriate time-points, the cells were lysed rapidly by adding 0.25 ml of 5× concentrated “solubilization buffer” (SDS-B) to 1.0 ml of the reaction mixture, and the samples were boiled for 4.0 min. The final composition of SDS-B after mixing was 2.3% (w/v) SDS, 62.5 mM Tris-HCl (pH 6.8), 5.0 mM EDTA, 10.0% (v/v) glycerol, 5.0% (v/v) 2-ME, and 0.002% (w/v) bromophenol blue. Aliquots of these samples were separated by SDS-PAGE (35 μg/lane) on 9.0% (v/v) polyacrylamide slab gels and transferred electrophoretically to Immobilon P-membranes. Protein phosphorylation was assayed by Western blotting with the pPKC (S) Ab. Membranes were incubated with the pAb (ca., 1:1000 dilution) overnight at 4°C. Phosphorylated proteins were visualized with a luminol-enhanced chemiluminescence detection system (Pierce). Conditions for Western blotting are detailed elsewhere [38]. At the end of these experiments, the immunodetection system and the bound antibodies were removed from the blot by incubating the membrane with ImmunoPure elution buffer (Pierce) for 30–60 min at room temperature followed by two washes in TBST. The blots were then stained with the mAb to p47-phox, which recognized the phosphorylated and nonphosphorylated forms of this protein.
Statistical analysis
Results are expressed as mean ± se. Analysis of covariance (ANCOVA) adjusting for age was used with the Statistical Analysis System. Multiple comparison procedures were used for group pairs, and comparison was controlling for overall type I error. Means were adjusted for possible confounding factors of age, smoking, and gender. Difference in the age-adjusted means between groups was considered significant for a P value less than 0.05. Correlation analysis was conducted using Spearman’s ρ test. We performed a trend analysis to examine whether the mean hemoglobin level increases as the severity of periodontitis increases.
RESULTS
O2− release by neutrophils from healthy individuals and diabetic subjects
Figure 1 presents a comparison of the rates of O2− release from neutrophils obtained from healthy, nondiabetic individuals and a variety of patients with diabetes. The diabetic patient population consisted of individuals with different levels of glycemic control, as defined by the American Diabetes Association (HbA1c <7.0%=good control; 7.0–8.0%=moderate control, consider altering treatment; >8.0%=poor control, requires treatment) [25]. Healthy controls were age-, race-, and gender-matched as closely as possible. Fifteen subjects (n=15) were monitored in each group. Neutrophils from diabetic patients with good glycemic control appeared to release increased amount of O2−, but this trend was not statistically significant. However, when glycemic control was moderate or poor, O2− release by unstimulated and stimulated cells was significantly elevated (Fig. 1). Neutrophils from diabetic patients with moderate or poor glycemic control release two to three times more O2− than normal neutrophils upon stimulation with 300 nM PMA or 1.0 μM fMLP. The enhanced release of O2− by neutrophils from diabetic patients persisted after the cells were washed several times during isolation and stored on ice for 2–4 h. Thus, the biochemical alteration(s) responsible for this “priming” effect is stable.
Fig. 1.
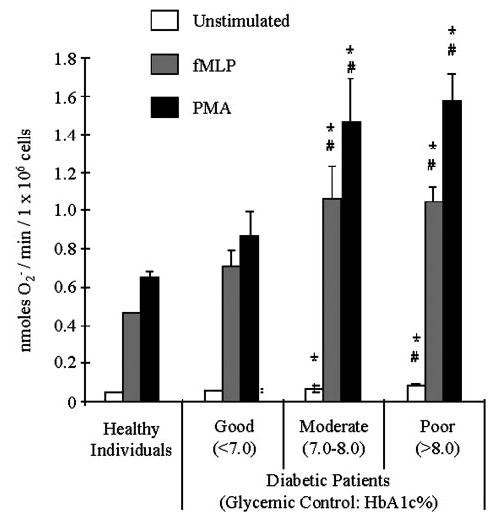
Neutrophils from diabetic subjects generate more O2− anion. Diabetic individuals were grouped into three categories based on glycemic control as defined by the American Diabetes Association. Rates of O2− release by neutrophils obtained from healthy individuals and patients with good (HbA1c 3 7.0%), moderate (HbA1c between 7.0% and 8.0%), and poor (HbA1c >8.0%) glycemic control are presented. Cells were stimulated with 1.0 μM fMLP or 300 nM PMA, and O2− release was measured as described in Materials and Methods. Neutrophils from diabetic patients with moderate and poor glycemic control exhibited greater release of O2− than neutrophils from patients with well-controlled diabetes or healthy individuals. (*, P<0.05, compared with healthy; #, P<0.05, compared with well-controlled diabetics.)
Neutrophils from patients with type 1 (n = 20) or type 2 (n = 20) diabetes released significantly more O2− than normal neutrophils when stimulated with 1.0 μM fMLP or 300 nM PMA (Fig. 2). Thus, the in vivo priming of neutrophils from these patients appears to be independent of their ability to respond to insulin. Data summarized in Figure 2 included patients with good, moderate, and poor glycemic control.
Fig. 2.

O2− release by diabetic neutrophils is not insulin-dependent. Cells were stimulated with 1.0 μM fMLP or 300 nM PMA, and O2− release was measured as described in Materials and Methods. No differences in O2− release were observed between patients with type 1 and type 2 DM. These data include patients with good, moderate, and poor glycemic control.
Levels of PKC activity in neutrophils from patients with diabetes
PKC plays a major role in stimulating O2− release from neutrophils by catalyzing phosphorylation of the 47-kDa subunit of the oxidase (p47-phox; e.g., ref. [7]). Figure 3 compares Ca2+-dependent PKC activity in the soluble and membrane fractions of neutrophils from healthy individuals and diabetic patients (N=60). As was the case with O2− release, neutrophils from patients with good glycemic control (n=15) exhibited a nonsignificant increase in PKC, whereas neutrophils from patients with moderate (n=15) or poor glycemic control (n=15) exhibited significantly higher PKC activity in the soluble and membrane fractions (P<0.001, one-way ANCOVA; Fig. 3A). There were no significant differences between the various diabetic groups when the ratio of soluble to membrane-bound PKC activity was compared. Ca2+-dependent PKC activity in neutrophil cell lysates from diabetic patients was approximately twofold higher than that in lysates from normal neutrophils (P<0.001; one-way ANCOVA).
Fig. 3.
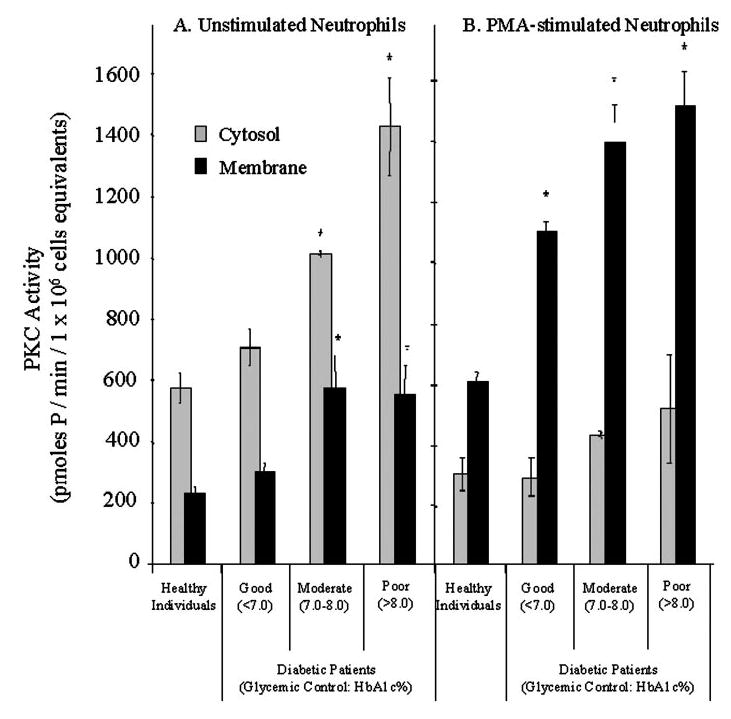
Ca2+-dependent PKC activity is increased in membranes of neutrophils from diabetic patients. Ca2+-dependent PKC activity was measured under optimal conditions in the 100,000 g soluble and membrane fractions of unstimulated (A) and stimulated neutrophils (B), which were stimulated with PMA (300 nM) for 10.0 min. (A) Unstimulated neutrophils from diabetic patients with moderate and poor glycemic control exhibited significantly higher PKC activity than neutrophils from healthy subjects and patients with well-controlled diabetes (#, P<0.05). (B) Redistribution of PKC activity from the soluble to the membrane fraction in PMA-stimulated neutrophils was significantly greater in cells from diabetic patients compared with healthy individuals, independent of glycemic control (*, P<0.05). Procedures for cell fractionation and the assaying Ca2+-dependent PKC activity are described in Materials and Methods.
Normal or diabetic neutrophils stimulated with PMA (300 nM) for 10 min significantly reduced soluble PKC activity and an increased membrane-bound activity (Fig. 3B). Neutrophils from all three groups of diabetic patients exhibited greater PKC activity in the membrane fraction after stimulation with PMA than cells from healthy individuals (P<0.05; one-way ANCOVA). Similar results were obtained with 1.0 μM fLMP (data not shown). Neutrophils from six nondiabetic patients with chronic periodontitis did not exhibit increased activity of PKC or enhanced O2− release upon stimulation with PMA or fMLP (data not shown).
To ascertain if the increased PKC activity resulted from activation of a static precursor pool or if there was an increase on total PKC protein that was being activated, total PKC was evaluated by Western blot. Figure 4 reveals that there is an increase in the total PKC pool with decreasing diabetic control.
Fig. 4.
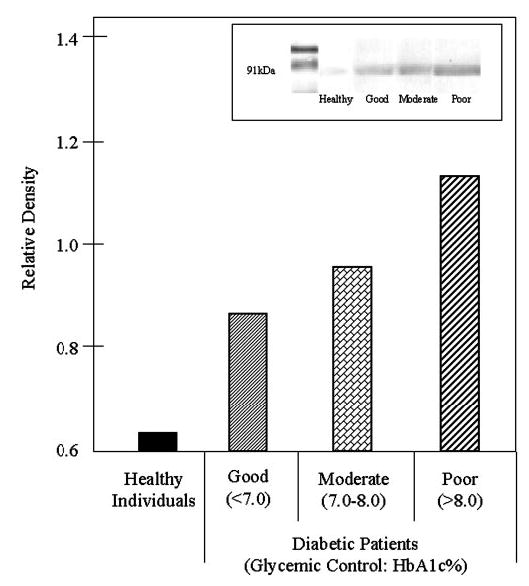
Total neutrophil PKC increases with poor glycemic control. Neutrophils were evaluated by Western blotting using an affinity-purified polyclonal antibody specific for PKC (inset). Band density was quantified by densitometry. The data reveal that PKC total protein is increased in diabetics, and the increase is associated with glycemic control.
To determine which isoform(s) of PKC was being up-regulated, a series of inhibitor experiments was performed. Neutrophils incubated with GF109203X, a selective inhibitor of PKC-α, -β1, and -β2, or staurosporine, a nonspecific and potent inhibitor of kinases, revealed that GF109203X returned O2− generation by primed diabetic neutrophils to normal levels, suggesting that the PKC-α/β isoforms are being up-regulated in diabetes (Fig. 5).
Fig. 5.
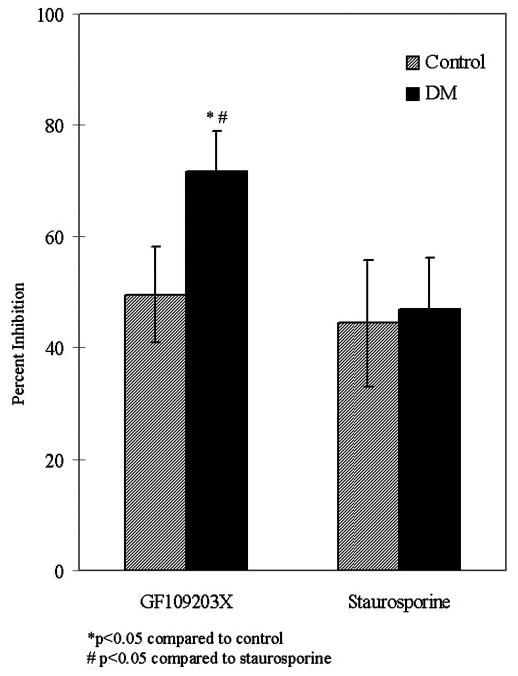
Inhibition of PKC suppresses O2− generation in healthy individuals and diabetic patients. Peripheral blood neutrophils were treated with GF109203X, a selective inhibitor of PKC-α,-β 1, and -β2 at 5 μM for 15 min, or staurosporine, a nonspecific, potent inhibitor of kinases at 0.1 μM for 10 min. O2− generation was then analyzed in response to fMLP (1.0 μM). GF109203X inhibited PKC activation ~50% more in diabetics compared with healthy individuals (*, P<0.05). Staurosporine inhibition was less effective, and there was no difference between patients and healthy control individuals. The effect of GF109203X on diabetic neutrophils was also significantly higher than the staurosporine-treated cells (#, P<0.05).
Diglyceride in neutrophils from healthy individuals and patients with diabetes
Certain diglycerides directly activate PKC and promote translocation of this kinase to the plasmalemma during cell stimulation (e.g., ref. [7]). Unstimulated neutrophils from diabetic patients were found to contain significantly more diglyceride than normal neutrophils, when cells from nine healthy individuals and nine diabetic patients were examined. Data summarized in Figure 6 included patients with good, moderate, and poor glycemic control. Diglyceride measured by this assay consists of a mixture of 1-O-acyl (i.e., diacylglycerol) and 1-O-alkyl diradylglycerols [39].
Fig. 6.
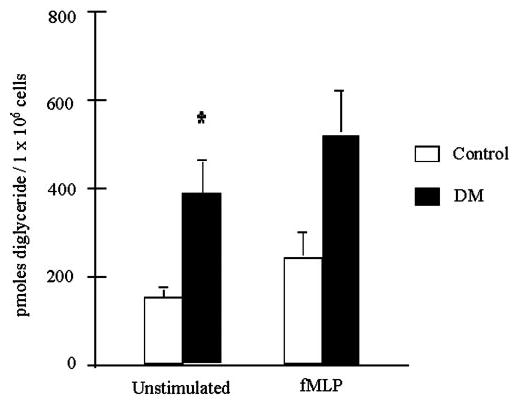
Diglycerides in neutrophils from diabetic patients and healthy, nondiabetic individuals. Diglyceride was measured in neutrophils from healthy individuals and diabetic patients before and after cell stimulation with 1.0 μM fMLP for 5.0 min. Unstimulated neutrophils from diabetic patients contained significantly more diglyceride than unstimulated neutrophils from healthy individuals (P<0.01). The diabetic patients used in these studies included individuals with good, moderate, and poor glycemic control. Diglyceride was measured as described in Materials and Methods.
Phosphorylation of p47-phox in neutrophils from normal individuals and patients with diabetes
Phosphorylation of p47-phox was examined in neutrophils from healthy individuals (control cells) and five diabetic patients with poor glycemic control. A pAb, which recognizes products of PKC [pPKC (S) Ab], was used in these studies to monitor the phosphorylation of p47-phox in PMA-stimulated neutrophils by Western blotting [38] (Fig. 7). The preferred sequence recognized by this pAb contained a phospho-Ser flanked by Lys or Arg at the −2 and +2 positions and a hydrophobic residue at the +1 position. This preferred sequence is similar to the optimal consensus sequence/phosphorylation sites recognized by the conventional Ca2+-dependent isoforms of the PKC family (α, βI, βII, γ) [40]. Although the magnitude of the response varied, four of the five patients exhibited enhanced phosphorylation of p47-phox at all time-points examined when compared with time-matched control cells (Fig. 7). The fifth patient exhibited a phosphorylation pattern similar to the control/normal neutrophils (data not shown). Western blotting with an antibody that recognizes phosphorylated and nonphosphorylated p47-phox indicated that the amounts of this protein were similar in neutrophils from the patients and controls (Fig. 7, lanes i and j). Immunoprecipitation studies have positively identified the band shown in Figure 7 as p47-phox [38].
Fig. 7.
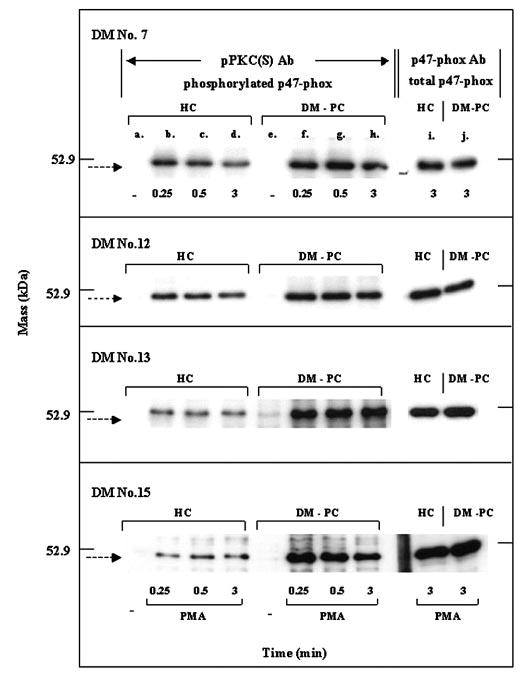
Phosphorylation of p47-phox in normal neutrophils and neutrophils from diabetic patients with poor glycemic control monitored by Western blotting with a pAb. Neutrophils from healthy individuals (HC; lanes a–d) and diabetes patients with poor glycemic control (DM-PC; lanes e–h) were stimulated with 300 nM PMA for the time periods indicated, and phosphorylation of p47-phox was measured by Western blotting with the pPKC (S) Ab. Neutrophils from the patients and healthy individuals were isolated and stimulated simultaneously. Lanes i and j show the total amount of p47-phox in these subjects monitored with an antibody that recognizes the phosphorylated and nonphosphorylated forms of this protein. Data from four different patients are presented. Conditions for cell stimulation and Western blotting are provided in Materials and Methods.
Severity of periodontal disease and diabetes
Out of 100 diabetic subjects that were evaluated, 23% showed moderate periodontitis, and 39% had severe periodontal tissue destruction as assessed by clinical and radiographic analyses. Further evaluation of 50 diabetic subjects showed that the HbA1c in erythrocytes from peripheral blood increased with the severity of periodontitis (Fig. 8). Within the diabetic group, there was a positive and statistically significant correlation between the levels of HbA1c and the severity of periodontitis (r = 0.71, P<0.001). Diabetic patients with moderate (DM-MP) and severe chronic periodontitis exhibited poor glycemic control (significantly higher HbA1c) compared with diabetic patients without periodontitis (DM-NP; *, P<0.001, compared with DM-NP; #, P<0.005, compared with DM-MP).
Fig. 8.
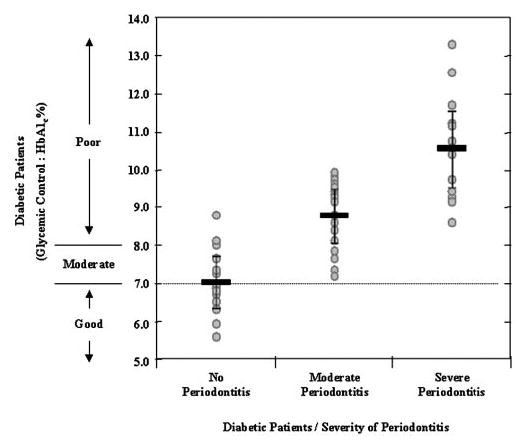
The severity of periodontitis is increased with poor glycemic control on diabetes. The mean level of HbA1c increases significantly as the severity of periodontitis increases (r = 0.71, P<0.001). The broken line indicates the threshold for “strict glycemic control” as the “treatment goal” recommended by American Diabetes Association. Diabetics with severe periodontitis have significantly higher HbA1c percent levels compared with diabetics with no periodontal disease (DM-NP; *, P<0.001) and those with moderate periodontitis (#, P<0.005). The severity of periodontitis was assessed by measuring “pocket depth” on two nonadjacent teeth as described in Materials and Methods. Chronic periodontitis alone did not lead to increased O2− production (data not shown). Data presented here were adjusted for the duration of diabetes.
DISCUSSION
Phagocytic leukocytes from diabetic patients release enhanced amounts of O2−/peroxide when compared with normal cells [8–11]. In this paper, we report that neutrophils from diabetic subjects exhibit increased activity of PKC, increased amount of diglyceride, and enhanced phosphorylation of p47-phox during cell stimulation, leading to increased O2− generation. This pathway represents a new form of neutrophil priming that clinically results in an increase in oxidative stress. In addition, we report a significant correlation between glycemic control (HbA1c levels) and the severity of periodontitis in diabetic patients, suggesting that the increased risk for inflammatory diseases, such as periodontitis in diabetes, is linked to increased inflammation and oxidative stress mediated by the neutrophil.
Certain biologically active lipids (e.g., platelet-activating factor; 5-hydroxyeicosatetraenoic acid), microbial products (lipopolysaccharide), cytokines (tumor necrosis factor α; granulocyte macrophage-colony stimulating factor), calcium ionophores, and AGE prime neutrophils to produce increased O2− in response to chemoattractants and suboptimal amounts of PMA [16–18, 41–46]. These agents enhance O2− through a variety of inter-related mechanisms, which include promoting association of PKC with the plasmalemma, increased generation of lipid-derived second messenger molecules [phosphoinositide-3 kinase, phospholipase A2 (PLA2) and PLD, sphingosine 1-kinase], and/or preassembly of the oxidase on the plasmalemma [18, 41–46]. To our knowledge, neutrophils from diabetic patients with moderate or poor glycemic control represent the first example where these cells are primed to respond to an optimal amount of PMA or fMLP and contain increased PKC activity in the cytosolic and membrane fractions. PMA binds to and activates PKC [7], which in turn, catalyzes the phosphorylation of p47-phox on multiple sites (e.g., ref. [6]). p47-phox forms a stable complex with the β-isoforms of PKC, and this interaction promotes phosphorylation of this substrate [47, 48]. The rate of O2− release from PMA-stimulated neutrophils is proportional to the amount of phosphorylated p47-phox [7, 49]. Data in the present study support a mechanism in which elevated PKC activity in diabetic neutrophils results in enhanced O2− release.
Hyperglycemia promotes de novo synthesis of diglyceride in various cells, which can trigger an increased translocation/redistribution of PKC to the membrane [12, 13]. Neutrophils from diabetic subjects contain elevated diglycerides (Fig. 6), which can explain the increases in membrane-bound PKC activity found in these cells (Fig. 3). However, an increase in translocation cannot explain the enhanced activity of soluble PKC, which was also observed in neutrophils from diabetic patients with moderate or poor glycemic control (Fig. 3). Data from Western blots indicate that these increases in PKC activity are a result of an increased content of enzyme protein. As the antibodies available are specific to PKC-α, -β1, and -β2, the data are consistent with previous reports suggesting an increase in PKC-β1 activity in diabetes. This was confirmed using α/β-specific inhibitors of PKC. Hyperglycemia can increase PKC activity in cultured THP-1 cells and in human monocytes in vivo [50, 51]. Increased PKC in diabetic neutrophils is consistent with our observation that the priming of these cells is stable and was not reversible simply by washing the cells. In contrast, washing might be expected to reverse priming events that are dependent solely on the constant occupation of certain receptors [17].
The NADPH-oxidase complex and its homologs have been identified in a variety of nonmyeloid cells/tissues, where they participate in a number of diverse physiological and pathological processes that include insulin signaling, blood vessel tonicity, and atherogenesis [52–54]. Hyperglycemia/chronic activation of PKC triggers activation of the oxidase, and the resulting increase in O2−/H2O2 has been implicated in the atherosclerosis and nephropathy that accompany diabetes [55, 56]. Thus, understanding how the NADPH oxidase is altered in neutrophils by diabetes may provide important insights into the dysregulation of this enzyme complex in nonmyeloid cells.
An interesting finding with respect to the clinical management of the diabetic patient is the relationship/correlationbetween elevated HbA1c, enhanced O2− release by neutrophils (Fig. 1), and the severity of periodontitis (Fig. 8). It is interesting that there were no differences amongst the diabetic groups in other laboratory values, such as mean cholesterol, triglyceride, or high-density lipoprotein (Table 1). This finding is consistent with previous studies suggesting a reciprocal relationship between diabetes and periodontal disease [21, 22, 24, 57]. O2− and/or H2O2 have been implicated in the destruction of periodontal tissues, and periodontitis has been characterized as an inflammatory disease [57]. Moreover, the increased risk for periodontitis in diabetics and the increased severity of diabetes in patients with periodontitis suggest a common inflammatory pathway leading to increased severity of both diseases. We have recently reported that lipoxins and their stable analogs, which block neutrophil chemotaxis and O2− production in vitro, dramatically reduce periodontitis in a rabbit model [58]. A selective antagonist of PKC-β can prevent some of the complications of type 2 diabetes in a rodent model [59], and clinical studies are now under way to evaluate the long-term usefulness of these compounds in humans [13]. It will be important to determine if treatments targeted at limiting the inflammatory response reduce the severity of periodontitis in diabetics without compromising the antimicrobial properties of phagocytic leukocytes in these patients.
Acknowledgments
These studies were supported by National Institutes of Health Grants PO1 DE13499, PO1 DE13191, R21 DE14478, RR00533, DE14066, DK50015, and AI23323. M. K. is a recipient of a scholarship provided by King Abdulaziz University, Jeddah, Saudi Arabia.
References
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 2.Gillum RF, Mussolino ME, Madans JH. Diabetes mellitus, coronary heart disease incidence, and death from all causes in African American and European American women: The NHANES I epidemiologic follow-up study. J Clin Epidemiol. 2000;53:511–518. doi: 10.1016/s0895-4356(99)00208-5. [DOI] [PubMed] [Google Scholar]
- 3.Robinson JM, Badwey JA. The NADPH oxidase complex of phagocytic leukocytes: a biochemical and cytochemical view. Histochem Cell Biol. 1995;103:163–180. doi: 10.1007/BF01454021. [DOI] [PubMed] [Google Scholar]
- 4.Allen LA, DeLeo FR, Gallois A, Toyoshima S, Suzuki K, Nauseef WM. Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood. 1999;93:3521–3530. [PubMed] [Google Scholar]
- 5.Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell. 2003;113:343–355. doi: 10.1016/s0092-8674(03)00314-3. [DOI] [PubMed] [Google Scholar]
- 6.Fontayne A, Dang PM, Gougerot-Pocidalo MA, El-Benna J. Phosphorylation of p47phox sites by PKCα, β II, δ, and ζ: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41:7743–7750. doi: 10.1021/bi011953s. [DOI] [PubMed] [Google Scholar]
- 7.Heyworth PG, Badwey JA. Protein phosphorylation associated with the stimulation of neutrophils. Modulation of superoxide production by protein kinase C and calcium. J Bioenerg Biomembr. 1990;22:1–26. doi: 10.1007/BF00762842. [DOI] [PubMed] [Google Scholar]
- 8.Hiramatsu K, Arimori S. Increased superoxide production by mononuclear cells of patients with hypertriglyceridemia and diabetes. Diabetes. 1988;37:832–837. doi: 10.2337/diab.37.6.832. [DOI] [PubMed] [Google Scholar]
- 9.Shah SV, Wallin JD, Eilen SD. Chemiluminescence and superoxide anion production by leukocytes from diabetic patients. J Clin Endocrinol Metab. 1983;57:402–409. doi: 10.1210/jcem-57-2-402. [DOI] [PubMed] [Google Scholar]
- 10.Cantero M, Parra T, Conejo JR. Increased hydrogen peroxide formation in polymorphonuclear leukocytes of IDDM patients. Diabetes Care. 1998;21:326–327. doi: 10.2337/diacare.21.2.326. [DOI] [PubMed] [Google Scholar]
- 11.Devaraj S, Jialal I. Low-density lipoprotein postsecretory modification, monocyte function, and circulating adhesion molecules in type 2 diabetic patients with and without macrovascular complications: the effect of α-tocopherol supplementation. Circulation. 2000;102:191–196. doi: 10.1161/01.cir.102.2.191. [DOI] [PubMed] [Google Scholar]
- 12.Sheetz MJ, King GL. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA. 2002;288:2579–2588. doi: 10.1001/jama.288.20.2579. [DOI] [PubMed] [Google Scholar]
- 13.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Festa A, Schmolzer B, Schernthaner G, Menzel EJ. Differential expression of receptors for advanced glycation end products on monocytes in patients with IDDM. Diabetologia. 1998;41:674–680. doi: 10.1007/s001250050967. [DOI] [PubMed] [Google Scholar]
- 16.Collison KS, Parhar RS, Saleh SS, Meyer BF, Kwaasi AA, Hammami MM, Schmidt AM, Stern DM, Al-Mohanna FA. RAGE-mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs) J Leukoc Biol. 2002;71:433–444. [PubMed] [Google Scholar]
- 17.Wong RK, Pettit AI, Davies JE, Ng LL. Augmentation of the neutrophil respiratory burst through the action of advanced glycation end products: a potential contributor to vascular oxidant stress. Diabetes. 2002;51:2846–2853. doi: 10.2337/diabetes.51.9.2846. [DOI] [PubMed] [Google Scholar]
- 18.Wong RK, Pettit AI, Quinn PA, Jennings SC, Davies JE, Ng LL. Advanced glycation end products stimulate an enhanced neutrophil respiratory burst mediated through the activation of cytosolic phospholipase A2 and generation of arachidonic acid. Circulation. 2003;108:1858–1864. doi: 10.1161/01.CIR.0000089372.64585.3B. [DOI] [PubMed] [Google Scholar]
- 19.Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, Kislinger T, Lu Y, Stern DM, Schmidt AM. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 2000;105:1117–1124. doi: 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lalla E, Lamster IB, Stern DM, Schmidt AM. Receptor for advanced glycation end products, inflammation, and accelerated periodontal disease in diabetes: mechanisms and insights into therapeutic modalities. Ann Periodontol. 2001;6:113–118. doi: 10.1902/annals.2001.6.1.113. [DOI] [PubMed] [Google Scholar]
- 21.Grossi SG, Genco RJ. Periodontal disease and diabetes mellitus: a two-way relationship. Ann Periodontol. 1998;3:51–61. doi: 10.1902/annals.1998.3.1.51. [DOI] [PubMed] [Google Scholar]
- 22.Grossi SG, Skrepcinski FB, DeCaro T, Robertson DC, Ho AW, Dunford RG, Genco RJ. Treatment of periodontal disease in diabetics reduces glycated hemoglobin. J Periodontol. 1997;68:713–719. doi: 10.1902/jop.1997.68.8.713. [DOI] [PubMed] [Google Scholar]
- 23.Loe H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care. 1993;16:329–334. [PubMed] [Google Scholar]
- 24. Diabetes and periodontal diseases. Committee on Research, Science and Therapy American Academy of Periodontology. J Periodontol. 2000;71:664–678. doi: 10.1902/jop.2000.71.4.664. [DOI] [PubMed] [Google Scholar]
- 25.Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the Expert Committee on the diagnosis and classification of diabetes mellitus. Diabetes Care. 2003;26 (Suppl 1):S5–S20. doi: 10.2337/diacare.26.2007.s5. [DOI] [PubMed] [Google Scholar]
- 26.American Diabetes Association. Standards of medical care for patients with diabetes mellitus. Diabetes Care. 2003;26 (Suppl 1):S33–S50. doi: 10.2337/diacare.26.2007.s33. [DOI] [PubMed] [Google Scholar]
- 27.Genco, R. J. (1990) Classification and clinical and radiographic features of periodontal disease. In Contemporary Periodontics (R. J. Genco, H. M. Goldman, D. W. Cohen, eds.), St. Louis, MO, C. V. Mosby.
- 28.Kalmar JR, Arnold RR, Warbington ML, Gardner MK. Superior leukocyte separation with a discontinuous one-step Ficoll-Hypaque gradient for the isolation of human neutrophils. J Immunol Methods. 1988;110:275–281. doi: 10.1016/0022-1759(88)90115-9. [DOI] [PubMed] [Google Scholar]
- 29.Cohen HJ, Chovaniec ME. Superoxide generation by digitonin-stimulated guinea pig granulocytes. A basis for a continuous assay for monitoring superoxide production and for the study of the activation of the generating system. J Clin Invest. 1978;61:1081–1087. doi: 10.1172/JCI109007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfson M, McPhail LC, Nasrallah VN, Snyderman R. Phorbol myristate acetate mediates redistribution of protein kinase C in human neutrophils: potential role in the activation of the respiratory burst enzyme. J Immunol. 1985;135:2057–2062. [PubMed] [Google Scholar]
- 31.Kurihara H, Murayama Y, Warbington ML, Champagne CM, Van Dyke TE. Calcium-dependent protein kinase C activity of neutrophils in localized juvenile periodontitis. Infect Immun. 1993;61:3137–3142. doi: 10.1128/iai.61.8.3137-3142.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bell RM, Hannun Y, Loomis C. Mixed micelle assay of protein kinase C. Methods Enzymol. 1986;124:353–359. doi: 10.1016/0076-6879(86)24027-6. [DOI] [PubMed] [Google Scholar]
- 33.Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Staurosporine, a potent inhibitor of phospholipid/Ca++-dependent protein kinase. Biochem Biophys Res Commun. 1986;135:397–402. doi: 10.1016/0006-291x(86)90008-2. [DOI] [PubMed] [Google Scholar]
- 34.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 35.Matsumoto H, Sasaki Y. Staurosporine, a protein kinase C inhibitor, interferes with proliferation of arterial smooth muscle cells. Biochem Biophys Res Commun. 1989;158:105–109. doi: 10.1016/s0006-291x(89)80183-4. [DOI] [PubMed] [Google Scholar]
- 36.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 37.Preiss J, Loomis CR, Bishop WR, Stein R, Niedel JE, Bell RM. Quantitative measurement of sn-1,2-diacylglycerols present in platelets, hepatocytes, and ras- and sis-transformed normal rat kidney cells. J Biol Chem. 1986;261:8597–8600. [PubMed] [Google Scholar]
- 38.Ohira T, Zhan Q, Ge Q, VanDyke T, Badwey JA. Protein phosphorylation in neutrophils monitored with phosphospecific antibodies. J Immunol Methods. 2003;281:79–94. doi: 10.1016/s0022-1759(03)00278-3. [DOI] [PubMed] [Google Scholar]
- 39.Tyagi SR, Burnham DN, Lambeth JD. On the biological occurrence and regulation of 1-acyl and 1-O-alkyl-diradylglycerols in human neutrophils. Selective destruction of diacyl species using Rhizopus lipase. J Biol Chem. 1989;264:12977–12982. [PubMed] [Google Scholar]
- 40.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 41.Badwey JA, Robinson JM, Horn W, Soberman RJ, Karnovsky MJ, Karnovsky ML. Synergistic stimulation of neutrophils. Possible involvement of 5-hydroxy-6,8,11,14-eicosatetraenoate in superoxide release. J Biol Chem. 1988;263:2779–2786. [PubMed] [Google Scholar]
- 42.Bass DA, McPhail LC, Schmitt JD, Morris-Natschke S, McCall CE, Wykle RL. Selective priming of rate and duration of the respiratory burst of neutrophils by 1,2-diacyl and 1-O-alkyl-2-acyl di-glycerides. Possible relation to effects on protein kinase C. J Biol Chem. 1988;263:19610–19617. [PubMed] [Google Scholar]
- 43.DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, Nauseef WM. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest. 1998;101:455–463. doi: 10.1172/JCI949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cadwallader KA, Condliffe AM, McGregor A, Walker TR, White JF, Stephens LR, Chilvers ER. Regulation of phosphatidyl-inositol 3-kinase activity and phosphatidylinositol 3,4,5-trisphosphate accumulation by neutrophil priming agents. J Immunol. 2002;169:3336–3344. doi: 10.4049/jimmunol.169.6.3336. [DOI] [PubMed] [Google Scholar]
- 45.Ward RA, Nakamura M, McLeish KR. Priming of the neutrophil respiratory burst involves p38 mitogen-activated protein kinase-dependent exocytosis of flavocytochrome b558-containing granules. J Biol Chem. 2000;275:36713–36719. doi: 10.1074/jbc.M003017200. [DOI] [PubMed] [Google Scholar]
- 46.MacKinnon AC, Buckley A, Chilvers ER, Rossi AG, Haslett C, Sethi T. Sphingosine kinase: a point of convergence in the action of diverse neutrophil priming agents. J Immunol. 2002;169:6394–6400. doi: 10.4049/jimmunol.169.11.6394. [DOI] [PubMed] [Google Scholar]
- 47.Curnutte JT, Erickson RW, Ding J, Badwey JA. Reciprocal interactions between protein kinase C and components of the NADPH oxidase complex may regulate superoxide production by neutrophils stimulated with a phorbol ester. J Biol Chem. 1994;269:10813–10819. [PubMed] [Google Scholar]
- 48.Reeves EP, Dekker LV, Forbes LV, Wientjes FB, Grogan A, Pappin DJ, Segal AW. Direct interaction between p47phox and protein kinase C: evidence for targeting of protein kinase C by p47phox in neutrophils. Biochem J. 1999;344:859–866. [PMC free article] [PubMed] [Google Scholar]
- 49.Okamura N, Curnutte JT, Roberts RL, Babior BM. Relationship of protein phosphorylation to the activation of the respiratory burst in human neutrophils. Defects in the phosphorylation of a group of closely related 48-kDa proteins in two forms of chronic granulomatous disease. J Biol Chem. 1988;263:6777–6782. [PubMed] [Google Scholar]
- 50.Venugopal SK, Devaraj S, Yang T, Jialal I. α-Tocopherol decreases superoxide anion release in human monocytes under hyperglycemic conditions via inhibition of protein kinase C-α. Diabetes. 2002;51:3049–3054. doi: 10.2337/diabetes.51.10.3049. [DOI] [PubMed] [Google Scholar]
- 51.Ceolotto G, Gallo A, Miola M, Sartori M, Trevisan R, Del Prato S, Semplicini A, Avogaro A. Protein kinase C activity is acutely regulated by plasma glucose concentration in human monocytes in vivo. Diabetes. 1999;48:1316–1322. doi: 10.2337/diabetes.48.6.1316. [DOI] [PubMed] [Google Scholar]
- 52.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ET, Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J Clin Invest. 2001;108:1513–1522. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86:E85–E90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- 54.Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, Lambeth JD, Goldstein BJ. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kitada M, Koya D, Sugimoto T, Isono M, Araki S, Kashiwagi A, Haneda M. Translocation of glomerular p47phox and p67phox by protein kinase C-β activation is required for oxidative stress in diabetic nephropathy. Diabetes. 2003;52:2603–2614. doi: 10.2337/diabetes.52.10.2603. [DOI] [PubMed] [Google Scholar]
- 56.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 57.Altman LC, Baker C, Fleckman P, Luchtel D, Oda D. Neutrophil-mediated damage to human gingival epithelial cells. J Periodontal Res. 1992;27:70–79. doi: 10.1111/j.1600-0765.1992.tb02088.x. [DOI] [PubMed] [Google Scholar]
- 58.Serhan CN, Jain A, Marleau S, Clish C, Kantarci A, Behbehani B, Colgan SP, Stahl GL, Merched A, Petasis NA, Chan L, Van Dyke TE. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J Immunol. 2003;171:6856–6865. doi: 10.4049/jimmunol.171.12.6856. [DOI] [PubMed] [Google Scholar]
- 59.Koya D, Haneda M, Nakagawa H, Isshiki K, Sato H, Maeda S, Sugimoto T, Yasuda H, Kashiwagi A, Ways DK, King GL, Kikkawa R. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC β inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J. 2000;14:439–447. doi: 10.1096/fasebj.14.3.439. [DOI] [PubMed] [Google Scholar]