

Abstract
Estrogen receptors (ERs) are localized to many sites within the cell, potentially contributing to overall estrogen action. In the nucleus, estrogen mainly modulates gene transcription, and the resulting protein products determine the cell biological actions of the sex steroid. In addition, a small pool of ERs localize to the plasma membrane and signal mainly though coupling, directly or indirectly, to G proteins. In response to steroid, signal transduction modulates both nontranscriptional and transcriptional events and impacts both the rapid and more prolonged actions of estrogen. Cross-talk from membrane-localized ERs to nuclear ERs can be mediated through growth factor receptor tyrosine kinases, such as epidermal growth factor receptor and IGF-I receptor. Growth factor receptors enact signal transduction to kinases such as ERK and phosphatidylinositol 3-kinase that phosphorylate and activate nuclear ERs, and this can also occur in the absence of sex steroid. A complex relationship between the membrane and nuclear effects of estrogen also involves membrane-initiated phosphorylation of coactivators, recruiting these proteins to the nuclear transcriptosome. Finally, large pools of cytoplasmic ERs exist, and some are localized to mitochondria. The integration of sex steroid effects at distinct cellular locations of its receptor leads to important cellular physiological outcomes and are manifest in both reproductive and nonreproductive organs.
Abbreviations: AP-1, Activator protein 1; E2, estradiol; EC, endothelial cell; ER, estrogen receptor; EGFR, epidermal growth factor receptor; eNOS, endothelial nitric oxide synthase; FKHRL1, Forkhead; GPR30, G protein-coupled receptor 30; GSK, glycogen synthase kinase; NLS, nuclear localization signal; PI3K, phosphatidylinositol 3-kinase; SERM, selective ER modulator
STEROID HORMONES HAVE traditionally been conceived to act exclusively through nuclear receptors (1). Regarding the estrogen receptor (ER), both nuclear and membrane/cytoplasmic pools of ERα and ERβ have been demonstrated (reviewed in Ref. 2), and the receptors have similar affinity for steroid ligand. Estrogen binding to nuclear receptors activates or represses gene transcription, resulting from the steroid-receptor complex binding to DNA at estrogen response elements in the promoters of target genes. A second, nonclassical mechanism involves the interaction of nuclear estradiol (E2)/ER with transcription factors, such as activator protein 1 (AP-1) or Sp-1, that in turn bind their cognate DNA elements (3). This leads to alteration of chromatin, histone unwinding, and interactions with components of the basal transcription machinery complex. In both scenarios, recruitment of coactivators (and displacement of core-pressors) to the sites of DNA binding modulates gene and subsequent protein expression.
In addition, steroid actions occur at the cell surface, a function that is conserved from plants to humans (2, 4, 5). It is widely appreciated that membrane ER signaling through kinase cascades, calcium, and other second messengers impacts transcription (6, 7). For instance, E2 stimulation of the cyclin D1 gene occurs through ERK or phosphatidylinositol 3-kinase (PI3K) activation, promoting G1/S cell cycle progression in breast cancer cells expressing endogenous ERs (8). Many sex steroid effects reflect the integration of actions at several receptor pools. Work over the past decade has identified important mechanisms whereby membrane-initiated signaling through growth factor receptors or membrane ERs impacts the function of nuclear ERs, as one model. Integrative signaling by E2 from several sites in the cell results both in rapid and prolonged actions, and provides plasticity for the cell response to the sex steroid.
In this review, I highlight the recent developments in understanding the nature of the membrane ER, and the emerging field of integration of membrane and nuclear receptor signaling. Applications of these mechanisms to physiological and pathophysiological models of estrogen action are emphasized, and new concepts of receptor cross-talk for both genomic and nongenomic actions are identified.
MEMBRANE ER
Much evidence favors the idea that the membrane-localized receptor is the same protein as the nuclear receptor, transported to the plasma membrane by unclear mechanisms. This idea is based on immunohistochemistry of the endogenous membrane ER, using a panel of antibodies directed against multiple epitopes of nuclear ERs (9), loss of endogenous ER protein detection at the membrane in cells transfected with an antisense oligonucleotide to nuclear ERα (10), and the codetection of membrane and nuclear ERs after nuclear ER cDNA expression in ER null cells (11). Chambliss et al. (12) have identified endogenous ERα and ERβ receptors of 67 and 54 kDa, respectively, in the caveolae and cell membranes from endothelial cells (ECs). This was done using antibodies against the classical nuclear ERα and ERβ. More recent data indicate that cells from the DERKO mouse (ERα and ERβ deleted) fail to show endogenous membrane or nuclear ERα or ERβ, by Western blot (13), E2 binding, and rapid signaling. Definitive proof that endogenous membrane and nuclear ERs are the same protein requires isolation and sequencing of the two receptor pools.
Work from the laboratory of Bender and associates (14) has reported a truncated, 46-kDa receptor as the predominant ERα isoform at the plasma membrane of an immortalized human EC line. However, several other laboratories working with isolated ECs and/or aorta have not identified the 46-kDa receptor to be abundant, including at the cell membrane (12, 13, 15).
Several reports have surfaced describing endogenous membrane ERs in neurons with slightly altered receptor pharmacology (16, 17). However, these receptors have not been isolated, and whether they exist in cells from DERKO mice is unknown at present. Non-ER or ER-indirectly mediated actions of E2 have also been proposed, often based on the lack of antagonism of E2 effects by ICI182780 (18). Perhaps this results from E2 facilitating the action of membrane-acting proteins (sex hormone-binding globulin) (19) or the allosteric modulation of receptors at the cell membrane (20). The full nature and relevance of these responses are unclear.
Work from several laboratories has affirmed the ability of endogenous or expressed ERs to activate G protein-related signaling at the plasma membrane (11, 21, 22). Membrane ERs possibly exist as a cytoplasmic pool tethered to the inner face of the plasma membrane bilayer through binding to proteins such as caveolin-1 (23), in conjunction with MNAR (24), or Shc and IGF-I (25). Alternatively, evidence exists for endogenous ERα within isolated caveolae vesicles shorn from the plasma membrane of ECs, as well as non-caveolar compartments of the membrane (12, 23, 26). Caveolae are typically spread throughout the cell membrane, and therefore ERs could habituate within the plasma membrane at times, like growth factor receptors. It is likely that in a dynamic fashion, both models are correct, and endogenous membrane-localized ERs also spend time in non-clathrin-coated endosomes in the cytoplasm (27).
Whether ERs span the plasma membrane or contain an extracellular ligand binding region is controversial. The membrane-impermeant estrogen conjugate, E2-BSA, has been used by many laboratories to support E2 action at membrane ERs. However, it has been reported that this compound dissociates into BSA and E2 (28), requiring filtration and careful handling of this reagent. Furthermore, BSA is well known to be taken up into caveolae in the plasma membrane (29), and therefore E2-BSA can probably access ERs in this plasma membrane raft. Thus, this compound cannot be used as definitive proof that there is externalization of a segment of ERs beyond the plasma membrane. Another approach to this issue is to carry out functional studies or immunohistochemistry with antibodies to ER (9). The presumption is that antibodies do not gain access to the cell interior and therefore identify a functional extracellular domain of ERs. However, it is not clear that cells are impermeable to the antibody intruding into the plasma membrane, and thus this reagent may suffer from the same limitations as E2-BSA.
Several reports suggest that the ability of E2 to activate G proteins is mediated through an orphan G protein-coupled receptor, GPR30. However, the pharmacology of this interaction is atypical, as 17β-E2, equimolar 17α-E2, and even ICI 182780 trigger the GPR30-related response (30). One report indicated that this interaction with GPR30 required micromolar E2, perhaps indicating an allosteric modulation with questionable physiological significance (31). Furthermore, antisense oligonucleotides to GPR30 failed to prevent E2 signaling through ERK to proliferation in breast cancer (32). A recent report from Thomas et al. (33) identified GPR30 as a low-capacity receptor for E2, one capable of supporting a modest generation of cAMP. It was undetermined whether E2-GPR30 interactions contribute to the overall signaling by E2 at the plasma membrane or the downstream physiology effects in breast cancer cells.
The endogenous membrane receptor assembles as part of a large signalsome complex that includes G proteins, receptor tyrosine kinases (EGFR, IGF-I receptor), and nonreceptor tyrosine kinases, such as Src (34). In a confined space, undetermined dynamics of signal molecule configuration lead to discrete G protein subunit activation. For instance, E2/ER coimmunoprecipitates with and activates Gsα and Gqα in transfected or endogenous ER cell models (11, 13), and Giα in ECs (21). Gα and Gβγ activation lead to downstream augmentation of kinase activities, such as ERK and PI3K in both breast cancer cells and ECs. This signaling impacts the cellular actions of E2 (21, 34, 35). In some instances, the association of ER with G proteins may be indirect. E2 signaling to endothelial nitric oxide synthase (eNOS) activation in ECs may involve the striatin protein, which binds to ER, Gαi, and caveolin-1 (36). This protein may help assemble and activate the eNOS-signaling cassette at the plasma membrane.
Recent work has defined motifs in the E domain of ERα that are critical to membrane localization and function (37). This includes E domain (ligand-binding domain) residues that are necessary for dimerization of the endogenous membrane ERα and ERβ (13). Mutation of these motifs prevents both receptor dimerization and signaling through ERK, PI3K, and cAMP: loss of the former signals prevents the cell survival action of E2 in breast cancer cells (13). In contrast, signaling to eNOS activation in ER-transfected COS cells may not require membrane ER dimerization (37). Another residue, S522 of ERα, is necessary for the physical association of this receptor with caveolin-1 protein, a protein the N-terminus scaffolding domain of which facilitates ER transport from the cytosol to the membrane (38). Interactions between the A/B domain of ERα and Shc may also contribute to membrane localization through unclear transport mechanisms (39).
Very recent studies have identified an important modification of ERα that facilitates both caveolin-1 binding and membrane localization. Marino and colleagues (40, 41) have shown that cystine 447 of transfected human ERα is crucial to steroid-independent palmitoylation of the receptor. This E domain amino acid is necessary for ERα to physically associate with the caveolin-1 protein and localize at the membrane. Mutation of this single amino acid or inhibition of palmitoylation with 2-Bromopalmitate results in a significant decrease of expressed receptors at the plasma membrane, compared with wild-type ER expression. Furthermore, cystine 447-mutated ERα does not support proliferative signaling through ERK and PI3K (41). Palmitoylation as a possible mechanism for membrane ER localization was also suggested from previous studies in endothelial cells (14). A partial model for membrane localization involves palmitoylation of ERα to promote binding to caveolin-1 (which itself is palmitoylated). Subsequently, the membrane localization sequence of caveolin-1 (scaffolding domain) is crucial for ER transport to and localization at the membrane (38).
Other proteins may also facilitate the membrane localization of ERs, including MNAR, Shc and growth factor receptors, and striatin (21, 22, 36). Shc and striatin have been reported to interact with the A/B domain of ERα (25, 36) whereas MNAR binds the E domain of the steroid receptor (24). Interestingly, A/B and C domain-deleted ERα localizes to the membrane and signals to ERK, in the same manner as to wild-type ERα (38, 41). This is consistent with the idea that the E domain contains most of the information for both localization and function of ERα at the membrane (42). Very recently, elements within the nuclear localization sequence (NLS) of ERα (D domain) were reported as required for E2-induced ERK and PI3K and nitric oxide production through nitric oxide synthase (eNOS) activation in transfected COS cells (37). Although this may be specific to eNOS activation, Zhang et al. (43) previously reported that expression of an NLS-deleted ERα, targeted to the cell membrane, supported ERK activation by E2. Thus the extent of the role of the NLS in membrane ER function is not clear.
Could nuclear ER also rapidly up-regulate kinase activity? As mentioned, NLS-deficient ERs have been reported to either affect or not affect rapid signaling by E2 (37, 43). When the E domain of ERα or the full-length receptor is targeted to the nucleus, no rapid activation of ERK is detected (8). Instead, when either construct is targeted exclusively to the plasma membrane, multiple rapid signals are generated in response to E2 (44). Thus, the preponderance of available evidence suggests that the membrane localization of ERs is necessary for kinase activation, consistent with the localization and signaling by growth factor and typical G protein-coupled receptors.
MEMBRANE ER CROSS-TALK TO THE NUCLEUS
Impact of Membrane/Cytoplasmic Signaling on Nuclear ERs
Signaling from the membrane ER to the nucleus potentially impacts various functions of the sex steroid (Fig. 1). In breast cancer, the endogenous membrane ER cross-talks to the trans activation of the EGF, ErbB2, and IGF-I receptors (30, 45, 46). This results in downstream signaling to ERK MAPK and PI3 /AKT kinases. Important nonreceptor tyrosine kinases such as Src are also involved as intermediary steps (34). ERK and PI3K phosphorylate discrete residues of the endogenous nuclear ERs, up-regulating its transcriptional activity or stability (47, 48). In response to EGF or IGF-I, the nuclear ER can be activated in steroid ligand-independent fashion (47). As an example, kinase-induced phosphorylation of nuclear ERα on serine 305 enhances cyclin D1 transcription in breast cancer (49). Activation from the membrane of PI3/AKT kinases can repress the downstream inhibitory actions of kinases. PI3K/AKT phosphorylation of glycogen synthase kinase (GSK)-3β (50) disinhibits the repressive effect of GSK-3β on ERα serine 118 phosphorylation. This results in enhanced transcriptional action of the nuclear ER.
Fig. 1.
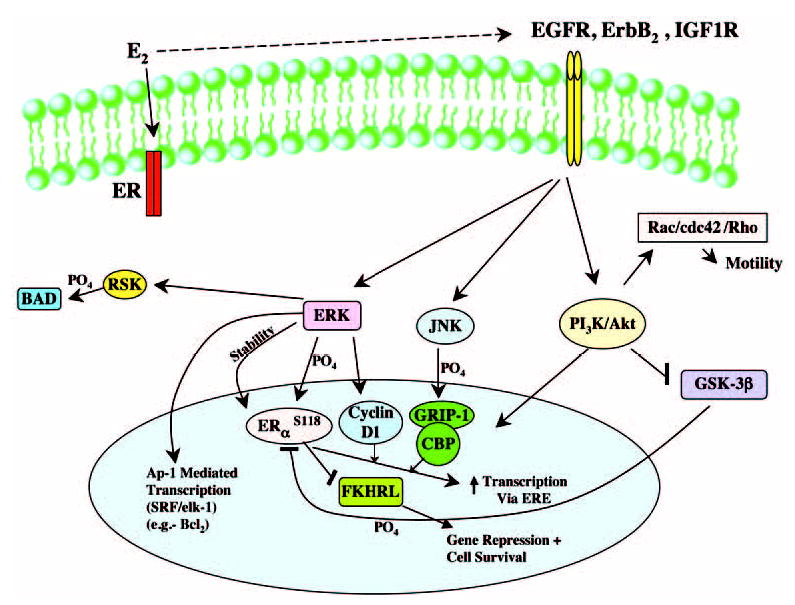
Integrative Signaling by Estrogen through Receptors (ER) at the Membrane and Nucleus in Breast Cancer IGF1R, IGF-I receptor; JNK, c-Jun N-terminal kinase; RSK, ribosomal S6 kinase; ERE, estrogen response element; CBP, cAMP response element binding protein (CREB)-binding protein; SRF, serum response factor.
Membrane signaling also impacts the phosphorylation and recruitment of coactivator proteins. This augments the unique participation of cyclin D1 (51), or more traditional steroid receptor coactivator proteins, such as glucocorticoid receptor-interacting protein 1 (52), steroid receptor coactivator 1, or cAMP response element-binding protein (CREB)-binding protein (53). One can envisage a carefully controlled modulation of nuclear ER-induced transcription, depending upon which signaling pathway(s) are activated by E2 in a given cell context. It is likely that discrete signaling pathways also (negatively) regulate the access of corepressor proteins to promoters of target genes, although this mechanism is not well studied. As a corollary to this, phosphorylation of coactivators at discrete motifs may be inhibitory as well.
Receptor phosphorylation also impacts selective ER modulator (SERM) effects. Tamoxifen has recently been reported to convert from an antagonist to an agonist in breast cancer, depending upon Ser 305 phosphorylation (54). This site can be phosphorylated by both protein kinase A (54) and p21-activated kinase 1 (49). Circulating or locally produced E2 or growth factors acting at the membrane could potentially stimulate this nuclear ER modification via protein kinase A (55) and contribute to tamoxifen resistance in women (56).
Membrane ER Signaling to Nuclear ER-Independent Functions
Activation of target gene transcription also occurs through membrane signaling, independently of nuclear ERs. ERK activation up-regulates AP-1 mediated genes (e.g. c-fos) (3, 52, 57). This results in part from serum response factor/elk-1 stimulation by E2, and in part by recruitment of nuclear ERs and coactivators to AP-1 sites on gene promoters. Similarly, PI3K activation by E2-induced signaling from the endogenous membrane ER rapidly up-regulates hundreds of genes in a target cell (58). One such E2-induced gene, cyclooxygenase-2, is regulated by signaling to nuclear factor-κB, potentially contributing to an important cell biological outcome of tumor angiogenesis (59).
The loss of inhibition produced by tumor suppressor gene mutation often underlies carcinogenesis (60). Such a role for membrane signaling can be proposed in steroid hormone-responsive cancers. PI3K/AKT inhibition of GSK-3β function allows β-catenin to translocate to the nucleus. β-Catenin cooperates with TCF/Lef transcription factors to up-regulate proliferation genes, such as cyclin D1 (61), contributing to carcinogenesis. PI3K/AKT activation also leads to decreased Forkhead (FKHRL1) transcription factor function, the inhibition of which results in cell survival (60). Thus, membrane signaling to the nucleus through these and other, undiscovered pathways contributes to estrogen-induced cell proliferation and survival, essential features of tumor biology. It has been estimated that more than 500 kinases are encoded within the human genome (62). The ability of membrane ERs and/or growth factor receptor tyrosine kinases to signal through multiple kinases to the nucleus undoubtedly impacts all aspects of cellular function.
NONTRANSCRIPTIONAL ACTIONS OF ER
Signaling from membrane ERs also induces the posttranslational modification of many existing proteins. This includes the phosphorylation and regulation of enzymes, such as kinases or phosphatases, that impact cell physiology. These enzymes act both within and outside the nucleus. As an example, E2 rapidly down-regulates MAPK phosphatase 1 activity, leading to the up-regulation of ERK activity in breast cancer cells within 10 min (8). However, E2 also cooperates with the BRCA1 tumor suppressor protein (63) to modulate MAPK phosphatase 1 production, stemming from nuclear action. The overall regulation impacts the proliferation and survival of the cancer and represents an important integration of E2 action, occurring in both membrane/cytoplasm and nucleus. Loss of restraint of kinase signaling underlies the interaction between BRCA1 and ER that may contribute to breast cancer when BRCA1 is mutated (8, 63).
Signaling by membrane ERs to PI3K reveals additional integrated functions in breast cancer. PI3K activation causes the phosphorylation of BAD, which is then sequestered by 14–3-3 proteins in the cytoplasm (64). Sequestration dissociates phosphorylated BAD from binding the antiapoptotic proteins, Bcl2 and Bcl-xl, which allows these proteins to maintain mitochondrial membrane integrity and prevent the release of cytochrome c and subsequent cell death. In some instances, downstream signaling by E2 to additional kinase substrates (mammalian target of rapamycin for PI3K/Akt or p70S6 kinase for AKT or ERK) is required, and these target signals expand the potential repertoire of E2 actions. For instance, the p70S6 kinase modulates protein translation and is important for sensing and responding to the nutrient status of the organism (65). The localization and duration of kinase signaling may importantly contribute to E2 actions in these respects. For instance, the ability of estrogen to rescue osteocytes or induce cell death in osteoclasts is dictated, in part, by the duration of ERK activation, and whether this kinase signals predominantly in cytoplasm or nucleus (66, 67).
E2 also promotes the motility of cells through signaling via p38 (35) and perhaps PI3K pathways (68). The latter signal activates the small GTP proteins Rac and Cdc42, which in conjunction with Rho kinase, modulate target cell migration (69). Cell motility is ultimately driven by the formation of lamellipodia and extension of filipodia, and estrogen promotes this (25). Motility depends on nucleated actin-related processes, the synthesis of capping proteins, and association of ARP 2/3 and Ena/VASP regulatory proteins with actin-barbed ends (70). These proteins are potential substrates for posttranslational modification by E2 signaling.
ERs IN CYTOPLASM
The ability of E2 to act at receptor pools throughout the cell brings into focus the issue of cytoplasmic ERs that are not proximal to the cell membrane or nucleus. One model is that this pool of receptors is in the process of translocating to other sites in the cell. Dynamic imaging using techniques such as fluorescence resonance energy transfer will be needed to determine whether this occurs. However, mitochondria have been identified to respond to estrogenic compounds, both in whole cell or isolated organelle preparations (71, 72). Saturation binding of sex steroid to putative ER in the mitochondria has been proposed for many years, but often these studies identified a low-affinity receptor, requiring micromolar E2 for the activation of a mitochondrial metabolic process. More recent studies have identified high-affinity E2 binding proteins (73, 74) that, by partial peptide degradation and MALDI-TOF mass spectrometry, are identical to nuclear ERs (74). Currently, the functions of this pool of ERs are unknown.
CELL PHYSIOLOGY OUTCOMES OF RAPID SIGNALING BY E2
Understanding how estrogen action at the membrane contributes to the overall cellular functions of the steroids has been facilitated by the development of reagents that act in a relatively specific fashion at the membrane ER. One such compound, estren, activates signal transduction to a variety of kinases. This signaling by estren or E2 subsequently activates transcription factors to rescue HeLa or bone cells from apoptotic cell death (44, 76, 77). Cell survival contributes to the ability of this weakly ER-binding steroid to prevent osteoporosis in vivo, yet estren does not stimulate breast or uterine proliferation (77). This compound apparently does not have the ability to activate genes through nuclear ERs.
From this work, it has been proposed that estrogenic compounds that only act at the cell membrane might not stimulate the proliferation of cells, and thus not promote breast or uterine neoplasia. This concept requires rigorous demonstration, because signaling through kinase activation strongly promotes breast cancer cell proliferation in vitro and in vivo (78, 79). Developing agonists or antagonists that act strictly at membrane ERs should allow better understanding of the contributions of this receptor pool to overall E2 action and provide possible targets for therapeutic intervention.
Analogous to this, the development of newer generations of SERMS should consider whether their actions result from binding membrane and/or nuclear ERs. It has been shown that raloxifene activates the same kinase signaling pathways as E2, to trigger nitric oxide production in ECs (80, 81). This leads to enhanced cardiac perfusion and contractility of the ischemic heart in vivo (82). This is consistent with the ability of E2 to prevent ischemia-reperfusion injury in mice, via PI3K signaling from the membrane receptor (83).
Estrogenic compounds also prevent neuronal cell death, induced by cerebrovascular ischemia in vivo. Several groups have shown that E2 rescues rodents from neuronal apoptosis, in part through ERα up-regulation of cell survival genes (84–87). This may be related, in part, to signaling though PI3K (88, 89). E2 also prevents alcohol-induced neuronal injury by modifying protein kinase C activity (90). These results suggest that membrane ER signaling contributes to the overall neuroprotective effects of the sex steroid. In this regard, rapidly signaling ERαs are localized to the neurites, and not the nuclei of neurons (91). These in vivo data have resulted in the development of estrogen compounds that protect neurons against insult-induced cell death but bind poorly to ERα or ERβ (87). ER-independent neuroprotective effects of estrogenic compounds during stroke may be related to the prevention of reactive oxygen species formation (92). In other work, estrogen rapidly signals to CREB phosphorylation and ERK activation in neurons. This is mediated by the classical ER (α or β) expressed in a specific brain region, and was shown using ER knockout mice (93).
Estrogen action certainly impacts ovarian follicle development, and this may reflect integrated functions of membrane and nuclear ERs. An important family of transcription factors in follicle development are the Forkhead proteins. In vivo and in vitro rodent models indicate that the transcription and actions of Foxo1, FKHRL1, and AFX are regulated by growth factors and estrogen and depend on kinase modulation (94). For instance, its is well recognized that PI3K/AKT phosphorylates and thus sequesters Forkhead proteins in the cytoplasm (50), thus limiting transcriptional activity. This is a pathway that E2 activates in virtually all ER-producing cell types investigated. Activation of this pathway in ovarian cancer by the sex steroid leads to increased telomerase gene expression and activity, thus contributing to tumor propagation (95). E2 activates p21-activated kinase in breast cancer cells, also leading to FKHR phosphorylation/sequestration (96). In breast cancer, hypersensitivity of estrogen-deprived cells corresponds to increased ERK activation and resulting Elk-1 transcription factor activation (97). Additional in vivo models utilizing physiological concentrations of estrogen will be necessary to define the pathophysiological roles of sex steroid signaling from the membrane to tumor promotion.
Tamoxifen resistance in breast cancer may be related to enhanced cross-talk between the membrane ER and EGFR family member receptors, most notably ErbB2 (HER-2). When ErbB2 is experimentally overexpressed, tamoxifen activates both ER and ErbB2 to signal downstream through ERK and PI3K to AIB1 phosphorylation (98). Tamoxifen acts as an agonist in this situation, promoting coactivator recruitment as a result of kinase signaling. EGFR tyrosine kinase inhibition reconverts tamoxifen to an antagonist and disassembles the positive transcription complex on the promoter of a target gene. An emerging therapeutic approach in this malignancy is estrogen purging, in which raloxifene-resistant cancer cells undergo E2-induced apoptosis (99). Thus, integrative signaling from the membrane through kinase-induced transcription, or posttranscriptional effects on protein function, contributes to the tumor-promoting actions of estrogen in vivo and may contribute to SERM resistance.
PERSPECTIVE
A complex interaction between ERs in various cellular locations affords the opportunity for exquisite control of E2 function. The cell context-specific environment impacts the integration of rapid signaling by E2 from the membrane, and subsequent nuclear transcription. This leads to different signal cascades or transcribed genes in response to the same steroid hormone, and a different cell biological outcome. In part, this is dictated by the different physiology and functions of diverse cell types.
Nevertheless, many cells divide, attempt to survive, migrate, and differentiate. These common functions allow us the opportunity to deduce cellular programs that are shared between steroid hormone-responsive cells. Such programs provide potential interventional targets to enhance or inhibit estrogen functions. Elucidating the details of these programs could provide a more informed approach to disorders resulting from reproductive organ dysfunction, the prevention of osteoporosis after the menopause, or sex steroid-responsive cancers. Estrogen-related reagents that target a particular receptor pool may be efficacious in these respects (25, 76), but we will have to better understand the integrative nature of hormonal action to avoid undesirable consequences of this approach.
Acknowledgments
I regret the inability to cite many fine contributions to this scientific area due to space limitations. I thank Christina Pedram for her illustration, and my scientific partners, Ali Pedram and Mahnaz Razandi, for their dedicated and excellent work.
Footnotes
This work was supported by grants from the Veterans Administration Research Service and the National Institutes of Health.
References
- 1.Jensen EV, Jacobson HI. Basic guides to the mechanism of estrogen action. Recent Prog Horm Res. 1962;18:387–414. [Google Scholar]
- 2.Levin ER. Cell Localization, physiology and non-genomic actions of estrogen receptors. J Applied Physiol. 2001;91:1860–1867. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- 3.Kushner PJ, Agard D, Feng WJ, Lopez G, Schiau A, Uht R, Webb P, Greene G. Oestrogen receptor function at classical and alternative response elements. Novartis Found Symp. 2000;230:20–26. doi: 10.1002/0470870818.ch3. [DOI] [PubMed] [Google Scholar]
- 4.Mandava MB. Plant growth-promoting brassinosteroids. Annu Rev Plant Physiol Plant Mol Biol. 1988;39:23–52. [Google Scholar]
- 5.Wang ZY, Seto H, Fujioka S, Yoshida S, Chory J. BRl1 is a critical component of a plasma-membrane receptor for plant steroids. Nature. 2001;410:380–383. doi: 10.1038/35066597. [DOI] [PubMed] [Google Scholar]
- 6.Qin C, Samudio I, Ngwenya S, Safe S. Estrogen-dependent regulation of ornithine decarboxylase in breast cancer cells through activation of nongenomic cAMP-dependent pathways. Mol Carcinog. 2004;40:160–170. doi: 10.1002/mc.20030. [DOI] [PubMed] [Google Scholar]
- 7.Watters JJ, Chun TY, Kim YN, Bertics PJ, Gorski J. Estrogen modulation of prolactin gene expression requires an intact mitogen-activated protein kinase signal transduction pathway in cultured rat pituitary cells. Mol Endocrinol. 2000;14:1872–1881. doi: 10.1210/mend.14.11.0551. [DOI] [PubMed] [Google Scholar]
- 8.Razandi M, Pedram A, Rosen E, Levin ER. BRCA1 inhibits membrane estrogen and growth factor receptor signaling to cell proliferation in breast cancer. Mol Cell Biol. 2004;24:5900–5913. doi: 10.1128/MCB.24.13.5900-5913.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pappas TC, Gametchu B, Yannariello-Brown J, Collins TJ, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded ligand binding. FASEB J. 1995;9:404–410. doi: 10.1096/fasebj.9.5.7896011. [DOI] [PubMed] [Google Scholar]
- 10.Norfleet AM, Thomas ML, Gametchu B, Watson CS. Estrogen receptor-α detected on the plasma membrane of aldehyde-fixed GH3/B6/F10 rat pituitary tumor cells by enzyme-linked immunocytochemistry. Endocrinology. 1999;140:3805–3814. doi: 10.1210/endo.140.8.6936. [DOI] [PubMed] [Google Scholar]
- 11.Razandi M, Pedram A, Greene GL, Levin ER. Cell membrane and nuclear estrogen receptors derive from a single transcript: studies of ERα and ERβ expressed in CHO cells. Mol Endocrinol. 1999;13:307–319. doi: 10.1210/mend.13.2.0239. [DOI] [PubMed] [Google Scholar]
- 12.Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, Shaul PW. ERβ has nongenomic action in caveolae. Mol Endocrinol. 2002;16:938–946. doi: 10.1210/mend.16.5.0827. [DOI] [PubMed] [Google Scholar]
- 13.Razandi M, Pedram A, Merchenthaler I, Greene GL, Levin ER. Plasma membrane estrogen receptors exist and function as dimers. Mol Endocrinol. 2004;18:2854–2865. doi: 10.1210/me.2004-0115. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Haynes MP, Bender JR. Plasma membrane localization and function of the estrogen receptor α variant (ER46) in human endothelial cells. Proc Natl Acad Sci USA. 2003;100:4807–4812. doi: 10.1073/pnas.0831079100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pendaries C, Darblade B, Rochaix P, Krust A, Chambon P, Korach KS, Bayard F, Arnal JF. The AF-1 activation-function of ERα may be dispensable to mediate the effect of estradiol on endothelial NO production in mice. Proc Natl Acad Sci USA. 2002;99:2205–2210. doi: 10.1073/pnas.042688499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu J, Bosch MA, Tobias SC, Grandy DK, Scanlan TS, Ronnekleiv OK, Kelly MJ. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J Neurosci. 2003;23:9529–9540. doi: 10.1523/JNEUROSCI.23-29-09529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly Jr ES, Nethrapalli IS, Tinnikov AA. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci. 2002;19:8391–8401. doi: 10.1523/JNEUROSCI.22-19-08391.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh M, Setalo G, Jr, Guan X, Warren M, Toran-Allerand CD.1999Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways J Neurosci 191179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakhla AM, Romas NA, Rosner W. Estradiol activates the prostate androgen receptor and prostate-specific antigen secretion through the intermediacy of sex hormone binding globulin. J Biol Chem. 1997;272:6838–6841. doi: 10.1074/jbc.272.11.6838. [DOI] [PubMed] [Google Scholar]
- 20.Filippi S, Luconi M, Granchi S, Vignozzi L, Bettuzzi S, Tozzi P, Ledda F, Forti G, Maggi M. Estrogens, but not androgens, regulate expression and functional activity of oxytocin receptor in rabbit epididymis. Endocrinology. 2002;143:4271–4280. doi: 10.1210/en.2002-220384. [DOI] [PubMed] [Google Scholar]
- 21.Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, Shaul PW. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Gα(i) J Biol Chem. 2001;276:27071–27076. doi: 10.1074/jbc.M100312200. [DOI] [PubMed] [Google Scholar]
- 22.Kelly MJ, Qiu J, Wagner EJ, Ronnekleiv OK. Rapid effects of estrogen on G protein-coupled receptor activation of potassium channels in the central nervous system (CNS) J Steroid Biochem Mol Biol. 2002;83:187–193. doi: 10.1016/s0960-0760(02)00249-2. [DOI] [PubMed] [Google Scholar]
- 23.Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER. Estrogen receptors associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol. 2002;16:100–115. doi: 10.1210/mend.16.1.0757. [DOI] [PubMed] [Google Scholar]
- 24.Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci USA. 2002;99:14783–14788. doi: 10.1073/pnas.192569699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Song RX, Barnes CJ, Zhang Z, Bao Y, Kumar R, Santen RJ. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor α to the plasma membrane. Proc Natl Acad Sci USA. 2004;101:2076–2081. doi: 10.1073/pnas.0308334100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HP, Lee JY, Jeong JK, Bae SW, Lee HK, Jo I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor α localized in caveolae. Biochem Biophys Res Commun. 1999;263:257–262. doi: 10.1006/bbrc.1999.1348. [DOI] [PubMed] [Google Scholar]
- 27.Benten WP, Stephan C, Lieberherr M, Wunderlich F. Estradiol signaling via sequestrable surface receptors. Endocrinology. 2001;142:1669–1677. doi: 10.1210/endo.142.4.8094. [DOI] [PubMed] [Google Scholar]
- 28.Stevis PE, Deecher DC, Suhadolnik L, Mallis LM, Frail DE. Differential effects of estradiol and estradiol-BSA conjugates. Endocrinology. 1999;140:5455–5458. doi: 10.1210/endo.140.11.7247. [DOI] [PubMed] [Google Scholar]
- 29.Tiruppathi C, Naqvi T, Wu Y, Vogel SM, Minshall RD, Malik AB. Albumin mediates the transcytosis of myeloperoxidase by means of caveolae in endothelial cells. Proc Natl Acad Sci USA. 2004;101:7699–7704. doi: 10.1073/pnas.0401712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 31.Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, Montanaro D, Musti AM, Picard D, Andò S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. 2004;279:27008–27016. doi: 10.1074/jbc.M403588200. [DOI] [PubMed] [Google Scholar]
- 32.Ahola TM, Manninen T, Alkio N, Ylikomi T. G protein-coupled receptor 30 is critical for a progestin-induced growth inhibition in MCF-7 breast cancer cells. Endocrinology. 2002;143:3376–3384. doi: 10.1210/en.2001-211445. [DOI] [PubMed] [Google Scholar]
- 33.Thomas P, Pang Y, Filardo EJ, Dong J 2004 Identity of an estrogen membrane receptor coupled to a G-protein in human breast cancer cells. Endocrinology, in press [DOI] [PubMed]
- 34.Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, Auricchio F. Steroid-induced androgen receptor-oestradiol receptor β-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000;19:5406–5417. doi: 10.1093/emboj/19.20.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Razandi M, Pedram A, Levin ER. Estrogen signals to preservation of endothelial cell form and function. J Biol Chem. 2000;275:38540–38546. doi: 10.1074/jbc.M007555200. [DOI] [PubMed] [Google Scholar]
- 36.Lu Q, Pallas DC, Surks HK, Baur WE, Mendelsohn ME, Karas RH. Striatin assembles a membrane signaling complex necessary for rapid, nongenomic activation of endothelial NO synthase by estrogen receptor α. Proc Natl Acad Sci USA. 2004;101:17126–17131. doi: 10.1073/pnas.0407492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chambliss KL, Simon L, Yuhanna IS, Mineo C, Shaul PW. Dissecting the basis of nongenomic activation of eNOS by estradiol: role of ERα domains with known nuclear functions. Mol Endocrinol. 2005;19:277–289. doi: 10.1210/me.2004-0008. [DOI] [PubMed] [Google Scholar]
- 38.Razandi M, Alton G, Pedram A, Ghonshani S, Webb D, Levin ER. Identification of a structural determinant for the membrane localization of ERα. Mol Cell Biol. 2003;23:1633–1646. doi: 10.1128/MCB.23.5.1633-1646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song RX, McPherson RA, Adam L, Bao Y, Shupnik M, Kumar R, Santen RJ. Linkage of rapid estrogen action to MAPK activation by ER α-Shc association and Shc pathway activation. Mol Endocrinol. 2002;16:116–127. doi: 10.1210/mend.16.1.0748. [DOI] [PubMed] [Google Scholar]
- 40.Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. S-palmitoylation modulates human estrogen receptor-α functions. Biochem Biophys Res Commun. 2004;316:878–883. doi: 10.1016/j.bbrc.2004.02.129. [DOI] [PubMed] [Google Scholar]
- 41.Acconcia F, Ascenzi P, Bocedi A, Spisni E, Tomasi V, Trentalance A, Visca P, Marino M. Palmitoylatin-dependent estrogen receptor α membrane localization regulation by 17 β-estradiol. Mol Biol Cell. 2005;16:231–237. doi: 10.1091/mbc.E04-07-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Razandi M, Pedram A, Parks S, Levin ER. Proximal events in ER signaling from the plasma membrane. J Biol Chem. 2003;278:2701–2712. doi: 10.1074/jbc.M205692200. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Z, Maier B, Santen RJ, Song RX. Membrane association of estrogen receptor α mediates estrogen effect on MAPK activation. Biochem Biophys Res Commun. 2002;294:926–933. doi: 10.1016/S0006-291X(02)00348-0. [DOI] [PubMed] [Google Scholar]
- 44.Kousteni S, Han L, Chen JR, Almeida M, Plotkin LI, Bellido T, Manolagas SC. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. 2003;111:1651–1664. doi: 10.1172/JCI17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keshamouni VG, Mattingly RR, Reddy KB. Mechanism of 17-β-estradiol-induced Erk1/2 activation in breast cancer cells. A role for HER-2 and PKC-β. J Biol Chem. 2002;277:22558–22565. doi: 10.1074/jbc.M202351200. [DOI] [PubMed] [Google Scholar]
- 46.Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor α rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;275:18447–18453. doi: 10.1074/jbc.M910345199. [DOI] [PubMed] [Google Scholar]
- 47.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 48.Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, McLemore MS, Olivo SE, Stoica A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000;141:4503–4511. doi: 10.1210/endo.141.12.7836. [DOI] [PubMed] [Google Scholar]
- 49.Balasenthil S, Barnes CJ, Rayala SK, Kumar R. Estrogen receptor activation at serine 305 is sufficient to upregulate cyclin D1 in breast cancer cells. FEBS Lett. 2004;567:243–247. doi: 10.1016/j.febslet.2004.04.071. [DOI] [PubMed] [Google Scholar]
- 50.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 51.Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–415. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]
- 52.Lopez GN, Turck CW, Schaufele F, Stallcup MR, Kushner PJ. Growth factors signal to steroid receptors through mitogen-activated protein kinase regulation of p160 coactivator activity. J Biol Chem. 2001;276:22177–22182. doi: 10.1074/jbc.M010718200. [DOI] [PubMed] [Google Scholar]
- 53.Iwase H. Molecular action of the estrogen receptor and hormone dependency in breast cancer. Breast Cancer. 2003;10:89–96. doi: 10.1007/BF02967632. [DOI] [PubMed] [Google Scholar]
- 54.Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, Floore A, Velds A, van‘t Veer L, Neefjes J. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell. 2004;5:597–605. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 55.Yamakawa K, Arita J. Cross-talk between the estrogen receptor-, protein kinase A-, and mitogen-activated protein kinase-mediated signaling pathways in the regulation of lactotroph proliferation in primary culture. J Steroid Biochem Mol Biol. 2004;88:123–130. doi: 10.1016/j.jsbmb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 56.Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer Cell. 2004;5:207–213. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 57.Duan R, Xie W, Li X, McDougal A, Safe S. Estrogen regulation of c-fos gene expression through phosphati-dylinositol-3-kinase-dependent activation of serum response factor in MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2002;294:384–394. doi: 10.1016/S0006-291X(02)00499-0. [DOI] [PubMed] [Google Scholar]
- 58.Pedram A, Razandi M, Aitkenhead M, Hughes CCW, Levin ER. Integration of the non-genomic and genomic actions of estrogen: membrane initiated signaling by steroid (MISS) to transcription and cell biology. J Biol Chem. 2002;277:50768–50775. doi: 10.1074/jbc.M210106200. [DOI] [PubMed] [Google Scholar]
- 59.Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol. 2004;31(2 Suppl 7):2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 60.Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle. 2003;2:339–345. [PubMed] [Google Scholar]
- 62.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 63.Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–72. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 64.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 65.Proud CG. Role of mTOR signalling in the control of translation initiation and elongation by nutrients. Curr Top Microbiol Immunol. 2004;279:215–244. doi: 10.1007/978-3-642-18930-2_13. [DOI] [PubMed] [Google Scholar]
- 66.Plotkin LI, Aguirre JI, Kousteni S, Manolagas SC, Bellido T. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of ERK activation. J Biol Chem. 2005;280:7317–7325. doi: 10.1074/jbc.M412817200. [DOI] [PubMed] [Google Scholar]
- 67.Chen JR, Plotkin LI, Aguirre JI, Han L, Jilka RL, Kousteni S, Bellido T, Manolagas SC. Transient versus sustained phosphorylation and nuclear accumulation of ERKs underlie anti- versus Pro-apoptotic effects of estrogens. J Biol Chem. 2005;280:4632–4638. doi: 10.1074/jbc.M411530200. [DOI] [PubMed] [Google Scholar]
- 68.Bartucci M, Morelli C, Mauro L, Ando’ S, Surmacz E. Differential insulin-like growth factor I receptor signaling and function in estrogen receptor (ER)-positive MCF-7 and ER-negative MDA-MB-231 breast cancer cells. Cancer Res. 2001;61:6747–6754. [PubMed] [Google Scholar]
- 69.Meyer G, Feldman EL. Signaling mechanisms that regulate actin-based motility processes in the nervous system. J Neurochem. 2002;83:490–503. doi: 10.1046/j.1471-4159.2002.01185.x. [DOI] [PubMed] [Google Scholar]
- 70.Schafer DA. Barbed ends rule. Nature. 2004;430:734–735. doi: 10.1038/430734a. [DOI] [PubMed] [Google Scholar]
- 71.Kipp JL, Ramirez VD. Effect of estradiol, diethylstilbestrol, and resveratrol on F0F1-ATPase activity from mitochondrial preparations of rat heart, liver, and brain. Endocrine. 2001;15:165–175. doi: 10.1385/ENDO:15:2:165. [DOI] [PubMed] [Google Scholar]
- 72.Stojanovski D, Johnston AJ, Streimann I, Hoogenraad NJ, Ryan MT. Import of nuclear-encoded proteins into mitochondria. Exp Physiol. 2003;88:57–64. doi: 10.1113/eph8802501. [DOI] [PubMed] [Google Scholar]
- 73.Chen JQ, Delannoy M, Cooke C, Yager JD. Mitochondrial localization of ERα and ERβ in human MCF7 cells. Am J Physiol Endocrinol Metab. 2004;286:E1011–E1022. doi: 10.1152/ajpendo.00508.2003. [DOI] [PubMed] [Google Scholar]
- 74.Yang SH, Liu R, Perez EJ, Wen Y, Stevens Jr SM, Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW. Mitochondrial localization of estrogen receptor β. Proc Natl Acad Sci USA. 2004;101:4130–4135. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang P, Oliff A. Signaling pathways in apoptosis as potential targets for cancer therapy. Trends Cell Biol. 2001;11:343–348. doi: 10.1016/s0962-8924(01)02063-3. [DOI] [PubMed] [Google Scholar]
- 76.Kousteni S, Bellido T, Plotkin LI, O‘Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, Roberson PK, Weinstein RS, Jilka RL, Manolagas SC. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- 77.Kousteni S, Chen JR, Bellido T, Han L, Ali AA, O’Brien CA, Plotkin L, Fu Q, Mancino AT, Wen Y, Vertino AM, Powers CC, Stewart SA, Ebert R, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Reversal of bone loss in mice by nongenotropic signaling of sex steroids. Science. 2002;298:843–846. doi: 10.1126/science.1074935. [DOI] [PubMed] [Google Scholar]
- 78.Stoica GE, Franke TF, Moroni M, Mueller S, Morgan E, Iann MC, Winder AD, Reiter R, Wellstein A, Martin MB, Stoica A. Effect of estradiol on estrogen receptor-α gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003;22:7998–8011. doi: 10.1038/sj.onc.1206769. [DOI] [PubMed] [Google Scholar]
- 79.Marquez DC, Pietras RJ. Membrane-associated binding sites for estrogen contribute to growth regulation of human breast cancer cells. Oncogene. 2001;20:5420–5430. doi: 10.1038/sj.onc.1204729. [DOI] [PubMed] [Google Scholar]
- 80.Simoncini T, Genazzani AR, Liao JK. Nongenomic mechanisms of endothelial nitric oxide synthase activation by the selective estrogen receptor modulator raloxifene. Circulation. 2002;105:1368–1373. doi: 10.1161/hc1102.105267. [DOI] [PubMed] [Google Scholar]
- 81.Hisamoto K, Ohmichi M, Kanda Y, Adachi K, Nishio Y, Hayakawa J, Mabuchi S, Takahashi K, Tasaka K, Miyamoto Y, Taniguchi N, Murata Y. Induction of endothelial nitric-oxide synthase phosphorylation by the raloxifene analog LY117018 is differentially mediated by Akt and extracellular signal-regulated protein kinase in vascular endothelial cells. J Biol Chem. 2001;276:47642–47649. doi: 10.1074/jbc.M103853200. [DOI] [PubMed] [Google Scholar]
- 82.Ogita H, Node K, Asanuma H, Sanada S, Kim J, Takashima S, Minamino T, Hori M, Kitakaze M. Raloxifene improves coronary perfusion, cardiac contractility, and myocardial metabolism in the ischemic heart: role of phosphatidylinositol 3-kinase/Akt pathway. J Cardiovasc Pharmacol. 2004;43:821–829. doi: 10.1097/00005344-200406000-00012. [DOI] [PubMed] [Google Scholar]
- 83.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM. Estradiol protects against ischemic injury. J Cereb Blood Flow Metab. 1998;18:1253–1258. doi: 10.1097/00004647-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 85.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor α, not β, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA. 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rau SW, Dubal DB, Bottner M, Gerhold LM, Wise PM. Estradiol attenuates programmed cell death after stroke-like injury. J Neurosci. 2003;23:11420–11426. doi: 10.1523/JNEUROSCI.23-36-11420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Simpkins JW, Yang SH, Liu R, Perez E, Cai ZY, Covey DF, Green PS. Estrogen-like compounds for ischemic neuroprotection. Stroke. 2004;35(Suppl 1):2648–2651. doi: 10.1161/01.STR.0000143734.59507.88. [DOI] [PubMed] [Google Scholar]
- 88.Yu X, Rajala RV, McGinnis JF, Li F, Anderson RE, Yan X, Li S, Elias RV, Knapp RR, Zhou X, Cao W. Involvement of insulin/phosphoinositide 3-kinase/Akt signal pathway in 17 β-estradiol-mediated neuroprotection. J Biol Chem. 2004;279:13086–13094. doi: 10.1074/jbc.M313283200. [DOI] [PubMed] [Google Scholar]
- 89.Wilson ME, Liu Y, Wise PM. Estradiol enhances Akt activation in cortical explant cultures following neuronal injury. Brain Res Mol Brain Res. 2002;102:48–54. doi: 10.1016/s0169-328x(02)00181-x. [DOI] [PubMed] [Google Scholar]
- 90.Jung ME, Watson DG, Wen Y, Simpkins JW. Role of protein kinase C in estrogen protection against apoptotic cerebellar cell death in ethanol-withdrawn rats. Alcohol. 2003;31:39–48. doi: 10.1016/j.alcohol.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 91.Xu Y, Traystman RJ, Hurn PD, Wang MM. Neurite-localized estrogen receptor-α mediates rapid signaling by estrogen. J Neurosci Res. 2003;74:1–11. doi: 10.1002/jnr.10725. [DOI] [PubMed] [Google Scholar]
- 92.Behl C. Oestrogen as a neuroprotective hormone. Nat Rev Neurosci. 2002;3:433–442. doi: 10.1038/nrn846. [DOI] [PubMed] [Google Scholar]
- 93.Abraham IM, Todman MG, Korach KS, Herbison AE. Critical in vivo roles for classical estrogen receptors in rapid estrogen actions on intracellular signaling in mouse brain. Endocrinology. 2004;145:3055–3061. doi: 10.1210/en.2003-1676. [DOI] [PubMed] [Google Scholar]
- 94.Richards JS, Sharma SC, Falender AE, Lo YH. Expression of FKHR, FKHRL1, and AFX genes in the rodent ovary: evidence for regulation by IGF-I, estrogen, and the gonadotropins. Mol Endocrinol. 2002;16:580–599. doi: 10.1210/mend.16.3.0806. [DOI] [PubMed] [Google Scholar]
- 95.Kimura A, Ohmichi M, Kawagoe J, Kyo S, Mabuchi S, Takahashi T, Ohshima C, Arimoto-Ishida E, Nishio Y, Inoue M, Kurachi H, Tasaka K, Murata Y. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene. 2004;23:4505–4515. doi: 10.1038/sj.onc.1207582. [DOI] [PubMed] [Google Scholar]
- 96.Mazumdar A, Kumar R. Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS Lett. 2003;535:6–10. doi: 10.1016/s0014-5793(02)03846-2. [DOI] [PubMed] [Google Scholar]
- 97.Santen RJ, Song RX, Zhang Z, Yue W, Kumar R. Adaptive hypersensitivity to estrogen: mechanism for sequential responses to hormonal therapy in breast cancer. Clin Cancer Res. 2004;10:337S–345S. doi: 10.1158/1078-0432.ccr-031207. [DOI] [PubMed] [Google Scholar]
- 98.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 99.Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, Osipo C, Loweth J, McKian K, De Los Reyes A, Wing L, Jordan VC. Apoptotic action of 17β-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95:1586–1597. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]