

Abstract
A hairpin RNA aptamer has been identified by in vitro selection against the transactivation-responsive element (TAR) of HIV-1. A nuclease-resistant N3′ → P5′ phosphoramidate isosequential analog of this aptamer also folds as a hairpin and forms with TAR a loop–loop “kissing” complex with a binding constant in the low nanomolar range as demonstrated by electrophoretic mobility-shift assays and surface plasmon resonance experiments. The key structural determinants, which contribute to the stability of the RNA aptamer–TAR complex, loop complementarity and the GA residues closing the aptamer loop, remain crucial for the N3′ → P5′ aptamer–TAR complex. Moreover, the N3′ → P5′ phosphoramidate aptamer specifically interferes with the binding of a peptide derived from the transactivator protein (Tat) peptide to TAR and selectively inhibits the Tat-mediated transcription in an in vitro assay, which marks this nuclease-resistant aptamer as a relevant candidate for experiments in cells.
Various RNA motifs play key roles in the regulation of gene expression. Thus, RNAs are involved in many regulatory processes including transcription, nucleocytoplasmic transport, RNA degradation, and translation through highly specific interactions with other RNAs, DNAs, or proteins (1). It was shown that transactivation of the transcription of the HIV-1 genome requires binding of the viral protein Tat to a structured RNA segment called the transactivation-responsive element (TAR). This element is transcribed from the R region of the long terminal repeat of the HIV-1 genome and present at the 5′ and 3′ ends of neo-synthesized viral mRNAs (for reviews see refs. 2–5). TAR, functional as a nascent RNA, folds into a stable hairpin with a tripyrimidine bulge and serves as a binding site for Tat. In the absence of Tat, initiation of the transcription is efficient, but the RNA polymerase II disengages rapidly from the template, leading to premature termination of HIV-1 genome transcription (6). Therefore TAR RNA constitutes a good target for artificial control of the replication of HIV-1 (7).
Different compounds have been evaluated as TAR binding molecules (8). Combinatorial approaches have been used to identify peptoids able to selectively recognize TAR and specifically inhibit the interaction of TAR with Tat in biochemical assays. In addition, this compound displayed antiviral properties in tissue culture (9). Recently, we used the RNA-based SELEX approach to identify efficient and selective DNA and RNA aptamers interacting with TAR (10, 11). The RNA aptamers with highest affinity for TAR fold as hairpins and interact with the RNA target primarily through loop–loop complementarity. The resulting kissing complex is further stabilized by stacking interactions by both stems through the loop–loop helix. Additional and crucial stabilizing interactions involve the GA residues closing the aptamer loop (12).
Feng and Holland (13) have shown that mutations in the apical loop of TAR that do not interfere with Tat binding modulate the transactivation, suggesting that the loop region acts as a binding site for essential cellular cofactors. Recent studies have indeed demonstrated that the transactivation domain of Tat interacts with the cyclin subunit (CycT1) of the positive transcription elongation factor (P-TEFb) complex and induces loop sequence-specific binding of the P-TEFb complex to TAR RNA (14, 15). Therefore, the selected RNA aptamers strongly interacting with the apical loop of the TAR hairpin could disrupt the ternary TAR–Tat–CycT1 complex, leading to abortive RNA synthesis. However, in a cellular environment RNAs are rapidly degraded by nucleases.
Numerous chemically modified oligonucleotides antisense to the TAR stem loop and with increased resistance to nuclease have shown activity in inhibition of Tat-dependent transactivation (16–19). Synthesis of N3′ → P5′ phosphoramidate oligodeoxynucleotides (NP-DNA), which are resistant to digestion by snake venom phosphodiesterase and by nucleases in HeLa cell nuclear extract, has been described (20, 21). NMR experiments showed that a NP-DNA duplex d(CGCGAATTCGCG)2 adopts an A-type helix conformation in solution (22), suggesting that NP-DNA could be used as RNA mimetics. To ascertain this possibility, a NP-DNA version of the anti-TAR RNA hairpin aptamer was synthesized.
We demonstrate here that the NP-DNA analog interacts specifically with the TAR of HIV-1 with an affinity of 1.5–3.4 nM. This analog likely forms with the RNA target a kissing complex, the overall conformation of which is close to that formed by the selected RNA sequence. Moreover, this nuclease-resistant aptamer derivative competes with binding to TAR of a Tat-derived peptide that includes the central basic region of the viral protein necessary for TAR RNA recognition (23) and inhibits the HIV-1 Tat-dependent in vitro transactivation with an IC50 of about 400 nM.
Materials and Methods
Oligonucleotides.
RNA molecules including the biotinylated mini-TAR RNA aptamer (Fig. 1) were purchased either from Xeragon (Zürich) and Eurogentec (Brussels) or were synthesized on an Expedite 8908 synthesizer (Applied Biosystems). Oligonucleotide N3′ → P5′ deoxyphosphoramidates were prepared as described (20, 21). All oligonucleotides were purified by electrophoresis on denaturing 20% polyacrylamide, 7 M urea gels and desalted on Sephadex G-25 spin columns.
Figure 1.

R0624 aptamers targeted to the HIV-1 mini-TAR RNA. (A) Sequence and secondary structure of the R0624 aptamer and mini-TAR RNAs used in this study. BRU or MAL refer to two HIV-1 strains differing by apical loop sequences (the mutated base is in italics). The consensus octamer obtained from in vitro selection (11) and the loop bases of the TAR element susceptible to base pair with the aptamer loop are shown in bold. The closing residues of the aptamer loop are underlined. The variant of R0624 in which a CU closing combination was substituted for the selected GA residues is indicated at the top (R0624 CU). (B) Schematic representation of the N3′ → P5′ deoxyphosphoramidate chemical modification.
UV Melting Experiments.
Thermal denaturation of mini-TAR with R0624 aptamer or phosphoramidate analogs were performed as described (12) with 1 μM final concentration of each oligomer in 20 mM cacodylate buffer, pH 7.3 at 20°C, containing 140 mM potassium chloride, 20 mM sodium chloride, and 0.3 mM magnesium chloride. Denaturation of the samples was achieved by increasing the temperature at 0.4°C/min from 5°C to 90°C. The melting temperature (Tm) was determined as the maximum of the first derivative of the UV melting curves.
Surface Plasmon Resonance Kinetic Measurements.
Surface plasmon resonance experiments were performed on a BIAcore 2000 apparatus (Biacore, Uppsala, Sweden). A total of 200–300 resonance units (RUs) of biotinylated mini-TAR RNA were immobilized on carboxymethylated dextran sensorchips (CM5, Biacore) coated with streptavidin according to the procedure described (12). Binding kinetics experiments were performed at 23°C in a 20 mM Hepes buffer, pH 7.3 at 20°C, containing 20 mM sodium acetate, 140 mM potassium acetate, and 3 mM magnesium acetate (R buffer). All oligonucleotides were prepared in this buffer and injected at a flow rate of 20 μl/min.
The kinetic parameters were determined by assuming a pseudofirst-order model according to Eqs. 1 and 2, for the association and dissociation phases, respectively,
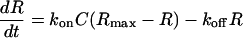 |
1 |
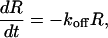 |
2 |
where R is the signal response, Rmax the maximum response level, C the molar concentration of the injected oligonucleotide, kon the association rate constant, and koff the dissociation rate constant.
Electrophoretic Mobility-Shift Assays (EMSAs).
Dissociation constants (Kd) for mini-TAR RNA-R0624 complexes were determined at room temperature. A total of 0.5 nM of [32P] 5′ end-labeled mini-TAR was incubated with increasing concentration of R0624 for 15 min at 23°C in R buffer. Binding reactions were loaded onto running native gels [15% (wt/vol) acrylamide and 75:1 acrylamide/bis(acrylamide) in 50 mM Tris-acetate (pH 7.3 at 20°C), 3 mM magnesium acetate] equilibrated at 4°C and electrophoresed overnight under 10 V/cm at 4°C. Complexes were quantitated with an Instant Imager apparatus (Hewlett–Packard). The Kd was deduced from data-point fitting with KALEIDAGRAPH 3.0 (Abelbeck Software, Reading, PA), according to Eq. 3,
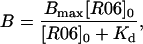 |
3 |
where B is the proportion of complex, Bmax the maximum of complex formed, and [R06]0 is the R0624 total concentration.
Aptamer/Tat Competition Assays.
32P-labeled mini-TAR (2 nM) was incubated with 100 nM of Tat peptide, Tat36(37–72) (Neosystem, Strasbourg, France) and with increasing concentrations of R0624 NP-DNA aptamer in R buffer containing 0.01% Triton X-100 and 1 mM DTT at 4°C for 30 min. Complexes were loaded onto running native gels [10% (wt/vol) acrylamide and 75:1 acrylamide/bis(acrylamide) in 44.5 mM Tris-borate (pH 8.3 at 20°C), 20 μM magnesium acetate] and electrophoresed for 2 h at 12 W, at 4°C. The gel was dried and visualized by autoradiography.
Tat-Dependent Transcription Assays.
Cell-free transcription assays were carried out essentially as described (16, 17). Before the transcription assays, modified aptamers were heated 1 min at 85°C in 20 mM Hepes (pH 7.9 at 20°C), 140 mM KCl, 3 mM MgCl2, 2 mM DTT, and 10 μM ZnSO4, and put on ice for 15 min. In vitro transcription reaction contained 15 μl HeLa cell nuclear extract, 20 mM Hepes (pH 7.9), 80 mM KCl, 3 mM MgCl2, 10 nM DNA template, 10 μM ZnSO4, 2 mM DTT, 10 mM creatine phosphate, 100 μg⋅ml−1 creatine kinase, 1 μg poly[d(I-C)], 50 μM ATP, GTP, and CTP, 5 μM UTP, [α-32P]UTP (10 μCi), 1 unit⋅μl−1 Rnasin, 200 ng recombinant Tat protein, and aptamers at increasing concentration. The samples were incubated at 30°C for 20 min. Transcription reaction was stopped with 50 μl of 150 mM sodium acetate solution, 0.5% SDS, 10 mM EDTA, and 20 μg⋅ml−1 tRNA. The transcripts were extracted by phenol/chloroform followed by ethanol precipitation. The reaction products were then analyzed on 6% polyacrylamide, 7 M urea gels and revealed by autoradiography. The full-length transcripts were quantified with National Institutes of Health IMAGE 1.62 software.
Results
Design of R0624-Derived Aptamer Sequences.
In vitro selection of RNA candidates, within a library of about 1015 sequences with a 60-nt variable region, against the TAR RNA structure of HIV-1 generated hairpins recognizing the RNA target through loop–loop interactions (11). Aptamer designed as R0624 and exhibiting the highest affinity for TAR RNA was chosen to evaluate the effects of chemical modifications on its properties. R0624 displayed the 5′-GUCCCAGA-3′ consensus sequence characteristic for the selected aptamers, the six central bases of which are complementary to the TAR loop (Fig. 1A). In this study, mini-TAR, a 27-nt long hairpin, which contains the minimal motif for in vivo responsiveness (24), was used instead of the full-length TAR. The selection was carried out against the TAR motif of the BRU strain of HIV-1. The MAL variant differs in particular by a U → C substitution in the loop (Fig. 1A). An aptamer selective for TAR MAL was previously derived from the selected candidates by introducing A → G compensatory mutation in the aptamer loop (11). Mini-TAR and R0624 are named BRU or MAL depending on their loop sequence. When unspecified, mini-TAR and R0624 refer to the BRU version as described (11, 12). R0624 2′-deoxy oligonucleotide N3′ → P5′ phosphoramidate (R0624 NP-DNA) was investigated in this study (Fig. 1B).
Melting Transition of TAR–Aptamer Complexes.
Complex formation was first characterized by thermal denaturation experiments monitored by UV absorption spectroscopy. As observed with mini-TAR and R0624 RNA hairpins, the NP-DNA analog displays a single transition (Tm > 80°C) independent of the oligonucleotide concentration (Fig. 2A). This finding indicates that it folds as a hairpin likely similar to the parent RNA aptamer. Therefore, the consensus 8-mer sequence is offered to the target TAR RNA hairpin in a structural context appropriate for loop–loop interaction. The effect of magnesium on kissing complexes is well documented (25–27) and was extensively characterized for the TAR–R0624 RNA complex: magnesium ions are required for stable loop–loop interactions (12). The stability of the bimolecular complexes decreases with the magnesium concentration but that of the corresponding monomolecular hairpins is not as sensitive. We took advantage of this fact to avoid overlapping of the monomolecular and bimolecular melting transitions. The melting experiments were then performed at 0.3 mM Mg2+ even though the selection was carried out at 3 mM. Two melting transitions were observed for complexes between mini-TAR and R0624 either in its RNA or NP-DNA form (Fig. 2B): a first one above 60°C resulted from the melting of the hairpin stems, whereas the second one below 40°C reflected the melting of bimolecular complexes (Table 1).
Figure 2.

UV-monitored melting transition of aptamer–mini-TAR complexes. (A) First derivative melting curves of individual mini-TAR RNA and R0624 variants: mini-TAR (thin line), R0624 RNA (dotted line), and NP-DNA (bold line). (B) First derivative melting curves of mini-TAR complex with R0624 variants: RNA (dotted line) and NP-DNA (bold line). Experiments were performed with 1 μM of each oligomer in 20 mM cacodylate buffer, pH 7.3 at 20°C, with 140 mM potassium chloride, 20 mM sodium chloride, and 0.3 mM magnesium chloride.
Table 1.
Tm, equilibrium, and rate constants for aptamer–mini-TAR complexes
| + Mini-TAR Bru
|
||||
|---|---|---|---|---|
| Tm, °C | kon, × 104 M−1⋅s−1 | koff, × 10−4 s−1 | Kd, nM | |
| R0624 RNA | 30.3 ± 1.4 | 3.9 ± 0.9 | 2.6 ± 0.5 | 6.9 ± 2.9 |
| R0624NP-DNA | 28.6 ± 0.4 | 10.2 ± 0.1 | 3.5 ± 0.3 | 3.4 ± 0.3 |
UV transitions of aptamer–mini-TAR complexes and individual hairpins were monitored as described in Materials and Methods in a buffer containing 0.3 mM Mg2+. Tms are the average and standard deviation of at least three independent experiments. Kd was calculated as koff/kon. Kd, kon, and koff are the average of five sensorgrams: surface plasmon resonance experiments were performed in R buffer (3 mM Mg2+) at 23°C as described in Materials and Methods.
N3′ → P5′ Phosphoramidate and RNA Aptamers Display a Similar Kinetic Behavior.
Surface plasmon resonance was used to follow the interaction of the sensor chip-immobilized mini-TAR with the different aptamers in the BRU series. Sensorgrams obtained with R0624 RNA or NP-DNA aptamer injected over the mini-TAR-functionalized surface are presented in Fig. 3. The elementary rate constants were obtained from direct curves fitting of the sensorgrams assuming a pseudofirst-order model. The NP-DNA aptamer–mini-TAR complex is slightly more stable than the RNA one because of a faster association process: kon was equal to 10.2 × 104 and 3.9 × 104 M−1⋅s−1 for NP-DNA–mini-TAR and RNA–mini-TAR complexes, respectively (Table 1).
Figure 3.

Sensorgrams of R0624–mini-TAR complexes. Increasing concentration of R0624, RNA (A) or NP-DNA (B) as indicated by the arrows were injected on a mini-TAR-functionalized sensor chip. Elementary rate constants, kon and koff, for bimolecular complex formation were deduced from direct fitting of these plots according to Eqs. 1 and 2 (see Materials and Methods). Experiments were carried out in R buffer (3 mM Mg2+) at 23°C.
Chemically Modified Aptamers Bind Specifically to Mini-TAR RNA.
Binding constants of R0624 RNA or NP-DNA with mini-TAR were determined by using EMSAs in the presence of 3 mM magnesium ions in the gel. Formation of the complex with radiolabeled mini-TAR results in the appearance of a single slowly moving band characteristic of a (1:1) complex stoichiometry (Fig. 4A). The apparent dissociation constants (Kd) for the R0624 RNA–mini-TAR complex, 6.2 ± 0.9 nM (Table 2), determined by direct fitting of the titration curves to Eq. 3 (Fig. 4B), agrees well with the one found previously in solution (2.0 ± 0.4 nM) (12). Binding affinity for the complexes with the NP-DNA analog is in the low nanomolar range as well (Kd = 1.5 ± 0.1 nM). In contrast, the unmodified DNA aptamer binds very poorly (Kd > 1,000 nM, data not shown). The stability of complexes formed by the BRU and MAL mini-TAR targets with the RNA and NP-DNA compounds (Table 2) resides primarily in the Watson–Crick base pairs involved in the loop–loop interaction (12). Indeed mismatched BRU/MAL complexes are less stable than the matched ones (Table 2): regardless of the oligonucleotide chemistry, the BRU aptamer hardly binds to mini-TAR MAL (Kd > 1,000 nM) because of the substitution of an A-U pair by an AC mismatch. The crossed complexes aptamers MAL/mini-TAR BRU are less unstable (Kd = 92 ± 17 nM in the RNA series), which is in good agreement with a GU mismatch being thermodynamically more stable than an AC one. If the loop complementarity between the aptamer and mini-TAR is restored, the complex with MAL is as stable as the one with BRU for either RNA or NP-DNA aptamers.
Figure 4.

Analysis of R0624–mini-TAR complexes at 23°C by EMSAs. (A) Radiolabeled mini-TAR was incubated in R buffer at 23°C with increasing amounts of R0624 variants as indicated above each lane (nM). Thin and bold arrows to the left indicate free mini-TAR and mini-TAR–R0624 complexes, respectively. (B) The percentage of the complex formed with mini-TAR [Bound (%)] was determined by Instant Imager analysis and binding constants (Kd) were deduced from direct fitting of the curves according to Eq. 3.
Table 2.
Apparent dissociation constants Kd (nM) of complexes formed by R0624 aptamers, in the RNA or NP-DNA series, with mini-TAR variants
| R0624 | Mini-TAR BRU | Mini-TAR MAL |
|---|---|---|
| RNA | ||
| BRU | 6.2 ± 0.9 | >1,000 |
| MAL | 92 ± 17 | 6.6 ± 1.5 |
| CU | 634 ± 94 | >1,000 |
| NP-DNA | ||
| BRU | 1.5 ± 0.1 | >1,000 |
| MAL | 12.3 ± 1.3 | 1.7 ± 0.2 |
| CU | 114 ± 38 | >1,000 |
Kd determined by EMSA at 23°C as described in Materials and Methods are the average and standard deviation of two or three experiments.
We also tested a mutation of the GA “pair” closing the loop of the aptamer that was previously shown to be crucial for the loop–loop complex stability (12). As described for RNA–RNA complexes (11, 12), the GA to CU substitution dramatically decreases the stability of the R0624 NP-DNA/mini-TAR complex: the Kd value for the CU variant is lowered by 2 orders of magnitude for the NP-DNA analog as well as for the RNA parent aptamer (Table 2). Therefore the loop closing GA combination of the NP-DNA is also crucial for the complex stability.
In Vitro Inhibition of Tat Binding by R0624 NP-DNA Aptamer.
We used a Tat peptide (Tat36) containing the amino acid residues 37–72, which includes the major part of the conserved hydrophobic core and the entire glutamine region of the Tat protein (HIV-1 BRU strain). This peptide was shown to mimic major properties of the intact protein for binding to TAR RNA (23). EMSA with the aptamer–TAR RNA complex are usually performed at 3 mM of magnesium ions. Tat–TAR interaction cannot be detected by EMSA under this condition. Studies of Tat peptide–TAR complexes by EMSA were actually generally performed without magnesium in the gel (23, 28). This effect was further characterized in solution (29). Magnesium concentration above 10 mM prevents Tat binding to TAR but the interaction is relatively unaffected by concentrations up to 1 mM. A systematic band-shift assay study was then conducted to determine the concentration allowing the binding of both aptamers and Tat to TAR. Both complexes could be detected at 20 μM Mg2+ (Fig. 5, lanes 2 and 3). Despite the reduced affinity of the R0624 NP-DNA compared with that under in vitro selection conditions (50% of band target RNA at 2 μM of aptamer) we observed specific inhibition of Tat binding to TAR (Fig. 5). The addition of increasing amounts of R0624 NP-DNA reduced the intensity of the band corresponding to the TAR–Tat peptide complex (Fig. 5, lanes 4–7). The presence of 500 nM R0624 NP-DNA aptamer caused almost complete dissociation of the Tat peptide–mini-TAR BRU complex. A much less efficient competition was observed when the aptamer with the CU closing combination was substituted to the “wild-type” GA sequence (Fig. 5, lanes 9–12). At high concentration of the CU variant (2 μM) we observed an inhibition of Tat–TAR interaction that was related to nonspecific interactions with the Tat peptide as no band corresponding to the TAR–aptamer complex was detected (Fig. 5, lane 12).
Figure 5.

Inhibition of Tat–TAR interaction by R0624 NP-DNA aptamers. The R0624 NP-DNA aptamer was used with either GA (Left) or CU closing nucleotides (Right). 32P-labeled mini-TAR (2 nM) was run in the absence (lane 1) or presence of 100 nM of Tat peptide (Tat36; lanes 2, 4–7, and 9–12). Aptamer variants were added either to mini-TAR alone (lanes 3 and 8) or the mini-TAR–Tat peptide complex at the concentration (nM) indicated at the top of each lane. Experiments were analyzed on a 10% polyacrylamide gel in Tris-borate buffer, pH 8.3 at 20°C, containing 20 μM magnesium acetate, after incubation in R buffer as described in Materials and Methods.
Inhibition of Tat-Dependent Transcription by Modified Kissing Aptamers.
The ability of the NP-DNA analog to inhibit the in vitro transcription of a DNA template containing the HIV-1 long terminal repeat (strain NL4–3) in the presence of HeLa cell nuclear extract and of the HIV Tat protein was then investigated (Fig. 6). Increasing the concentration of R0624 NP-DNA(GA) decreased the production of the ρ and τ transcripts with an IC50 of about 400 nM (Fig. 6A). The observed effect is specific as the NP-DNA(CU) variant does not significantly inhibit the transcription compared with the NP-DNA(GA) aptamer (Fig. 6B). The low inhibitory effect observed with the RNA aptamer (IC50 > 4 μM) likely results from rapid degradation of the RNA aptamer by nucleases present in the HeLa nuclear extract (data not shown). The specific inhibitory effect of R0624 on the in vitro transcription was further assessed by using a DNA control template containing a cytomegalovirus promoter. None of the NP-DNA aptamers inhibits the transcription of this control template even at a concentration of 4 μM (data not shown).
Figure 6.

Inhibition of Tat-dependent in vitro transcription in HeLa cell nuclear extract. (A) HIV-1 template containing long terminal repeat (LTR) obtained from plasmid p10SLT (34) showing two termination sites (τ and ρ) responsible for the synthesis of two transcripts 325 and 524 nt long. (B) Autoradiograph of transcription products produced from the HIV template in the presence (lane 1) or absence (lane 2) of Tat and of increasing concentration of phosphoramidate aptamers GA (lanes 3–8) and CU (lanes 9–13). The run-off (ρ) and internal artificially terminated (τ) products are marked. (C) Normalized amounts of full-length transcripts as a function of aptamer concentration: NP-DNA(GA) (bold line), NP-DNA(CU) (dotted line).
Discussion
Hydrolytic cleavage by cellular nucleases drastically restricts the use of RNA as a therapeutic agent (30). Numerous analogs that circumvent this problem have been proposed, and several promising candidates have been recently described for antisense studies (31). One way to increase RNA resistance is to modify the internucleoside phosphodiester bond by introducing N3′ → P5′ phosphoramidate linkages. We investigated the properties of a NP-DNA analog of RNA aptamer, R0624, raised against the TAR RNA hairpin of HIV-1 (11). This modification was shown to be thermodynamically favorable with respect to duplex formation with complementary RNA strands that mimic the conformation of RNA–RNA complexes.
As previously reported (12), there is no direct correlation between the thermal stability of the aptamer stem and the complex formed with the TAR RNA. Indeed, despite the high stability of the NP-DNA analog (Tm > 80°C; Fig. 2) over the RNA aptamer, the R0624 NP-DNA–TAR complex is slightly less stable than the parent one. NMR studies of two different RNA kissing complexes showed a continuous stacking from one hairpin stem to the other one, through the loop–loop helix (27, 32). The loop complementarity is a key feature for RNA kissing complex formation. Indeed, all mismatched RNA–RNA TAR–aptamer complexes formed between BRU and MAL hairpins are destabilized compared with the matched BRU–BRU and MAL–MAL complexes, which is likely caused by the presence of a non-Watson–Crick base pair in the loop–loop duplex. This finding is also true for the NP-DNA aptamer–TAR complexes. The nucleotides closing the loop of this phosphoramidate aptamer are crucial for kissing complex stability as for the parent RNA aptamer. The replacement of the selected GA by CU nucleotides is thermodynamically unfavorable, leading to a ΔΔG°23°C loss of +10.7 kJ/mol in the case of the NP-DNA–BRU complex. All of the structural determinants that have been shown to play a key role for RNA–RNA kissing complex stability (namely a loop complementary to the TAR loop, closed by GA residues, followed by a stable double-stranded stem) therefore are also crucial for the complex formed by the N3′ → P′5 phosphoramidate aptamer and mini-TAR. This finding suggests that both RNA–RNA and RNA–NP-DNA complexes may adopt close overall conformations at least at the aptamer stem/loop–loop helix junctions, keeping similar stacking interaction, thus accounting for comparable thermodynamic stability. This conclusion is further supported by CD experiments on both complexes (data not shown). Only slight differences in CD spectra are observed, which likely originate from those reported in the structure of an A-type helix NP-DNA duplex (22).
The binding sites for the R0624 aptamer and the Tat peptide on TAR RNA do not overlap: the aptamer binds to the 6-nt apical loop whereas the Tat protein recognizes the pyrimidine bulge and adjacent nucleic acid bases. Nevertheless the NP-DNA aptamer specifically competes with Tat binding to the mini-TAR target through a teleospecific effect (Fig. 5B). R0624 NP-DNA binding likely induces or prevents conformational changes that interfere with Tat binding to the bulge. Indeed, structural studies have shown that binding of Tat to TAR reduces the conformational flexibility of the TAR RNA and induces structural rearrangements in the trinucleotide bulge, resulting in new contacts between Tat and the nucleoside adjacent to the bulge (33). The Tat peptide is actually sensitive to the geometry of the TAR loop, even though no direct interaction is involved: its binding to a TAR element in which the three guanosines of the loop have been substituted by three adenosines is weakened (data not shown). Even though the structural features brought by the modification do not affect the stability the loop–loop interaction, binding of the NP-DNA aptamer likely induces a conformation of the TAR bulge neighborhood different from the RNA aptamer as only the former competes with the Tat peptide for TAR binding.
Recent studies have shown that the transcriptional transactivation of the HIV-1 genome required the specific binding of the positive transcription elongation factor (P-TEFb)–cyclin T1–CDK9 kinase ternary complex to the TAR loop together with Tat binding to the bulge (14). In vitro assays reported here clearly demonstrated that the NP-DNA kissing aptamer specifically inhibited the HIV-1 Tat-mediated transcription with an IC50 of about 400 nM. This finding might result from competition with both Tat to the bulge (see above) and components of the P-TEFb complex to the TAR loop. Anti-TAR oligonucleotides were shown to specifically block the interaction of Tat with TAR in vitro and inhibit the Tat-mediated transactivation of the HIV-1 transcription (16, 17, 19). Therefore, kissing aptamers that engage at most 6 bp with TAR rival longer antisense oligonucleotides complementary to both the bulge and the loop. We have shown that a compound targeted only to the loop efficiently inhibits the Tat-mediated transcription.
In conclusion, we showed that a posteriori chemical modifications of nucleic acid-based aptamer targeted to RNA structure can extend the range of molecules that interfere with interactions or processes mediated by functionally important RNA elements.
Acknowledgments
We thank Krisztina Pongracz for the help with oligonucleotide N3′ → P5′ phosphoramidates synthesis and Fabien Aussenac for technical assistance in CD experiments. We also thank Jonathan Karn and Tony Lowe for HeLa cell nuclear extract and HIV-1 Tat protein. F.D. is the recipient of a fellowship from the Agence Nationale de la Recherche sur le SIDA.
Abbreviations
- TAR
transactivation-responsive element
- EMSA
electrophoretic mobility-shift assay
- RU
resonance unit
- Tm
melting temperature
- NP-DNA
N3′ → P5′ phosphoramidate oligodeoxynucleotide
References
- 1.Toulmé J J, Di Primo C, Moreau S. Prog Nucleic Acid Res Mol Biol. 2001;69:1–46. doi: 10.1016/s0079-6603(01)69043-3. [DOI] [PubMed] [Google Scholar]
- 2.Karn J. J Mol Biol. 1999;293:235–254. doi: 10.1006/jmbi.1999.3060. [DOI] [PubMed] [Google Scholar]
- 3.Rana T M, Jeang K T. Arch Biochem Biophys. 1999;365:175–185. doi: 10.1006/abbi.1999.1206. [DOI] [PubMed] [Google Scholar]
- 4.Taube R, Fujinaga K, Wimmer J, Barboric M, Peterlin B M. Virology. 1999;264:245–253. doi: 10.1006/viro.1999.9944. [DOI] [PubMed] [Google Scholar]
- 5.Jeang K T, Xiao H, Rich E A. J Biol Chem. 1999;274:28837–28840. doi: 10.1074/jbc.274.41.28837. [DOI] [PubMed] [Google Scholar]
- 6.Kao S Y, Calman A F, Luciw P A, Peterlin B M. Nature (London) 1987;330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- 7.Wilson W D, Li K. Curr Med Chem. 2000;7:73–98. doi: 10.2174/0929867003375434. [DOI] [PubMed] [Google Scholar]
- 8.Lapidot A, Litovchick A. Drug Dev Res. 2000;50:502–515. [Google Scholar]
- 9.Hamy F, Felder E R, Heizmann G, Lazdins J, Aboul-ela F, Varani G, Karn J, Klimkait T. Proc Natl Acad Sci USA. 1997;94:3548–3553. doi: 10.1073/pnas.94.8.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boiziau C, Dausse E, Yurchenko L, Toulmé J J. J Biol Chem. 1999;274:12730–12737. doi: 10.1074/jbc.274.18.12730. [DOI] [PubMed] [Google Scholar]
- 11.Ducongé F, Toulmé J J. RNA. 1999;5:1605–1614. doi: 10.1017/s1355838299991318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ducongé F, Di Primo C, Toulmé J J. J Biol Chem. 2000;275:21287–21294. doi: 10.1074/jbc.M002694200. [DOI] [PubMed] [Google Scholar]
- 13.Feng S, Holland E C. Nature (London) 1988;334:165–167. doi: 10.1038/334165a0. [DOI] [PubMed] [Google Scholar]
- 14.Bieniasz P D, Grdina T A, Bogerd H P, Cullen B R. Proc Natl Acad Sci USA. 1999;96:7791–7796. doi: 10.1073/pnas.96.14.7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei P, Garber M E, Fang S M, Fischer W H, Jones K A. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 16.Arzumanov A, Walsh A P, Liu X, Rajwanshi V K, Wengel J, Gait M J. Nucleosides Nucleotides Nucleic Acids. 2001;20:471–480. doi: 10.1081/NCN-100002321. [DOI] [PubMed] [Google Scholar]
- 17.Arzumanov A, Walsh A P, Rajwanshi V K, Kumar R, Wengel J, Gait M J. Biochemistry. 2001;40:14645–14654. doi: 10.1021/bi011279e. [DOI] [PubMed] [Google Scholar]
- 18.Mayhood T, Kaushik N, Pandey P K, Kashanchi F, Deng L, Pandey V N. Biochemistry. 2000;39:11532–11539. doi: 10.1021/bi000708q. [DOI] [PubMed] [Google Scholar]
- 19.Mestre B, Arzumanov A, Singh M, Boulmé F, Litvak S, Gait M J. Biochim Biophys Acta. 1999;1445:86–98. doi: 10.1016/s0167-4781(99)00019-6. [DOI] [PubMed] [Google Scholar]
- 20.Nelson J S, Fearon K L, Nguyen M Q, McCurdy S N, Frediani J E, Foy M F, Hirschbein B L. J Org Chem. 1997;62:7278–7287. doi: 10.1021/jo970801t. [DOI] [PubMed] [Google Scholar]
- 21.Schultz R G, Gryaznov S M. Nucleic Acids Res. 1996;24:2966–2973. doi: 10.1093/nar/24.15.2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding D, Gryaznov S M, Wilson W D. Biochemistry. 1998;37:12082–12093. doi: 10.1021/bi980711y. [DOI] [PubMed] [Google Scholar]
- 23.Churcher M J, Lamont C, Hamy F, Dingwall C, Green S M, Lowe A D, Butler J G, Gait M, Karn J. J Mol Biol. 1993;230:90–110. doi: 10.1006/jmbi.1993.1128. [DOI] [PubMed] [Google Scholar]
- 24.Selby M J, Bain E S, Luciw P A, Peterlin B M. Genes Dev. 1989;3:547–558. doi: 10.1101/gad.3.4.547. [DOI] [PubMed] [Google Scholar]
- 25.Jossinet F, Paillart J C, Westhof E, Hermann T, Skripkin E, Lodmell J S, Ehresmann C, Ehresmann B, Marquet R. RNA. 1999;5:1222–1234. doi: 10.1017/s1355838299990982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gregorian R S, Jr, Crothers D M. J Mol Biol. 1995;248:968–984. doi: 10.1006/jmbi.1995.0275. [DOI] [PubMed] [Google Scholar]
- 27.Chang K Y, Tinoco I. J Mol Biol. 1997;269:52–66. doi: 10.1006/jmbi.1997.1021. [DOI] [PubMed] [Google Scholar]
- 28.Weeks K M, Ampe C, Schultz S C, Steitz T A, Crothers D M. Science. 1990;249:1281–1285. doi: 10.1126/science.2205002. [DOI] [PubMed] [Google Scholar]
- 29.Arzumanov A, Godde F, Moreau S, Toulmé J-J, Weeds A, Gait M J. Helv Chim Acta. 2000;83:1424–1436. [Google Scholar]
- 30.Brody E N, Gold L. J Biotechnol. 2000;74:5–13. doi: 10.1016/s1389-0352(99)00004-5. [DOI] [PubMed] [Google Scholar]
- 31.Toulmé J J. Nat Biotechnol. 2001;19:17–18. doi: 10.1038/83451. [DOI] [PubMed] [Google Scholar]
- 32.Lee A J, Crothers D M. Structure (London) 1998;6:993–1005. doi: 10.1016/s0969-2126(98)00101-4. [DOI] [PubMed] [Google Scholar]
- 33.Aboul-ela F, Karn J, Varani G. J Mol Biol. 1995;253:313–332. doi: 10.1006/jmbi.1995.0555. [DOI] [PubMed] [Google Scholar]
- 34.Keen N J, Gait M J, Karn J. Proc Natl Acad Sci USA. 1996;93:2505–2510. doi: 10.1073/pnas.93.6.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]