

The synthesis and crystal structure of (E)-N-[(2-bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline are reported.
Keywords: crystal structure, trifluoromethyl, aryl imine ligands
Abstract
The title compound C15H8BrF6N, was prepared by a condensation reaction of 2-bromobenzaldehyde and 3,5-bis(trifluoromethyl)aniline in the presence of anhydrous magnesium sulfate. The compound readily crystallizes from a concentrated hexanes solution in high yield. The imine bond is nearly planar with the 2-bromophenyl group, while the N-(3,5-bis(trifluoromethyl)phenyl moiety is significantly twisted from the imine group [49.61 (5)°]. The crystal packing involves short intermolecular C—H⋯Br contacts linking zigzag ribbons flanked by fluorous layers.
1. Chemical context
Arylimine-type ligands have increasingly been used as ancillary ligands for a variety of metal complexes. (Dostál et al., 2011 ▸; Hejda et al., 2012 ▸; Hejda et al., 2013 ▸, 2017 ▸; Joseph et al., 2018 ▸; Kingsley et al., 2016 ▸; Kořenková et al., 2016 ▸; Kremláček et al., 2018 ▸; Mungwe et al., 2011 ▸; Novák et al., 2014 ▸, 2016 ▸; Šimon et al., 2013 ▸; Urbanová et al., 2013 ▸, 2014 ▸; Vrána et al., 2013 ▸; Zhao et al., 2010 ▸, 2011 ▸) Synthesis of these ligands is typically performed with alkyl-substituted anilines or alkyl amines reacting with 2-brombenzaldehyde. There is interest in modifying the electronic structure of the ligands but this has been limited to substitutions on the aromatic ring of the benzaldehyde starting material. (Chen et al., 2004 ▸; Li et al., 2019 ▸). Our group is interested in developing arylimine ligands with electron-withdrawing groups on the aromatic ring of the N-imine portion of the ligand. Herein we report the synthesis and crystal structure of (E)-N-[(2-bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline (I).
2. Structural commentary
A displacement ellipsoid plot of compound I is shown in Fig. 1 ▸. The imine bond length C7—N1 [1.280 (4) Å] and C7—N1—C8 bond angle [119.1 (2)°] are consistent with atom N1 being sp2 hybridized. The imine bond is oriented nearly coplanar with the C1–C6 phenyl ring [angle between least squares planes formed by C1–C6 and N1,C7,C1 = 5.9 (3)°]. Torsion angles (Table 1 ▸) near the imine bond demonstrate this as well as the twist of the C8–C13 phenyl ring relative to the imine bond plane formed by C8/N1/C7 is 42.0 (3)°). The angle between the least-squares planes formed by C1–C8/N1/Br1 and C8–C15/N1 of 49.61 (5)° is consistent with steric limitations influenced by atom positions of H7 and H9. Short intramolecular interactions involving C—H bonds with acceptors are also present. A short C7—H7⋯Br1 contact (2.82 Å, length − vdW radii sums = −0.244 Å) and another contact involving C6—H6⋯N1 (2.52 Å, length − vdW sums = −0.342 Å) help define the observed conformation of I (Table 2 ▸).
Figure 1.

The molecular structure of I. Only one of the three CF3 disorder model conformations shown. Thermal displacement ellipsoids are drawn at the 50% probability level.
Table 1. Selected torsion angles (°).
| C2—C1—C7—N1 | 174.5 (2) | C7—N1—C8—C13 | 139.5 (3) |
| C6—C1—C7—N1 | −5.9 (4) | C8—N1—C7—C1 | 176.7 (2) |
| C7—N1—C8—C9 | −43.7 (4) |
Table 2. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C3—H3⋯F2Ai | 0.95 | 2.46 | 3.365 (7) | 160 |
| C3—H3⋯F7Ai | 0.95 | 2.25 | 3.063 (7) | 143 |
| C4—H4⋯F1Aii | 0.95 | 2.54 | 3.411 (6) | 153 |
| C4—H4⋯F9Aii | 0.95 | 2.36 | 3.152 (9) | 140 |
| C6—H6⋯N1 | 0.95 | 2.52 | 2.828 (4) | 99 |
| C7—H7⋯Br1 | 0.95 | 2.82 | 3.233 (3) | 108 |
| C13—H13⋯Br1iii | 0.95 | 2.97 | 3.851 (3) | 155 |
Symmetry codes: (i) 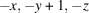 ; (ii)
; (ii)  ; (iii)
; (iii)  .
.
3. Supramolecular features
While no conventional hydrogen-bonding interactions are present, several short C—H⋯F contacts with fluorine atoms of the disordered CF3 group and hydrogen atoms H3 and H4 are present (Table 2 ▸, Fig. 2 ▸). A contact involving C13—H13⋯Br1 is also present, forming zigzag infinite ribbons of molecules along (001) as shown in Fig. 3 ▸. The packing diagram of I (Fig. 4 ▸) demonstrates how the CF3 groups associate in fluorous layers in the bc plane at x = 0.5 with the ribbons of C—H⋯Br interactions between the CF3-rich layers. Weak π–π dimer intermolecular interactions between a neighboring brominated phenyl ring (less electron poor) and the trifluoromethylated phenyl ring (more electron poor) are evident, with a 3.730 (2) Å centroid-to-centroid distance.
Figure 2.

Intermolecular interactions of I including centroids of the weak π–π dimer.
Figure 3.

Packing diagram of I in a view along the a-axis direction. C—H⋯Br interactions are shown as dashed lines.
Figure 4.

Packing diagram of I in a view along the c-axis direction. C—H⋯Br interactions are shown as dashed lines.
4. Database survey
A recent search in the CSD (version 6.00, last update April 2025; Groom et al., 2016 ▸) using ConQuest (Bruno et al., 2002 ▸) revealed the most closely related structures to I include a single meta-fluoro substitution (in place of a bis-trifluoromethyl-substituted phenyl ring). The structure of 1-(2-bromophenyl)-N-(3-fluorophenyl)methanimine (FOBLUF; Kaur & Choudhury, 2014 ▸) demonstrates similar intramolecular features to I, including the imine C—H⋯Br and phenyl ortho C—H⋯N distances (2.76 and 2.53 Å, respectively) as well as the twist angle between phenyl least-squares planes [40.97 (8)°]. Another related structure reported with a single electron-withdrawing meta substituent on the N-imine ring is N-(2-iodobenzylidene)-3-nitroaniline (MOPPOW; Wardell et al., 2002 ▸). Similar to I, this mono-nitro-substituted structure also demonstrates analogous phenyl ortho C—H⋯N distances (2.58 Å) but the twist angle between phenyl least-squares planes (64.2°) is significantly different and may be related to the presence of the intermolecular halogen bonding that is present (I⋯N distance = 3.487 Å, C8—I1⋯N1 = 160.1°). The structure of 2-({[3,5-bis(trifluoromethyl)phenyl]imino}methyl)-5-(dimethylamino)phenol (BIDCUQ; Zhao et al., 2023 ▸) possesses the same 3,5-bis(trifluoromethyl)phenyl moiety as I, but the imine-linked 2-hydroxy phenyl group engages an intramolecular interaction (O1—H1⋯N2 = 1.84 Å), inducing a coplanar orientation of the phenyl rings. Also, the CF3 groups of BIDCUQ associate in fluorous layers (similar to I) between infinite single-distance π–π stacked aromatic regions (3.47 Å).
5. Synthesis and crystallization
2-Bromobenzaldehyde (4.50 g, 24.3 mmol) was stirred in hexanes (250 mL) in the presence of anhydrous MgSO4 (1.0 g). 3,5-Bis(trifluoromethyl)aniline (5.575 g, 24.3 mmol) was slowly added and the solution was allowed to stir for 2 h. MgSO4 was then removed via vacuum filtration and the filtrate cooled to 243 K for 48 h. The yellow precipitate was collected via vacuum filtration and purified through recrystallization from hexanes. The resulting material was light-yellow and crystalline. Yield: 7.62 g, 79%. 1H NMR (CDCl3, 400 MHz): δ 8.86 (s, 1H, CH=N), 8.22 (dd, 3JHH = 7.7 Hz, 4JHH = 1.9 Hz 1H, aryl H6), 7.76 (br s, 1H, aryl H11), 7.66 (dd, 3JHH = 7.7 Hz, 4JHH = 1.9 Hz 1H, aryl H3), 7.63 (br s, 2H, aryl H9, H13), 7.44 (m, 1H, aryl H5), 7.38 (m, 1H, aryl H4). 13C NMR {1H} (CDCl3, 100 MHz): δ 162.30 (s, C7), 153.15 (s, aryl C8), 133.81 (s, aryl C1), 133.62 (s, aryl C4), 133.58 (s, aryl C3), 132.79 (q, 2JCF = 33.6 Hz, aryl C10, C12), 129.41 (s, aryl C6), 128.05 (s, aryl C5), 126.71 (s, aryl C2), 123.32 (q, 1JCF = 272.8 Hz, C14, C15), 121.40 (q, 3JCF = 2.7 Hz, aryl C9, C13), 119.71 (sept, 3JCF = 3.8 Hz, aryl C11). M.p.: 361–363 K. X-ray quality crystals were grown from a concentrated solution in warm hexanes followed by slow cooling to room temperature and storage at 243 K for 96 h.
6. Refinement
Crystal data, data collection and structure refinement details are summarized in Table 3 ▸. Hydrogen atoms were refined with riding coordinates with fixed Uiso(H) at 1.2 times of the riding C atom. Several restraints and constraints were used to model disorder in one of the CF3 groups. Distance restraints were employed for C14—F1A, C14—F2A, C14—F3A, C14—F4A, C14—F5A, C14—F6A, C14—F7A, C14—F8A, and C14—F9A with sigma of 0.02; F1A—F2A, F2A—F3A, F3A—F1A, F4A—F5A, F5A—F6A, F6A—F4A, F7A—F8A, F8A—F9A, and F9A—F7A with sigma of 0.04; and C10—F1A, C10—F2A, C10—F3A, C10—F4A, C10—F5A, C10—F6A, C10—F7A, C10—F8A, and C10—F9A with sigma of 0.1. Uaniso restraints were applied to C10, C14, F1A, F2A, F3A, F4A, F5A, F6A, F7A, F8A, and F9A within 2 Å with sigma of 0.04 and sigma for terminal atoms of 0.08 within 2 Å. Rigid body (RIGU) restraints were applied to C10, C14, F1A, F2A, F3A, F4A, F5A, F6A, F7A, F8A, F9A with sigma for 1–2 distances of 0.004 and sigma for 1–3 distances of 0.004. The three orientations of the CF3 group were fixed at sof values of 0.37 (F1A, F2A, F3A), 0.38 (F4A, F5A, F6A), and 0.25 (F7A, F8A, F9A).
Table 3. Experimental details.
| Crystal data | |
| Chemical formula | C15H8BrF6N |
| M r | 396.13 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 100 |
| a, b, c (Å) | 13.261 (2), 7.7224 (12), 14.176 (2) |
| β (°) | 93.394 (2) |
| V (Å3) | 1449.2 (4) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 2.90 |
| Crystal size (mm) | 0.25 × 0.20 × 0.15 |
| Data collection | |
| Diffractometer | Bruker APEXII CCD |
| Absorption correction | Multi-scan (SADABS; Krause et al., 2015 ▸) |
| Tmin, Tmax | 0.575, 0.747 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 23866, 5193, 3166 |
| R int | 0.053 |
| (sin θ/λ)max (Å−1) | 0.766 |
| Refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.046, 0.137, 1.06 |
| No. of reflections | 5193 |
| No. of parameters | 217 |
| No. of restraints | 189 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.89, −0.68 |
Supplementary Material
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989025007650/ej2015sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989025007650/ej2015Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989025007650/ej2015Isup3.cml
CCDC reference: 2483197
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
Special acknowledgement is given to Dr. Chris Gianopoulos for assistance in data collection and structure refinement, Elizabeth Grabowski at the University of Michigan-Flint for assistance in characterization and to the University of Toledo Instrumentation Center for the use of their Bruker APEXII diffractometer.
supplementary crystallographic information
(E)-N-[(2-Bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline . Crystal data
| C15H8BrF6N | F(000) = 776 |
| Mr = 396.13 | Dx = 1.816 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| a = 13.261 (2) Å | Cell parameters from 6800 reflections |
| b = 7.7224 (12) Å | θ = 2.9–31.3° |
| c = 14.176 (2) Å | µ = 2.90 mm−1 |
| β = 93.394 (2)° | T = 100 K |
| V = 1449.2 (4) Å3 | Irregular brick, yellow |
| Z = 4 | 0.25 × 0.20 × 0.15 mm |
(E)-N-[(2-Bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline . Data collection
| Bruker APEXII CCD diffractometer | 3166 reflections with I > 2σ(I) |
| Radiation source: sealed tube | Rint = 0.053 |
| φ and ω scans | θmax = 33.0°, θmin = 2.9° |
| Absorption correction: multi-scan (SADABS; Krause et al., 2015) | h = −20→19 |
| Tmin = 0.575, Tmax = 0.747 | k = −11→11 |
| 23866 measured reflections | l = −21→21 |
| 5193 independent reflections |
(E)-N-[(2-Bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline . Refinement
| Refinement on F2 | Primary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.046 | H-atom parameters constrained |
| wR(F2) = 0.137 | w = 1/[σ2(Fo2) + (0.0521P)2 + 1.1524P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.06 | (Δ/σ)max = 0.001 |
| 5193 reflections | Δρmax = 0.89 e Å−3 |
| 217 parameters | Δρmin = −0.68 e Å−3 |
| 189 restraints |
(E)-N-[(2-Bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline . Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
(E)-N-[(2-Bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline . Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| Br1 | −0.04663 (2) | 0.75027 (4) | −0.00485 (2) | 0.03627 (10) | |
| N1 | 0.05952 (17) | 0.5457 (3) | 0.28171 (18) | 0.0335 (5) | |
| C1 | −0.08432 (19) | 0.5863 (3) | 0.1738 (2) | 0.0288 (5) | |
| C2 | −0.12674 (19) | 0.6484 (3) | 0.08793 (19) | 0.0293 (5) | |
| C3 | −0.2302 (2) | 0.6421 (4) | 0.0656 (2) | 0.0363 (6) | |
| H3 | −0.257428 | 0.686307 | 0.006931 | 0.044* | |
| C4 | −0.2931 (2) | 0.5709 (4) | 0.1295 (2) | 0.0409 (7) | |
| H4 | −0.363907 | 0.565656 | 0.114812 | 0.049* | |
| C5 | −0.2530 (2) | 0.5069 (4) | 0.2152 (3) | 0.0415 (7) | |
| H5 | −0.296289 | 0.457257 | 0.258939 | 0.050* | |
| C6 | −0.1504 (2) | 0.5155 (3) | 0.2369 (2) | 0.0349 (6) | |
| H6 | −0.123955 | 0.472455 | 0.296075 | 0.042* | |
| C7 | 0.02492 (19) | 0.5917 (3) | 0.1993 (2) | 0.0289 (5) | |
| H7 | 0.070172 | 0.629651 | 0.154103 | 0.035* | |
| C8 | 0.16523 (19) | 0.5438 (3) | 0.3021 (2) | 0.0297 (5) | |
| C9 | 0.23302 (19) | 0.4776 (3) | 0.2399 (2) | 0.0288 (5) | |
| H9 | 0.209397 | 0.434980 | 0.179772 | 0.035* | |
| C10 | 0.33567 (19) | 0.4746 (3) | 0.26672 (19) | 0.0290 (5) | |
| C11 | 0.3715 (2) | 0.5378 (3) | 0.35389 (19) | 0.0317 (5) | |
| H11 | 0.441795 | 0.536850 | 0.371320 | 0.038* | |
| C12 | 0.3035 (2) | 0.6022 (4) | 0.41483 (19) | 0.0346 (6) | |
| C13 | 0.2009 (2) | 0.6037 (4) | 0.3903 (2) | 0.0350 (6) | |
| H13 | 0.154798 | 0.645660 | 0.433828 | 0.042* | |
| C14 | 0.4097 (2) | 0.4018 (4) | 0.2017 (2) | 0.0355 (6) | |
| F1A | 0.4543 (5) | 0.5294 (7) | 0.1579 (5) | 0.0421 (12)* | 0.37 |
| F2A | 0.3653 (4) | 0.2903 (8) | 0.1396 (4) | 0.0366 (12)* | 0.37 |
| F3A | 0.4837 (4) | 0.3151 (8) | 0.2533 (3) | 0.0359 (11)* | 0.37 |
| F4A | 0.4225 (4) | 0.4969 (7) | 0.1218 (4) | 0.0469 (12)* | 0.38 |
| F5A | 0.3808 (4) | 0.2437 (6) | 0.1641 (4) | 0.0375 (11)* | 0.38 |
| F6A | 0.5052 (4) | 0.3815 (7) | 0.2361 (4) | 0.0438 (12)* | 0.38 |
| F7A | 0.3694 (5) | 0.3701 (10) | 0.1145 (4) | 0.0354 (14)* | 0.25 |
| F8A | 0.4543 (7) | 0.2606 (11) | 0.2394 (6) | 0.049 (2)* | 0.25 |
| F9A | 0.4841 (7) | 0.5119 (11) | 0.1888 (7) | 0.055 (2)* | 0.25 |
| C15 | 0.3438 (3) | 0.6664 (6) | 0.5095 (2) | 0.0547 (9) | |
| F4 | 0.27725 (19) | 0.7607 (3) | 0.55397 (15) | 0.0614 (6) | |
| F5 | 0.4281 (2) | 0.7610 (4) | 0.50321 (19) | 0.0861 (10) | |
| F6 | 0.3718 (2) | 0.5370 (4) | 0.56638 (16) | 0.0989 (10) |
(E)-N-[(2-Bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline . Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.03181 (15) | 0.04145 (16) | 0.03619 (15) | 0.00118 (12) | 0.00736 (10) | 0.00104 (12) |
| N1 | 0.0269 (11) | 0.0333 (12) | 0.0412 (12) | 0.0038 (9) | 0.0078 (9) | 0.0063 (10) |
| C1 | 0.0238 (11) | 0.0230 (11) | 0.0405 (14) | −0.0009 (9) | 0.0086 (10) | −0.0062 (10) |
| C2 | 0.0259 (12) | 0.0251 (11) | 0.0377 (14) | 0.0000 (9) | 0.0088 (10) | −0.0087 (10) |
| C3 | 0.0284 (13) | 0.0351 (13) | 0.0452 (16) | 0.0030 (11) | 0.0011 (11) | −0.0102 (12) |
| C4 | 0.0215 (12) | 0.0402 (15) | 0.062 (2) | −0.0021 (11) | 0.0078 (12) | −0.0148 (14) |
| C5 | 0.0310 (15) | 0.0332 (14) | 0.063 (2) | −0.0032 (11) | 0.0217 (14) | −0.0041 (14) |
| C6 | 0.0315 (14) | 0.0265 (12) | 0.0479 (16) | −0.0001 (10) | 0.0120 (12) | −0.0021 (11) |
| C7 | 0.0257 (11) | 0.0238 (11) | 0.0380 (14) | −0.0006 (9) | 0.0086 (10) | −0.0016 (10) |
| C8 | 0.0265 (12) | 0.0274 (12) | 0.0357 (13) | 0.0010 (9) | 0.0049 (10) | 0.0085 (10) |
| C9 | 0.0253 (12) | 0.0276 (12) | 0.0339 (13) | −0.0014 (9) | 0.0041 (10) | 0.0035 (10) |
| C10 | 0.0269 (12) | 0.0269 (11) | 0.0336 (13) | −0.0008 (9) | 0.0042 (10) | 0.0052 (10) |
| C11 | 0.0309 (13) | 0.0309 (13) | 0.0329 (13) | −0.0003 (10) | −0.0005 (10) | 0.0104 (10) |
| C12 | 0.0421 (15) | 0.0353 (14) | 0.0262 (12) | 0.0044 (11) | 0.0008 (11) | 0.0072 (10) |
| C13 | 0.0380 (15) | 0.0351 (14) | 0.0324 (13) | 0.0082 (11) | 0.0074 (11) | 0.0085 (11) |
| C14 | 0.0257 (12) | 0.0401 (15) | 0.0406 (15) | −0.0003 (10) | 0.0004 (11) | 0.0021 (12) |
| C15 | 0.059 (2) | 0.072 (2) | 0.0316 (16) | 0.0147 (19) | −0.0048 (15) | 0.0031 (16) |
| F4 | 0.0617 (14) | 0.0846 (17) | 0.0374 (10) | 0.0156 (11) | −0.0001 (9) | −0.0144 (10) |
| F5 | 0.0564 (14) | 0.144 (3) | 0.0563 (15) | −0.0185 (15) | −0.0079 (11) | −0.0325 (15) |
| F6 | 0.139 (3) | 0.119 (2) | 0.0364 (12) | 0.058 (2) | −0.0165 (14) | 0.0149 (13) |
(E)-N-[(2-Bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline . Geometric parameters (Å, º)
| Br1—C2 | 1.908 (3) | C10—C11 | 1.386 (4) |
| N1—C7 | 1.280 (4) | C10—C14 | 1.496 (4) |
| N1—C8 | 1.414 (3) | C11—H11 | 0.9500 |
| C1—C2 | 1.396 (4) | C11—C12 | 1.378 (4) |
| C1—C6 | 1.400 (4) | C12—C13 | 1.384 (4) |
| C1—C7 | 1.473 (4) | C12—C15 | 1.499 (4) |
| C2—C3 | 1.391 (4) | C13—H13 | 0.9500 |
| C3—H3 | 0.9500 | C14—F1A | 1.323 (6) |
| C3—C4 | 1.382 (4) | C14—F2A | 1.342 (5) |
| C4—H4 | 0.9500 | C14—F3A | 1.364 (5) |
| C4—C5 | 1.388 (5) | C14—F4A | 1.369 (5) |
| C5—H5 | 0.9500 | C14—F5A | 1.377 (5) |
| C5—C6 | 1.379 (4) | C14—F6A | 1.338 (5) |
| C6—H6 | 0.9500 | C14—F7A | 1.339 (6) |
| C7—H7 | 0.9500 | C14—F8A | 1.336 (7) |
| C8—C9 | 1.392 (4) | C14—F9A | 1.323 (8) |
| C8—C13 | 1.390 (4) | C15—F4 | 1.331 (4) |
| C9—H9 | 0.9500 | C15—F5 | 1.343 (5) |
| C9—C10 | 1.392 (3) | C15—F6 | 1.324 (4) |
| C7—N1—C8 | 119.1 (2) | C12—C11—C10 | 118.9 (3) |
| C2—C1—C6 | 117.2 (2) | C12—C11—H11 | 120.5 |
| C2—C1—C7 | 122.9 (2) | C11—C12—C13 | 121.1 (3) |
| C6—C1—C7 | 119.9 (3) | C11—C12—C15 | 117.9 (3) |
| C1—C2—Br1 | 122.07 (19) | C13—C12—C15 | 121.0 (3) |
| C3—C2—Br1 | 116.1 (2) | C8—C13—H13 | 120.0 |
| C3—C2—C1 | 121.8 (3) | C12—C13—C8 | 120.0 (3) |
| C2—C3—H3 | 120.3 | C12—C13—H13 | 120.0 |
| C4—C3—C2 | 119.4 (3) | F1A—C14—C10 | 109.7 (3) |
| C4—C3—H3 | 120.3 | F1A—C14—F2A | 111.2 (4) |
| C3—C4—H4 | 120.0 | F1A—C14—F3A | 106.9 (4) |
| C3—C4—C5 | 120.1 (3) | F2A—C14—C10 | 111.5 (3) |
| C5—C4—H4 | 120.0 | F2A—C14—F3A | 108.1 (4) |
| C4—C5—H5 | 120.0 | F3A—C14—C10 | 109.3 (3) |
| C6—C5—C4 | 120.0 (3) | F4A—C14—C10 | 115.2 (3) |
| C6—C5—H5 | 120.0 | F4A—C14—F5A | 101.6 (4) |
| C1—C6—H6 | 119.2 | F5A—C14—C10 | 113.2 (3) |
| C5—C6—C1 | 121.6 (3) | F6A—C14—C10 | 117.4 (3) |
| C5—C6—H6 | 119.2 | F6A—C14—F4A | 101.7 (4) |
| N1—C7—C1 | 120.7 (2) | F6A—C14—F5A | 105.9 (4) |
| N1—C7—H7 | 119.7 | F7A—C14—C10 | 113.4 (3) |
| C1—C7—H7 | 119.7 | F8A—C14—C10 | 110.5 (4) |
| C9—C8—N1 | 123.0 (3) | F8A—C14—F7A | 111.3 (5) |
| C13—C8—N1 | 117.4 (2) | F9A—C14—C10 | 111.4 (4) |
| C13—C8—C9 | 119.6 (2) | F9A—C14—F7A | 104.5 (6) |
| C8—C9—H9 | 120.3 | F9A—C14—F8A | 105.3 (6) |
| C10—C9—C8 | 119.4 (3) | F4—C15—C12 | 113.2 (3) |
| C10—C9—H9 | 120.3 | F4—C15—F5 | 108.2 (3) |
| C9—C10—C14 | 120.4 (2) | F5—C15—C12 | 112.0 (3) |
| C11—C10—C9 | 121.0 (2) | F6—C15—C12 | 111.6 (3) |
| C11—C10—C14 | 118.6 (2) | F6—C15—F4 | 107.1 (3) |
| C10—C11—H11 | 120.6 | F6—C15—F5 | 104.3 (3) |
| Br1—C2—C3—C4 | −179.7 (2) | C9—C10—C14—F5A | −47.6 (4) |
| N1—C8—C9—C10 | −177.6 (2) | C9—C10—C14—F6A | −171.5 (4) |
| N1—C8—C13—C12 | 178.9 (2) | C9—C10—C14—F7A | 10.2 (5) |
| C1—C2—C3—C4 | −0.6 (4) | C9—C10—C14—F8A | −115.5 (5) |
| C2—C1—C6—C5 | 0.2 (4) | C9—C10—C14—F9A | 127.7 (5) |
| C2—C1—C7—N1 | 174.5 (2) | C10—C11—C12—C13 | 0.3 (4) |
| C2—C3—C4—C5 | 0.2 (4) | C10—C11—C12—C15 | 178.7 (3) |
| C3—C4—C5—C6 | 0.4 (4) | C11—C10—C14—F1A | −79.9 (4) |
| C4—C5—C6—C1 | −0.7 (4) | C11—C10—C14—F2A | 156.5 (4) |
| C6—C1—C2—Br1 | 179.45 (19) | C11—C10—C14—F3A | 37.0 (4) |
| C6—C1—C2—C3 | 0.4 (4) | C11—C10—C14—F4A | −111.5 (4) |
| C6—C1—C7—N1 | −5.9 (4) | C11—C10—C14—F5A | 132.2 (3) |
| C7—N1—C8—C9 | −43.7 (4) | C11—C10—C14—F6A | 8.3 (5) |
| C7—N1—C8—C13 | 139.5 (3) | C11—C10—C14—F7A | −170.0 (4) |
| C7—C1—C2—Br1 | −0.9 (3) | C11—C10—C14—F8A | 64.3 (5) |
| C7—C1—C2—C3 | −179.9 (2) | C11—C10—C14—F9A | −52.5 (6) |
| C7—C1—C6—C5 | −179.5 (3) | C11—C12—C13—C8 | −1.7 (4) |
| C8—N1—C7—C1 | 176.7 (2) | C11—C12—C15—F4 | 165.6 (3) |
| C8—C9—C10—C11 | −0.6 (4) | C11—C12—C15—F5 | 43.0 (4) |
| C8—C9—C10—C14 | 179.1 (2) | C11—C12—C15—F6 | −73.5 (4) |
| C9—C8—C13—C12 | 2.0 (4) | C13—C8—C9—C10 | −0.8 (4) |
| C9—C10—C11—C12 | 0.9 (4) | C13—C12—C15—F4 | −16.0 (5) |
| C9—C10—C14—F1A | 100.3 (4) | C13—C12—C15—F5 | −138.6 (3) |
| C9—C10—C14—F2A | −23.2 (5) | C13—C12—C15—F6 | 104.9 (4) |
| C9—C10—C14—F3A | −142.7 (4) | C14—C10—C11—C12 | −178.9 (2) |
| C9—C10—C14—F4A | 68.7 (4) | C15—C12—C13—C8 | 179.9 (3) |
(E)-N-[(2-Bromophenyl)methylidene]-3,5-bis(trifluoromethyl)aniline . Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···F2Ai | 0.95 | 2.46 | 3.365 (7) | 160 |
| C3—H3···F7Ai | 0.95 | 2.25 | 3.063 (7) | 143 |
| C4—H4···F1Aii | 0.95 | 2.54 | 3.411 (6) | 153 |
| C4—H4···F9Aii | 0.95 | 2.36 | 3.152 (9) | 140 |
| C6—H6···N1 | 0.95 | 2.52 | 2.828 (4) | 99 |
| C7—H7···Br1 | 0.95 | 2.82 | 3.233 (3) | 108 |
| C13—H13···Br1iii | 0.95 | 2.97 | 3.851 (3) | 155 |
Symmetry codes: (i) −x, −y+1, −z; (ii) x−1, y, z; (iii) x, −y+3/2, z+1/2.
Funding Statement
Funding for this research was provided by: University of Michigan-Flint Office of Research and Economic Development.
References
- Bruker (2017). APEX2 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002). Acta Cryst. B58, 389–397. [DOI] [PubMed]
- Chen, C.-L., Liu, Y.-H., Peng, S.-M. & Liu, S.-T. (2004). J. Organomet. Chem.689, 1806–1815.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst.42, 339–341.
- Dostál, L., Jambor, R., Růžička, A. & Šimon, P. (2011). Eur. J. Inorg. Chem.2011, 2380–2386.
- Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. (2016). Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed]
- Hejda, M., Dostál, L., Jambor, R., Růžička, A., Jirásko, R. & Holeček, J. (2012). Eur. J. Inorg. Chem.2012, 2578–02587.
- Hejda, M., Lork, E., Mebs, S., Dostál, L. & Beckmann, J. (2017). Eur. J. Inorg. Chem.2017, 3435–3445.
- Hejda, M., Lyčka, A., Jambor, R., Růžička, A. & Dostál, L. (2013). Dalton Trans.42, 6417–6428. [DOI] [PubMed]
- Joseph, M. C., Swarts, A. J. & Mapolie, S. F. (2018). Dalton Trans.47, 12209–12217. [DOI] [PubMed]
- Kaur, G. & Choudhury, A. R. (2014). Cryst. Growth Des.14, 1600–1616.
- Kingsley, N. B., Doyon, T. J. & Shephard, L. E. (2016). J. Organomet. Chem.801, 48–53.
- Kořenková, M., Jambor, R., Růžičková, Z. & Dostál, L. (2016). Inorg. Chem. Commun.69, 28–30.
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst.48, 3–10. [DOI] [PMC free article] [PubMed]
- Kremláček, V., Hyvl, J., Yoshida, W. Y., Růžička, A., Rheingold, A. L., Turek, J., Hughes, R. P., Dostál, L. & Cain, M. F. (2018). Organometallics37, 2481–2490.
- Li, F., Zhou, Y., Yang, H., Wang, Z., Yu, Q. & Zhang, F.-L. (2019). Org. Lett.21, 3692–3695. [DOI] [PubMed]
- Macrae, C. F., Sovago, I., Cottrell, S. J., Galek, P. T. A., McCabe, P., Pidcock, E., Platings, M., Shields, G. P., Stevens, J. S., Towler, M. & Wood, P. A. (2020). J. Appl. Cryst.53, 226–235. [DOI] [PMC free article] [PubMed]
- Mungwe, N., Swarts, A. J., Mapolie, S. F. & Westman, G. (2011). J. Organomet. Chem.696, 3527–3535.
- Novák, M., Bouška, M., Dostál, L., Růžička, A. & Jambor, R. (2014). Organometallics33, 6778–6784.
- Novák, M., Dostál, L., Turek, J., Alonso, M., De Proft, F., Růžička, A. & Jambor, R. (2016). Chem. A Eur. J.22, 5620–5628. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Šimon, P., Jambor, R., Růžička, A. & Dostál, L. (2013). Organometallics32, 239–248.
- Urbanová, I., Erben, M., Jambor, R., Růžička, A., Jirásko, R. & Dostál, L. (2013). J. Organomet. Chem.743, 156–162.
- Urbanová, I., Jambor, R., Růžička, A., Jirásko, R. & Dostál, L. (2014). Dalton Trans.43, 505–512. [DOI] [PubMed]
- Vrána, J., Jambor, R., Růžička, A., Lyčka, A., De Proft, F. & Dostál, L. (2013). J. Organomet. Chem.723, 10–14.
- Wardell, J. L., Wardell, S. M. S. V., Skakle, J. M. S., Low, J. N. & Glidewell, C. (2002). Acta Cryst. C58, o428–o430. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst.43, 920–925.
- Zhao, D., Gao, B., Gao, W., Luo, X., Tang, D., Mu, Y. & Ye, L. (2011). Inorg. Chem.50, 30–36. [DOI] [PubMed]
- Zhao, D., Gao, W., Mu, Y. & Ye, L. (2010). Chem. A Eur. J.16, 4394–4401. [DOI] [PubMed]
- Zhao, H., Zhang, X., Chen, K., Wu, W., Li, S., Wang, T., Huang, X., Wang, N., Zhou, L. & Hao, H. (2023). CrystEngComm25, 2600–2606.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I. DOI: 10.1107/S2056989025007650/ej2015sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989025007650/ej2015Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989025007650/ej2015Isup3.cml
CCDC reference: 2483197
Additional supporting information: crystallographic information; 3D view; checkCIF report